

β-Adrenoceptor Agonists Attenuate Thrombin-Induced Impairment of Human Lung Endothelial Cell Barrier Function and Protect the Lung Vascular Barrier during Resuscitation from Hemorrhagic Shock
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cells and Cell Culture
2.3. In Vitro Endothelial Cell Permeability Assays
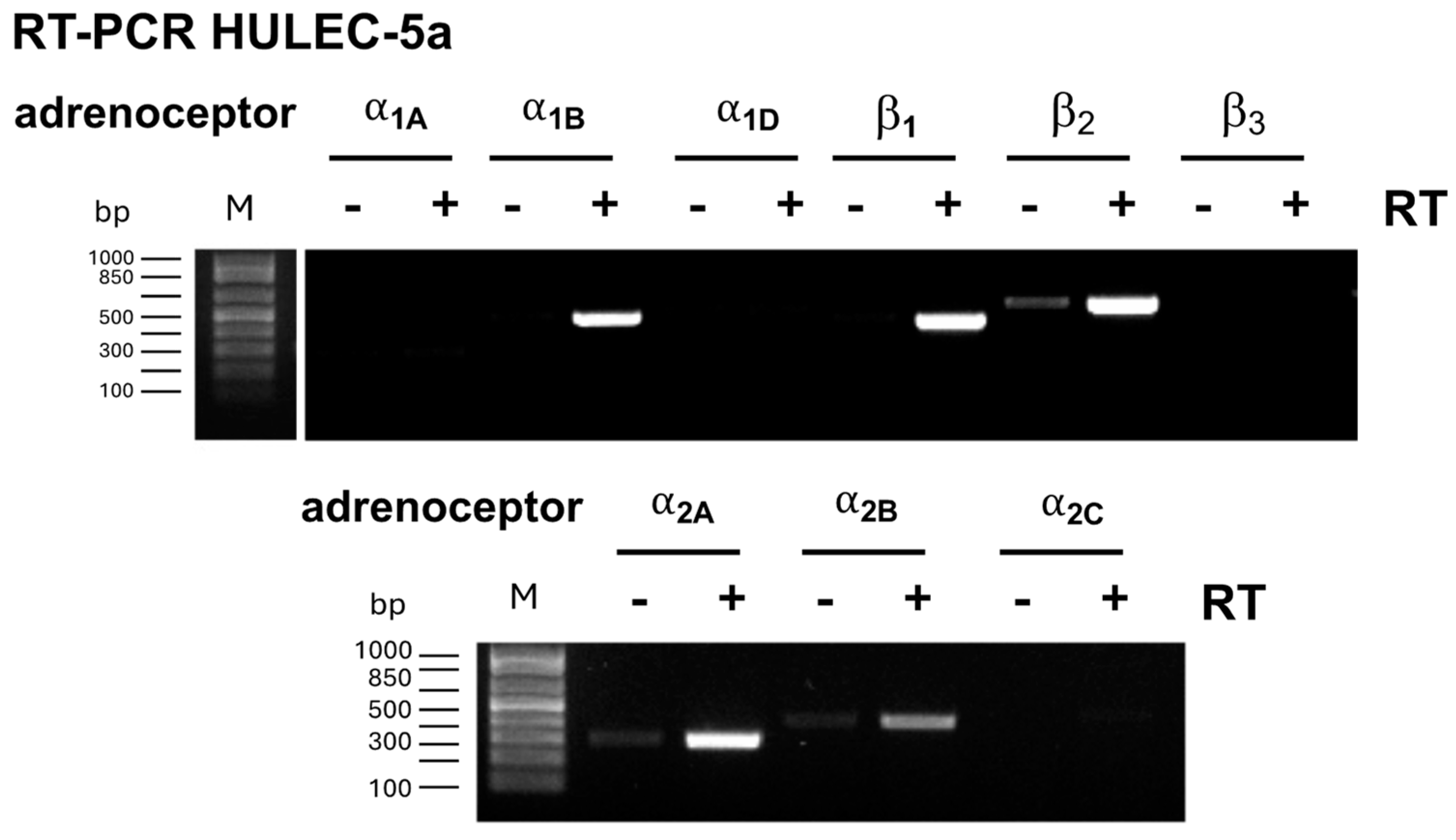
2.4. Reverse Transcription Polymerase Chain Reaction (RT-PCR)
2.5. Gαs Activation Assay
2.6. Hemorrhagic Shock and Fluid Resuscitation Model
2.7. Measurements of Evans Blue Concentrations
2.8. Data Analyses and Statistics
3. Results and Discussion
3.1. Adrenoceptor Agonists Improve Normal and Attenuate Thrombin-Induced Impairment of HULEC-5a Barrier Function
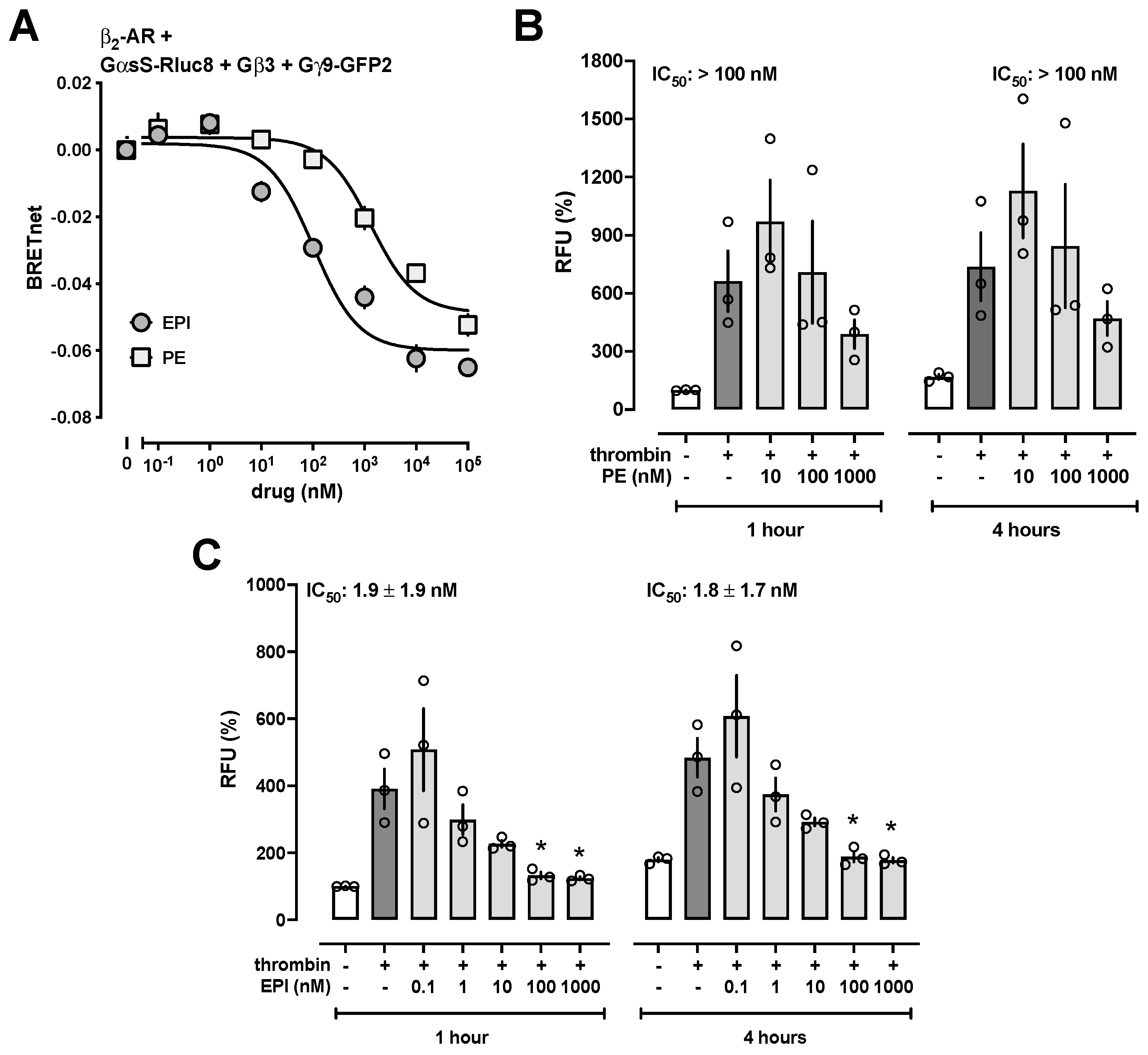
3.2. β-Adrenoceptor Agonists Attenuate Thrombin-Induced Impairment of HULEC-5a Barrier Function with High Potency
3.3. Low-Dose EPI Treatment Protects the Lung Vascular Barrier during Resuscitation from Hemorrhagic Shock
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ware, L.B. Pathophysiology of acute lung injury and the acute respiratory distress syndrome. Semin. Respir. Crit. Care Med. 2006, 27, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Farina, N.; Bixby, A.; Alaniz, C. Angiotensin II Brings More Questions Than Answers. P T 2018, 43, 685–687. [Google Scholar]
- Bendicksen, L.; Kesselheim, A.S.; Rome, B.N. The Vexing Voyage of Vasopressin: The Consequences of Granting Market Exclusivity to Unapproved Drugs. Chest 2022, 162, 433–435. [Google Scholar] [CrossRef]
- Allen, M.J.; Coleman, R.A. Beta 2-adrenoceptors mediate a reduction in endothelial permeability in vitro. Eur. J. Pharmacol. 1995, 274, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Minnear, F.L.; DeMichele, M.A.; Leonhardt, S.; Andersen, T.T.; Teitler, M. Isoproterenol antagonizes endothelial permeability induced by thrombin and thrombin receptor peptide. J. Appl. Physiol. 1993, 75, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Minnear, F.L.; DeMichele, M.A.; Moon, D.G.; Rieder, C.L.; Fenton, J.W., 2nd. Isoproterenol reduces thrombin-induced pulmonary endothelial permeability in vitro. Am. J. Physiol. 1989, 257, H1613–H1623. [Google Scholar] [CrossRef]
- Minnear, F.L.; Johnson, A.; Malik, A.B. Beta-adrenergic modulation of pulmonary transvascular fluid and protein exchange. J. Appl. Physiol. 1986, 60, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Kasa, A.; Csortos, C.; Verin, A.D. Cytoskeletal mechanisms regulating vascular endothelial barrier function in response to acute lung injury. Tissue Barriers 2015, 3, e974448. [Google Scholar] [CrossRef] [PubMed]
- Welty-Wolf, K.E.; Carraway, M.S.; Ortel, T.L.; Piantadosi, C.A. Coagulation and inflammation in acute lung injury. Thromb. Haemost. 2002, 88, 17–25. [Google Scholar]
- Glas, G.J.; Van Der Sluijs, K.F.; Schultz, M.J.; Hofstra, J.J.; Van Der Poll, T.; Levi, M. Bronchoalveolar hemostasis in lung injury and acute respiratory distress syndrome. J. Thromb. Haemost. 2013, 11, 17–25. [Google Scholar] [CrossRef]
- Babu, F.S.; LaPorte, H.M.; Nassoiy, S.P.; Majetschak, M. Chemokine (C-X-C motif) receptor 4 regulates lung endothelial barrier permeability during resuscitation from hemorrhagic shock. Physiol. Res. 2019, 68, 675–679. [Google Scholar] [CrossRef]
- Cheng, Y.H.; Eby, J.M.; LaPorte, H.M.; Volkman, B.F.; Majetschak, M. Effects of cognate, non-cognate and synthetic CXCR4 and ACKR3 ligands on human lung endothelial cell barrier function. PLoS ONE 2017, 12, e0187949. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Cheng, Y.H.; Enten, G.A.; DeSantis, A.J.; Gaponenko, V.; Majetschak, M. Regulation of the thrombin/protease-activated receptor 1 axis by chemokine (CXC motif) receptor 4. J. Biol. Chem. 2020, 295, 14893–14905. [Google Scholar] [CrossRef] [PubMed]
- McGee, M.Y.; Enten, G.A.; Boshra, S.N.; Ogunsina, O.; Gaponenko, V.; Gao, X.; Majetschak, M. Ethanol promotes protease activated receptor 1: Chemokine (C-X-C motif) receptor 4 heteromerization and enhances thrombin-induced impairment of human lung endothelial cell barrier function. Biochim. Biophys. Acta Mol. Basis Dis. 2024, 1870, 167335. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.H.J.; DiBerto, J.F.; English, J.G.; Glaudin, A.M.; Krumm, B.E.; Slocum, S.T.; Che, T.; Gavin, A.C.; McCorvy, J.D.; Roth, B.L.; et al. TRUPATH, an open-source biosensor platform for interrogating the GPCR transducerome. Nat. Chem. Biol. 2020, 16, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Enten, G.A.; McGee, M.Y.; Weche, M.; Majetschak, M. alpha(1)-adrenoceptor ligands inhibit chemokine receptor heteromerization partners of alpha(1B/D)-adrenoceptors via interference with heteromer formation. Pharmacol. Res. 2023, 190, 106730. [Google Scholar] [CrossRef] [PubMed]
- Enten, G.A.; Gao, X.; McGee, M.Y.; Weche, M.; Majetschak, M. Chemokine receptor hetero-oligomers regulate monocyte chemotaxis. Life Sci. Alliance 2024, 7, e202402657. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Majetschak, M. G protein activation via chemokine (C-X-C motif) receptor 4 and alpha(1b)-adrenoceptor is ligand and heteromer-dependent. FEBS Lett. 2023, 597, 2017–2027. [Google Scholar] [CrossRef]
- Moitra, J.; Sammani, S.; Garcia, J.G. Re-evaluation of Evans Blue dye as a marker of albumin clearance in murine models of acute lung injury. Transl. Res. 2007, 150, 253–265. [Google Scholar] [CrossRef]
- Alexander, S.P.; Christopoulos, A.; Davenport, A.P.; Kelly, E.; Marrion, N.V.; Peters, J.A.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Sharman, J.L.; et al. The Concise Guide to Pharmacology 2017/18: G protein-coupled receptors. Br. J. Pharmacol. 2017, 174 (Suppl. S1), S17–S129. [Google Scholar] [CrossRef]
- Torp, K.D.; Tschakovsky, M.E.; Halliwill, J.R.; Minson, C.T.; Joyner, M.J. beta-Receptor agonist activity of phenylephrine in the human forearm. J. Appl. Physiol. 2001, 90, 1855–1859. [Google Scholar] [CrossRef] [PubMed]
- Hakim, T.S.; Minnear, F.L.; van der Zee, H.; Barie, P.S.; Malik, A.B. Adrenoceptor control of lung fluid and protein exchange. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1981, 51, 68–72. [Google Scholar] [CrossRef]
- Vandenbroucke, E.; Mehta, D.; Minshall, R.; Malik, A.B. Regulation of endothelial junctional permeability. Ann. N. Y. Acad. Sci. 2008, 1123, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Huang, F.; Lum, H. PKA inhibits RhoA activation: A protection mechanism against endothelial barrier dysfunction. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 284, L972–L980. [Google Scholar] [CrossRef]
- Sayner, S.L. Emerging themes of cAMP regulation of the pulmonary endothelial barrier. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L667–L678. [Google Scholar] [CrossRef] [PubMed]
- Joffre, J.; Lloyd, E.; Wong, E.; Chung-Yeh, C.; Nguyen, N.; Xu, F.; Legrand, M.; Hellman, J. Catecholaminergic Vasopressors Reduce Toll-Like Receptor Agonist-Induced Microvascular Endothelial Cell Permeability But Not Cytokine Production. Crit. Care Med. 2021, 49, e315–e326. [Google Scholar] [CrossRef]
- Garcia de Lomana, A.L.; Vilhjalmsson, A.I.; McGarrity, S.; Sigurethardottir, R.; Anuforo, O.; Viktorsdottir, A.R.; Kotronoulas, A.; Bergmann, A.; Franzson, L.; Halldorsson, H.; et al. Metabolic Response in Endothelial Cells to Catecholamine Stimulation Associated with Increased Vascular Permeability. Int. J. Mol. Sci. 2022, 23, 3162. [Google Scholar] [CrossRef] [PubMed]
- Farnebo, L.O.; Hallman, H.; Hamberger, B.; Jonsson, G. Catecholamines and hemorrhagic shock in awake and anesthetized rats. Circ. Shock 1979, 6, 109–118. [Google Scholar] [PubMed]
- Lee, H.B.; Blaufox, M.D. Blood volume in the rat. J. Nucl. Med. 1985, 26, 72–76. [Google Scholar]
- Dewachter, P.; Jouan-Hureaux, V.; Lartaud, I.; Bello, G.; de Talancé, N.; Longrois, D.; Mertes, P.M. Comparison of arginine vasopressin, terlipressin, or epinephrine to correct hypotension in a model of anaphylactic shock in anesthetized brown Norway rats. Anesthesiology 2006, 104, 734–741. [Google Scholar] [CrossRef]
- Gu, X.; Simons, F.E.; Simons, K.J. Epinephrine absorption after different routes of administration in an animal model. Biopharm. Drug Dispos. 1999, 20, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Weche, M.; DeSantis, A.J.; McGee, M.Y.; Enten, G.A.; Gao, X.; Majetschak, M. Effects of chemokine (C-C motif) receptor 2 and 3 antagonists in rat models of hemorrhagic shock. PLoS ONE 2023, 18, e0284472. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McGee, M.Y.; Ogunsina, O.; Boshra, S.N.; Gao, X.; Majetschak, M. β-Adrenoceptor Agonists Attenuate Thrombin-Induced Impairment of Human Lung Endothelial Cell Barrier Function and Protect the Lung Vascular Barrier during Resuscitation from Hemorrhagic Shock. Biomedicines 2024, 12, 1813. https://doi.org/10.3390/biomedicines12081813
McGee MY, Ogunsina O, Boshra SN, Gao X, Majetschak M. β-Adrenoceptor Agonists Attenuate Thrombin-Induced Impairment of Human Lung Endothelial Cell Barrier Function and Protect the Lung Vascular Barrier during Resuscitation from Hemorrhagic Shock. Biomedicines. 2024; 12(8):1813. https://doi.org/10.3390/biomedicines12081813
Chicago/Turabian StyleMcGee, Michelle Y., Ololade Ogunsina, Sadia N. Boshra, Xianlong Gao, and Matthias Majetschak. 2024. "β-Adrenoceptor Agonists Attenuate Thrombin-Induced Impairment of Human Lung Endothelial Cell Barrier Function and Protect the Lung Vascular Barrier during Resuscitation from Hemorrhagic Shock" Biomedicines 12, no. 8: 1813. https://doi.org/10.3390/biomedicines12081813
APA StyleMcGee, M. Y., Ogunsina, O., Boshra, S. N., Gao, X., & Majetschak, M. (2024). β-Adrenoceptor Agonists Attenuate Thrombin-Induced Impairment of Human Lung Endothelial Cell Barrier Function and Protect the Lung Vascular Barrier during Resuscitation from Hemorrhagic Shock. Biomedicines, 12(8), 1813. https://doi.org/10.3390/biomedicines12081813