
Advances in Melanoma: From Genetic Insights to Therapeutic Innovations
 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Molecular Mechanisms
2.1. MAPK Signaling Pathway
2.2. PI3K/Akt/mTOR Signaling Pathway
2.3. Cell Cycle Dysregulation
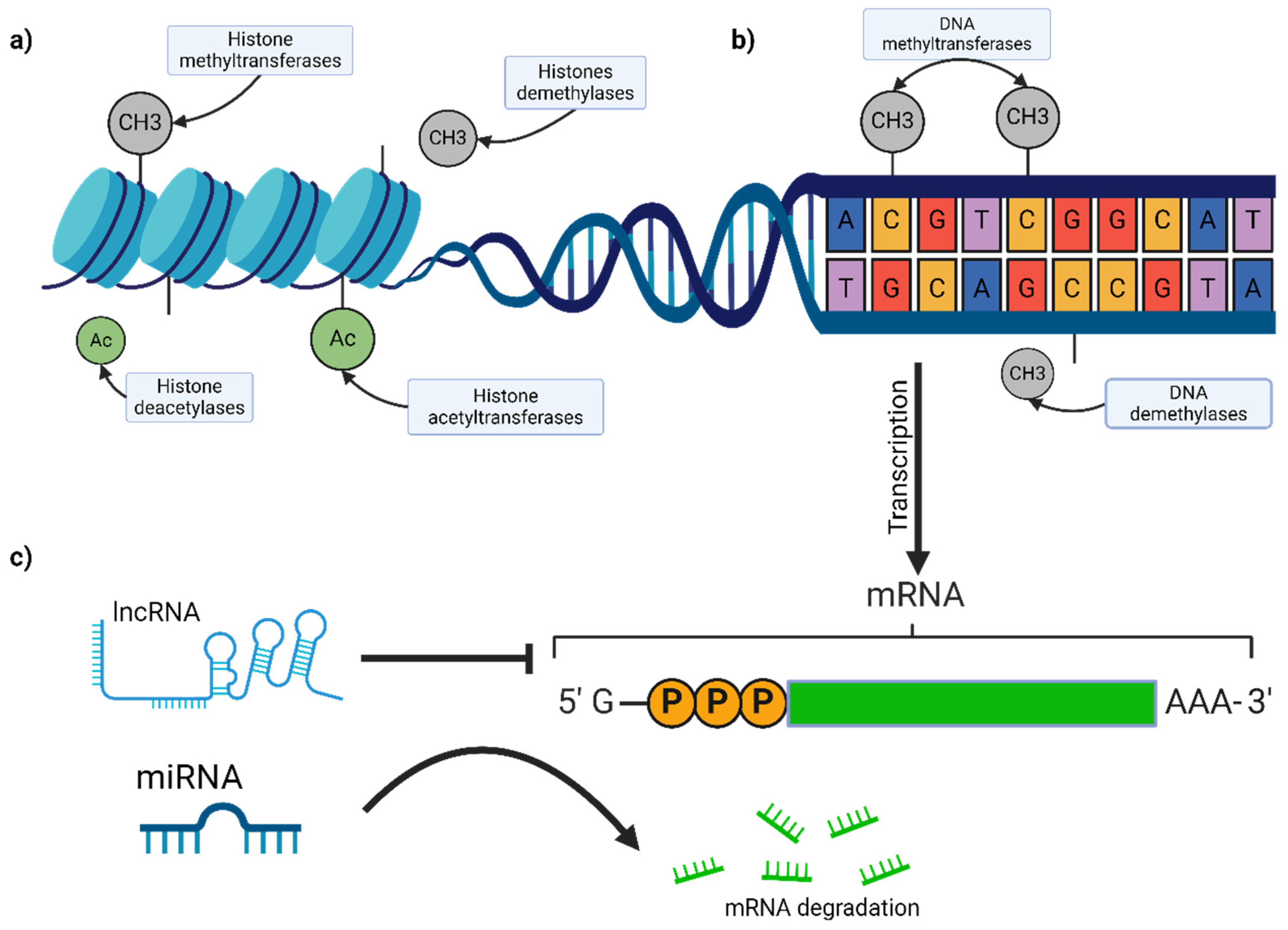
2.4. Epigenetic Modifications
2.4.1. DNA Methylation
2.4.2. Histone Modifications
2.4.3. Non-Coding RNAs
MicroRNAs (miRNAs)
Long Non-Coding RNAs (lncRNAs)
- MALAT1 (Metastasis-Associated Lung Adenocarcinoma Transcript 1): MALAT1 promotes melanoma metastasis through interactions with chromatin-modifying complexes and the regulation of gene expression involved in cell migration and invasion. It modulates the alternative splicing of pre-mRNAs, influencing cellular processes critical for tumor progression [69,70,71].
- Survival-Associated Mitochondrial Melanoma-Specific Oncogenic Non-Coding RNA (SAMMSON): A melanoma-specific lncRNA, SAMMSON, is essential for melanoma cell survival. It interacts with p32, a mitochondrial protein, to regulate mitochondrial function and energy production, highlighting its potential as a therapeutic target [74,75].
2.4.4. Implications for Diagnosis and Treatment
2.5. Tumor Microenvironment
2.5.1. Immune Cells
Tumor-Infiltrating Lymphocytes (TILs)
Macrophages
Myeloid-Derived Suppressor Cells (MDSCs)
Dendritic Cells
2.5.2. Stromal Cells
Cancer-Associated Fibroblasts (CAFs)
Endothelial Cells
Adipocytes
2.5.3. Extracellular Matrix (ECM)
Matrix Metalloproteinases (MMPs)
Integrins
2.5.4. Soluble Factors
Cytokines and Chemokines
Growth Factors
2.6. Heterogeneity and Plasticity
2.6.1. Genetic Heterogeneity
Clonal Evolution
Therapeutic Resistance
2.6.2. Phenotypic Plasticity
Epithelial–Mesenchymal Transition (EMT)
Stem Cell-like Properties
3. Other Genetic Biomarkers
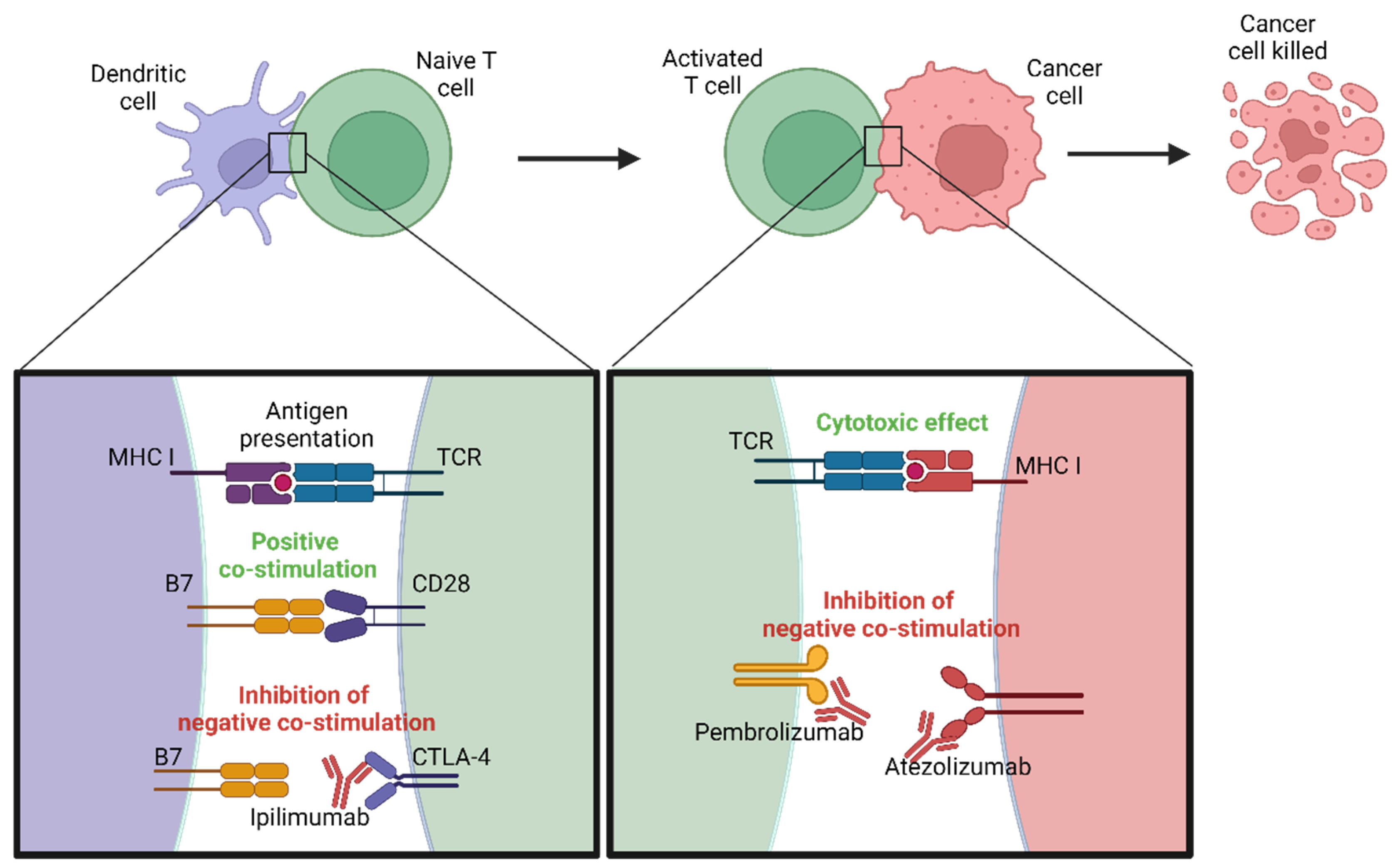
4. Immunotherapy
4.1. Immune Checkpoint Inhibitors
4.1.1. PD-1/PD-L1 Inhibitors
4.1.2. CTLA-4 Inhibitors
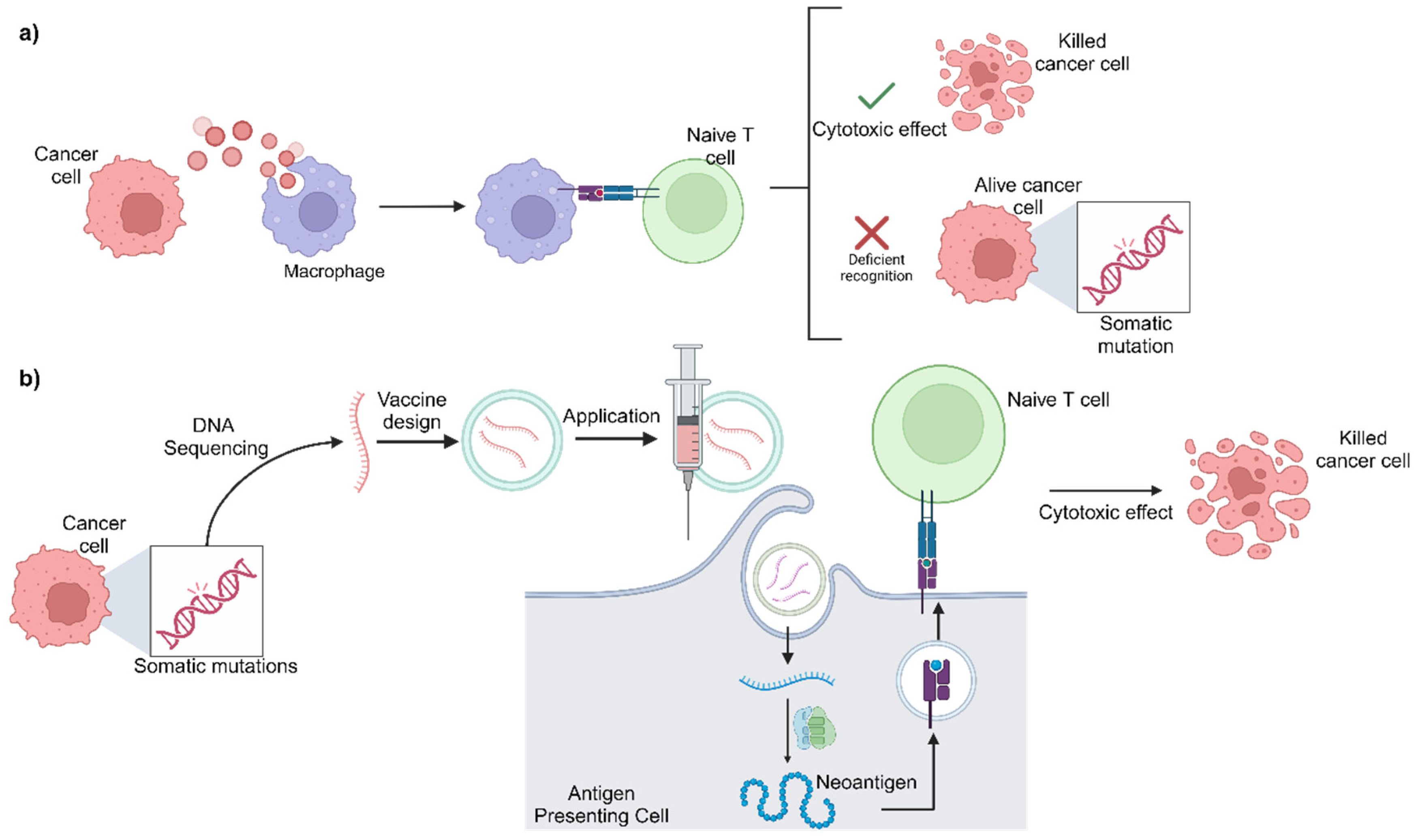
4.2. mRNA Vaccines
5. Emerging Technologies
6. Future Directions
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Switzer, B.; Puzanov, I.; Skitzki, J.J.; Hamad, L.; Ernstoff, M.S. Managing Metastatic Melanoma in 2022: A Clinical Review. JCO Oncol. Pract. 2022, 18, 335–351. [Google Scholar] [CrossRef]
- Teixido, C.; Castillo, P.; Martinez-Vila, C.; Arance, A.; Alos, L. Molecular Markers and Targets in Melanoma. Cells 2021, 10, 2320. [Google Scholar] [CrossRef]
- Ferlay, J.; Ervik, M.; Lam, F.; Laversanne, M.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; et al.; Global Cancer Observatory: Cancer Today Lyon, France: International Agency for Research on Cancer. 2024. Available online: https://gco.iarc.who.int/today (accessed on 22 June 2024).
- Gosman, L.M.; Țăpoi, D.-A.; Costache, M. Cutaneous Melanoma: A Review of Multifactorial Pathogenesis, Immunohistochemistry, and Emerging Biomarkers for Early Detection and Management. Int. J. Mol. Sci. 2023, 24, 15881. [Google Scholar] [CrossRef]
- Laskar, R.; Ferreiro-Iglesias, A.; Bishop, D.T.; Iles, M.M.; Kanetsky, P.A.; Armstrong, B.K.; Law, M.H.; Goldstein, A.M.; Aitken, J.F.; Giles, G.G.; et al. Risk factors for melanoma by anatomical site: An evaluation of aetiological heterogeneity. Br. J. Dermatol. 2021, 184, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Saginala, K.; Barsouk, A.; Aluru, J.S.; Rawla, P.; Barsouk, A. Epidemiology of Melanoma. Med. Sci. 2021, 9, 63. [Google Scholar] [CrossRef]
- Zambrano-Román, M.; Padilla-Gutiérrez, J.R.; Valle, Y.; Muñoz-Valle, J.F.; Valdés-Alvarado, E. Non-Melanoma Skin Cancer: A Genetic Update and Future Perspectives. Cancers 2022, 14, 2371. [Google Scholar] [CrossRef]
- Woo, Y.R.; Cho, S.H.; Lee, J.D.; Kim, H.S. The Human Microbiota and Skin Cancer. Int. J. Mol. Sci. 2022, 23, 1813. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Pellegrini, C.; Cardelli, L.; Ciciarelli, V.; Di Nardo, L.; Fargnoli, M.C. Familial Melanoma: Diagnostic and Management Implications. Dermatol. Pract. Concept. 2019, 9, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Zocchi, L.; Lontano, A.; Merli, M.; Dika, E.; Nagore, E.; Quaglino, P.; Puig, S.; Ribero, S. Familial Melanoma and Susceptibility Genes: A Review of the Most Common Clinical and Dermoscopic Phenotypic Aspect, Associated Malignancies and Practical Tips for Management. J. Clin. Med. 2021, 10, 3760. [Google Scholar] [CrossRef]
- Landi, M.T.; Bishop, D.T.; MacGregor, S.; Machiela, M.J.; Stratigos, A.J.; Ghiorzo, P.; Brossard, M.; Calista, D.; Choi, J.; Fargnoli, M.C.; et al. Genome-wide association meta-analyses combining multiple risk phenotypes provide insights into the genetic architecture of cutaneous melanoma susceptibility. Nat. Genet. 2020, 52, 494–504. [Google Scholar] [CrossRef]
- Salgado, C.; Gruis, N.; BIOS Consortium; Heijmans, B.T.; Oosting, J.; van Doorn, R. Genome-wide analysis of constitutional DNA methylation in familial melanoma. Clin. Epigenet. 2020, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Funchain, P.; Ni, Y.; Heald, B.; Bungo, B.; Arbesman, M.; Behera, T.R.; McCormick, S.; Song, J.M.; Kennedy, L.B.; Kennedy, L.B.; et al. Germline cancer susceptibility in individuals with melanoma. J. Am. Acad. Dermatol. 2024, 91, 267. [Google Scholar] [CrossRef] [PubMed]
- Beigi, Y.Z.; Lanjanian, H.; Fayazi, R.; Salimi, M.; Hoseyni, B.H.M.; Noroozizadeh, M.H.; Masoudi-Nejad, A. Heterogeneity and molecular landscape of melanoma: Implications for targeted therapy. Mol. Biomed. 2024, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Isaak, A.J.; Clements, G.R.; Buenaventura, R.G.M.; Merlino, G.; Yu, Y. Development of Personalized Strategies for Precisely Battling Malignant Melanoma. Int. J. Mol. Sci. 2024, 25, 5023. [Google Scholar] [CrossRef]
- Park, J.-I. MAPK-ERK Pathway. Int. J. Mol. Sci. 2023, 24, 9666. [Google Scholar] [CrossRef]
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O.; et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers 2019, 11, 1618. [Google Scholar] [CrossRef]
- Haugh, A.M.; Johnson, D.B. Management of V600E and V600K BRAF-Mutant Melanoma. Curr. Treat. Options Oncol. 2019, 20, 81. [Google Scholar] [CrossRef] [PubMed]
- Randic, T.; Kozar, I.; Margue, C.; Utikal, J.; Kreis, S. NRAS mutant melanoma: Towards better therapies. Cancer Treat. Rev. 2021, 99, 102238. [Google Scholar] [CrossRef]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef]
- Vendramini, E.; Bomben, R.; Pozzo, F.; Bittolo, T.; Tissino, E.; Gattei, V.; Zucchetto, A. KRAS and RAS-MAPK Pathway Deregulation in Mature B Cell Lymphoproliferative Disorders. Cancers 2022, 14, 666. [Google Scholar] [CrossRef]
- Pasmant, E.; Vidaud, D.; Ballerini, P. RAS MAPK inhibitors deregulation in leukemia. Oncoscience 2015, 2, 930–931. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.T.; Vyse, S.; Huang, P.H. Rare epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer. Semin. Cancer Biol. 2020, 61, 167–179. [Google Scholar] [CrossRef]
- Cohen, J.V.; Sullivan, R.J. Developments in the Space of New MAPK Pathway Inhibitors for BRAF-Mutant Melanoma. Clin. Cancer Res. 2019, 25, 5735–5742. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef] [PubMed]
- Tehranian, C.; Fankhauser, L.; Harter, P.N.; Ratcliffe, C.D.H.; Zeiner, P.S.; Messmer, J.M.; Hoffmann, D.C.; Frey, K.; Westphal, D.; Ronellenfitsch, M.W.; et al. The PI3K/Akt/mTOR pathway as a preventive target in melanoma brain metastasis. Neuro Oncol. 2022, 24, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Geng, Y.; Liu, X.; Chai, Q.; Li, X.; Ren, T.; Shang, Q. PI3K/AKT/mTOR pathway-derived risk score exhibits correlation with immune infiltration in uveal melanoma patients. Front. Oncol. 2023, 13, 1167930. [Google Scholar] [CrossRef]
- Popova, N.V.; Jücker, M. The Role of mTOR Signaling as a Therapeutic Target in Cancer. Int. J. Mol. Sci. 2021, 22, 1743. [Google Scholar] [CrossRef] [PubMed]
- Huang, S. mTOR Signaling in Metabolism and Cancer. Cells 2020, 9, 2278. [Google Scholar] [CrossRef]
- Prvanović, M.; Nedeljković, M.; Tanić, N.; Tomić, T.; Terzić, T.; Milovanović, Z.; Maksimović, Z.; Tanić, N. Role of PTEN, PI3K, and mTOR in Triple-Negative Breast Cancer. Life 2021, 11, 1247. [Google Scholar] [CrossRef]
- Tsai, P.-J.; Lai, Y.-H.; Manne, R.K.; Tsai, Y.-S.; Sarbassov, D.; Lin, H.-K. Akt: A key transducer in cancer. J. Biomed. Sci. 2022, 29, 76. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct. Target Ther. 2021, 6, 425. [Google Scholar] [CrossRef]
- Shariati, M.; Meric-Bernstam, F. Targeting AKT for cancer therapy. Expert Opin. Investig. Drugs 2019, 28, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Sun, E.J.; Wankell, M.; Palamuthusingam, P.; McFarlane, C.; Hebbard, L. Targeting the PI3K/Akt/mTOR Pathway in Hepatocellular Carcinoma. Biomedicines 2021, 9, 1639. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Pyun, W.Y.; Park, H.W. Cancer Metabolism: Phenotype, Signaling and Therapeutic Targets. Cells 2020, 9, 2308. [Google Scholar] [CrossRef]
- Peng, X.; Huang, X.; Lulu, T.B.; Jia, W.; Zhang, S.; Cohen, L.; Huang, S.; Fan, J.; Chen, X.; Liu, S.; et al. A novel pan-PI3K inhibitor KTC1101 synergizes with anti-PD-1 therapy by targeting tumor suppression and immune activation. Mol. Cancer 2024, 23, 54. [Google Scholar] [CrossRef] [PubMed]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol. Cancer 2023, 22, 138. [Google Scholar] [CrossRef] [PubMed]
- Uzbekov, R.; Prigent, C. A Journey through Time on the Discovery of Cell Cycle Regulation. Cells 2022, 11, 704. [Google Scholar] [CrossRef]
- Engeland, K. Cell cycle regulation: p53-p21-RB signaling. Cell Death Differ. 2022, 29, 946–960. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, Y.; Ma, X.; Hu, H. The Influence of Cell Cycle Regulation on Chemotherapy. Int. J. Mol. Sci. 2021, 22, 6923. [Google Scholar] [CrossRef]
- Trembath, D.G.; Ivanova, A.; Krauze, M.T.; Kirkwood, J.M.; Nikolaishvilli-Feinberg, N.; Moschos, S.J. Melanoma-specific expression of the tumor suppressor proteins p16 and PTEN is a favorable prognostic factor in established melanoma brain metastases. Melanoma Res. 2021, 31, 264–267. [Google Scholar] [CrossRef] [PubMed]
- Pandya, P.; Kublo, L.; Stewart-Ornstein, J. p53 Promotes Cytokine Expression in Melanoma to Regulate Drug Resistance and Migration. Cells 2022, 11, 405. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, E.S.; Singh, A.T.K. Cell-cycle Checkpoints and Aneuploidy on the Path to Cancer. In Vivo 2018, 32, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.M.; Ahmed, M.; Lai, T.H.; Bahar, M.E.; Hwang, J.S.; Maulidi, R.F.; Ngo, Q.N.; Kim, D.R. Regulation of Cell Cycle Progression through RB Phosphorylation by Nilotinib and AT-9283 in Human Melanoma A375P Cells. Int. J. Mol. Sci. 2024, 25, 2956. [Google Scholar] [CrossRef] [PubMed]
- Garutti, M.; Targato, G.; Buriolla, S.; Palmero, L.; Minisini, A.M.; Puglisi, F. CDK4/6 Inhibitors in Melanoma: A Comprehensive Review. Cells 2021, 10, 1334. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ba, J.; Kang, Y.; Gong, Z.; Liang, T.; Zhang, Y.; Qi, J.; Wang, J. Recent Progress in CDK4/6 Inhibitors and PROTACs. Molecules 2023, 28, 8060. [Google Scholar] [CrossRef] [PubMed]
- Vilgelm, A.E.; Saleh, N.; Shattuck-Brandt, R.; Riemenschneider, K.; Slesur, L.; Chen, S.-C.; Johnson, C.A.; Yang, J.; Blevins, A.; Yan, C.; et al. MDM2 antagonists overcome intrinsic resistance to CDK4/6 inhibition by inducing p21. Sci. Transl. Med. 2019, 11, eaav7171. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xu, J.; Wang, W.; Zhang, B.; Yu, X.; Shi, S. Epigenetic regulation in the tumor microenvironment: Molecular mechanisms and therapeutic targets. Signal Transduct. Target Ther. 2023, 8, 210. [Google Scholar] [CrossRef]
- Yang, Z.-K.; Yang, J.-Y.; Xu, Z.-Z.; Yu, W.-H. DNA Methylation and Uveal Melanoma. Chin. Med. J. 2018, 131, 845–851. [Google Scholar] [CrossRef]
- McKenna, S.; García-Gutiérrez, L. Resistance to Targeted Therapy and RASSF1A Loss in Melanoma: What Are We Missing? Int. J. Mol. Sci. 2021, 22, 5115. [Google Scholar] [CrossRef]
- Park, H.S.; Kim, J.H.; Cho, M.Y.; Chung, K.Y.; Roh, M.R. PTEN Promoter Hypermethylation Is Associated with Breslow Thickness in Acral Melanoma on the Heel, Forefoot, and Hallux. Ann. Dermatol. 2021, 33, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Rodger, E.J.; Ahn, A.; Stockwell, P.A.; Parry, M.; Motwani, J.; Gallagher, S.J.; Shklovskaya, E.; Tiffen, J.; Eccles, M.R.; et al. Marked Global DNA Hypomethylation Is Associated with Constitutive PD-L1 Expression in Melanoma. iScience 2018, 4, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Rius, F.E.; Papaiz, D.D.; Azevedo, H.F.Z.; Ayub, A.L.P.; Pessoa, D.O.; Oliveira, T.F.; Loureiro, A.P.M.; Andrade, F.; Fujita, A.; Reis, E.M.; et al. Genome-wide promoter methylation profiling in a cellular model of melanoma progression reveals markers of malignancy and metastasis that predict melanoma survival. Clin. Epigenet. 2022, 14, 68. [Google Scholar] [CrossRef] [PubMed]
- Ressler, J.M.; Tomasich, E.; Hatziioannou, T.; Ringl, H.; Heller, G.; Silmbrod, R.; Gottmann, L.; Starzer, A.M.; Zila, N.; Tschandl, P.; et al. DNA Methylation Signatures Correlate with Response to Immune Checkpoint Inhibitors in Metastatic Melanoma. Target Oncol. 2024, 19, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhang, H.; Gao, P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell 2022, 13, 877–919. [Google Scholar] [CrossRef] [PubMed]
- Emran, A.A.; Chatterjee, A.; Rodger, E.J.; Tiffen, J.C.; Gallagher, S.J.; Eccles, M.R.; Hersey, P. Targeting DNA Methylation and EZH2 Activity to Overcome Melanoma Resistance to Immunotherapy. Trends Immunol. 2019, 40, 328–344. [Google Scholar] [CrossRef]
- Drzewiecka, M.; Gajos-Michniewicz, A.; Hoser, G.; Jaśniak, D.; Barszczewska-Pietraszek, G.; Sitarek, P.; Czarny, P.; Piekarski, J.; Radek, M.; Czyż, M.; et al. Histone Deacetylases (HDAC) Inhibitor-Valproic Acid Sensitizes Human Melanoma Cells to Dacarbazine and PARP Inhibitor. Genes 2023, 14, 1295. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, M.; Czyz, M. lncRNAs-EZH2 interaction as promising therapeutic target in cutaneous melanoma. Front. Mol. Biosci. 2023, 10, 1170026. [Google Scholar] [CrossRef]
- Emmons, M.F.; Bennett, R.L.; Riva, A.; Gupta, K.; Carvalho, L.A.D.C.; Zhang, C.; Macaulay, R.; Dupéré-Richér, D.; Fang, B.; Seto, E.; et al. HDAC8-mediated inhibition of EP300 drives a transcriptional state that increases melanoma brain metastasis. Nat. Commun. 2023, 14, 7759. [Google Scholar] [CrossRef]
- Yan, H.; Bu, P. Non-coding RNA in cancer. Essays Biochem. 2021, 65, 625–639. [Google Scholar] [CrossRef]
- Vitiello, M.; D’Aurizio, R.; Poliseno, L. Biological role of miR-204 and miR-211 in melanoma. Oncoscience 2018, 5, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Vand-Rajabpour, F.; Savage, M.; Belote, R.L.; Judson-Torres, R.L. Critical Considerations for Investigating MicroRNAs during Tumorigenesis: A Case Study in Conceptual and Contextual Nuances of miR-211-5p in Melanoma. Epigenomes 2023, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Wu, Z.; Chen, J.; Guo, S.; You, W.; Wang, S.; Ma, J.; Wang, H.; Wang, X.; Wang, H.; et al. Nanoparticle delivery of miR-21-3p sensitizes melanoma to anti-PD-1 immunotherapy by promoting ferroptosis. J. Immunother. Cancer 2022, 10, e004381. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Liao, B.; Xiang, X.; Ke, S. miR-21-5p promotes cell proliferation and G1/S transition in melanoma by targeting CDKN2C. FEBS Open Bio 2020, 10, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Orso, F.; Quirico, L.; Virga, F.; Penna, E.; Dettori, D.; Cimino, D.; Coppo, R.; Grassi, E.; Elia, A.R.; Brusa, D.; et al. miR-214 and miR-148b Targeting Inhibits Dissemination of Melanoma and Breast Cancer. Cancer Res. 2016, 76, 5151–5162. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Llorente, L.; Ruiz-Rodríguez, M.J.; Savini, C.; González-Muñoz, T.; Riveiro-Falkenbach, E.; Rodríguez-Peralto, J.L.; Peinado, H.; Bernabeu, C. Correlation between Endoglin and Malignant Phenotype in Human Melanoma Cells: Analysis of hsa-mir-214 and hsa-mir-370 in Cells and Their Extracellular Vesicles. In Advances in Molecular Pathology; Springer Nature: Cham, Switzerland, 2023; pp. 253–272. [Google Scholar] [CrossRef]
- Prabhakar, K.; Rodrίguez, C.I.; Jayanthy, A.S.; Mikheil, D.M.; Bhasker, A.I.; Perera, R.J.; Setaluri, V. Role of miR-214 in regulation of β-catenin and the malignant phenotype of melanoma. Mol. Carcinog. 2019, 58, 1974–1984. [Google Scholar] [CrossRef] [PubMed]
- Safa, A.; Gholipour, M.; Dinger, M.E.; Taheri, M.; Ghafouri-Fard, S. The critical roles of lncRNAs in the pathogenesis of melanoma. Exp. Mol. Pathol. 2020, 117, 104558. [Google Scholar] [CrossRef]
- Yu, X.; Zheng, H.; Tse, G.; Chan, M.T.; Wu, W.K. Long non-coding RNAs in melanoma. Cell Prolif. 2018, 51, e12457. [Google Scholar] [CrossRef] [PubMed]
- Feichtenschlager, V.; Zheng, Y.J.; Ho, W.; Chen, L.; Callanan, C.; Chen, C.; Lee, A.; Ortiz, J.; Rappersberger, K.; Ortiz-Urda, S. Deconstructing the role of MALAT1 in MAPK-signaling in melanoma: Insights from antisense oligonucleotide treatment. Oncotarget 2023, 14, 543–560. [Google Scholar] [CrossRef]
- Liu, X.; Hao, J.; Xie, T.; Pant, O.P.; Lu, C.; Lu, C.; Zhou, D. The BRAF activated non-coding RNA: A pivotal long non-coding RNA in human malignancies. Cell Prolif. 2018, 51, e12449. [Google Scholar] [CrossRef]
- Hussen, B.M.; Azimi, T.; Abak, A.; Hidayat, H.J.; Taheri, M.; Ghafouri-Fard, S. Role of lncRNA BANCR in Human Cancers: An Updated Review. Front. Cell Dev. Biol. 2021, 9, 689992. [Google Scholar] [CrossRef]
- Han, S.; Yan, Y.; Ren, Y.; Hu, Y.; Wang, Y.; Chen, L.; Zhi, Z.; Zheng, Y.; Shao, Y.; Liu, J. LncRNA SAMMSON Mediates Adaptive Resistance to RAF Inhibition in BRAF-Mutant Melanoma Cells. Cancer Res. 2021, 81, 2918–2929. [Google Scholar] [CrossRef] [PubMed]
- Ghasemian, M.; Babaahmadi-Rezaei, H.; Khedri, A.; Selvaraj, C. The oncogenic role of SAMMSON lncRNA in tumorigenesis: A comprehensive review with especial focus on melanoma. J. Cell. Mol. Med. 2023, 27, 3966–3973. [Google Scholar] [CrossRef] [PubMed]
- Terrell, J.R.; Rybak, I.; Lyu, Y.; Konia, T.; Fung, M.A.; Qi, L.; Kiuru, M. The influence of p16 immunohistochemistry on diagnosis and management recommendation of melanocytic neoplasms by dermatopathologists: A prospective study. J. Cutan. Pathol. 2021, 48, 1094–1097. [Google Scholar] [CrossRef] [PubMed]
- Penter, L.; Liu, Y.; Wolff, J.O.; Yang, L.; Taking, L.; Jhaveri, A.; Southard, J.; Patel, M.; Cullen, N.; Pfaff, K.L.; et al. Mechanisms of response and resistance to combined decitabine and ipilimumab for advanced myeloid disease. Blood 2023, 141, 1817–1830. [Google Scholar] [CrossRef] [PubMed]
- Celesia, A.; Notaro, A.; Franzò, M.; Lauricella, M.; D’Anneo, A.; Carlisi, D.; Giuliano, M.; Emanuele, S. The Histone Deacetylase Inhibitor ITF2357 (Givinostat) Targets Oncogenic BRAF in Melanoma Cells and Promotes a Switch from Pro-Survival Autophagy to Apoptosis. Biomedicines 2022, 10, 1994. [Google Scholar] [CrossRef] [PubMed]
- Eraky, A.M. Advances in Brain Metastases Diagnosis: Non-coding RNAs As Potential Biomarkers. Cureus 2023, 15, e36337. [Google Scholar] [CrossRef] [PubMed]
- Marzagalli, M.; Ebelt, N.D.; Manuel, E.R. Unraveling the crosstalk between melanoma and immune cells in the tumor microenvironment. Semin. Cancer Biol. 2019, 59, 236–250. [Google Scholar] [CrossRef] [PubMed]
- Hodis, E.; Torlai Triglia, E.; Kwon, J.Y.H.; Biancalani, T.; Zakka, L.R.; Parkar, S.; Hütter, J.; Buffoni, L.; Delorey, T.; Phillips, D.; et al. Stepwise-edited, human melanoma models reveal mutations’ effect on tumor and microenvironment. Science 2022, 376, eabi8175. [Google Scholar] [CrossRef]
- Simiczyjew, A.; Dratkiewicz, E.; Mazurkiewicz, J.; Ziętek, M.; Matkowski, R.; Nowak, D. The Influence of Tumor Microenvironment on Immune Escape of Melanoma. Int. J. Mol. Sci. 2020, 21, 8359. [Google Scholar] [CrossRef]
- Dolina, J.S.; Van Braeckel-Budimir, N.; Thomas, G.D.; Salek-Ardakani, S. CD8+ T Cell Exhaustion in Cancer. Front. Immunol. 2021, 12, 715234. [Google Scholar] [CrossRef] [PubMed]
- van der Leun, A.M.; Thommen, D.S.; Schumacher, T.N. CD8+ T cell states in human cancer: Insights from single-cell analysis. Nat. Rev. Cancer 2020, 20, 218–232. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Vogelzang, A.; Miyajima, M.; Sugiura, Y.; Wu, Y.; Chamoto, K.; Nakano, R.; Hatae, R.; Menzies, R.; Sonomura, K.; et al. B cell-derived GABA elicits IL-10+ macrophages to limit anti-tumour immunity. Nature 2021, 599, 471–476. [Google Scholar] [CrossRef]
- Liu, L.; Mo, M.; Chen, X.; Chao, D.; Zhang, Y.; Chen, X.; Wang, Y.; Zhang, N.; He, N.; Yuan, X.; et al. Targeting inhibition of prognosis-related lipid metabolism genes including CYP19A1 enhances immunotherapeutic response in colon cancer. J. Exp. Clin. Cancer Res. 2023, 42, 85. [Google Scholar] [CrossRef]
- Rohaan, M.W.; Borch, T.H.; van den Berg, J.H.; Met, Ö.; Kessels, R.; Geukes Foppen, M.H.; Granhøj, J.S.; Nuijen, B.; Nijenhuis, C.; Jedema, I.; et al. Tumor-Infiltrating Lymphocyte Therapy or Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2022, 387, 2113–2125. [Google Scholar] [CrossRef] [PubMed]
- Kashfi, K.; Kannikal, J.; Nath, N. Macrophage Reprogramming and Cancer Therapeutics: Role of iNOS-Derived NO. Cells 2021, 10, 3194. [Google Scholar] [CrossRef] [PubMed]
- Boutilier, A.J.; Elsawa, S.F. Macrophage Polarization States in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 6995. [Google Scholar] [CrossRef]
- Kerneur, C.; Cano, C.E.; Olive, D. Major pathways involved in macrophage polarization in cancer. Front. Immunol. 2022, 13, 1026954. [Google Scholar] [CrossRef]
- Ji, S.; Shi, Y.; Yin, B. Macrophage barrier in the tumor microenvironment and potential clinical applications. Cell Commun. Signal. 2024, 22, 74. [Google Scholar] [CrossRef]
- Bied, M.; Ho, W.W.; Ginhoux, F.; Blériot, C. Roles of macrophages in tumor development: A spatiotemporal perspective. Cell Mol. Immunol. 2023, 20, 983–992. [Google Scholar] [CrossRef]
- Wattenberg, M.M.; Coho, H.; Herrera, V.M.; Graham, K.; Stone, M.L.; Xue, Y.; Chang, R.B.; Cassella, C.; Liu, M.; Choi-Bose, S.; et al. Cancer immunotherapy via synergistic coactivation of myeloid receptors CD40 and Dectin-1. Sci. Immunol. 2023, 8, eadj5097. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Teng, D.; Yang, L.; Xu, X.; Chen, J.; Jiang, T.; Feng, A.Y.; Zhang, Y.; Frederick, D.T.; Gu, L.; et al. Myeloid-derived itaconate suppresses cytotoxic CD8+ T cells and promotes tumour growth. Nat. Metab. 2022, 4, 1660–1673. [Google Scholar] [CrossRef] [PubMed]
- Umansky, V.; Blattner, C.; Gebhardt, C.; Utikal, J. CCR5 in recruitment and activation of myeloid-derived suppressor cells in melanoma. Cancer Immunol. Immunother. 2017, 66, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Ozbay Kurt, F.G.; Lasser, S.; Arkhypov, I.; Utikal, J.; Umansky, V. Enhancing immunotherapy response in melanoma: Myeloid-derived suppressor cells as a therapeutic target. J. Clin. Investig. 2023, 133, 6. [Google Scholar] [CrossRef] [PubMed]
- Sieminska, I.; Baran, J. Myeloid-Derived Suppressor Cells in Colorectal Cancer. Front. Immunol. 2020, 11, 1526. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Fernández de Córdoba, B.; Moreno, H.; Valencia, K.; Perurena, N.; Ruedas, P.; Walle, T.; Pezonaga-Torres, A.; Hinojosa, J.; Guruceaga, E.; Pineda-Lucena, A.; et al. Tumor ENPP1 (CD203a)/Haptoglobin Axis Exploits Myeloid-Derived Suppressor Cells to Promote Post-Radiotherapy Local Recurrence in Breast Cancer. Cancer Discov. 2022, 12, 1356–1377. [Google Scholar] [CrossRef]
- Tomela, K.; Pietrzak, B.; Galus, Ł.; Mackiewicz, J.; Schmidt, M.; Mackiewicz, A.A.; Kaczmarek, M. Myeloid-Derived Suppressor Cells (MDSC) in Melanoma Patients Treated with Anti-PD-1 Immunotherapy. Cells 2023, 12, 789. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.L.; Murphy, K.M. Dendritic cells in cancer immunology. Cell Mol. Immunol. 2022, 19, 3–13. [Google Scholar] [CrossRef]
- Gong, Z.; Li, Q.; Shi, J.; Wei, J.; Li, P.; Chang, C.-H.; Shultz, L.D.; Ren, G. Lung fibroblasts facilitate pre-metastatic niche formation by remodeling the local immune microenvironment. Immunity 2022, 55, 1483–1500.e9. [Google Scholar] [CrossRef]
- Han, Z.; Dong, Y.; Lu, J.; Yang, F.; Zheng, Y.; Yang, H. Role of hypoxia in inhibiting dendritic cells by VEGF signaling in tumor microenvironments: Mechanism and application. Am. J. Cancer Res. 2021, 11, 3777–3793. [Google Scholar]
- Mazurkiewicz, J.; Simiczyjew, A.; Dratkiewicz, E.; Ziętek, M.; Matkowski, R.; Nowak, D. Stromal Cells Present in the Melanoma Niche Affect Tumor Invasiveness and Its Resistance to Therapy. Int. J. Mol. Sci. 2021, 22, 529. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Shen, M.; Wu, L.; Yang, H.; Yao, Y.; Yang, Q.; Du, J.; Liu, L.; Li, Y.; Bai, Y. Stromal cells in the tumor microenvironment: Accomplices of tumor progression? Cell Death Dis. 2023, 14, 587. [Google Scholar] [CrossRef]
- Zhang, J.; Song, C.; Tian, Y.; Yang, X. Single-Cell RNA Sequencing in Lung Cancer: Revealing Phenotype Shaping of Stromal Cells in the Microenvironment. Front. Immunol. 2021, 12, 802080. [Google Scholar] [CrossRef]
- Papaccio, F.; Kovacs, D.; Bellei, B.; Caputo, S.; Migliano, E.; Cota, C.; Picardo, M. Profiling Cancer-Associated Fibroblasts in Melanoma. Int. J. Mol. Sci. 2021, 22, 7255. [Google Scholar] [CrossRef]
- Napoli, S.; Scuderi, C.; Gattuso, G.; Bella, V.D.; Candido, S.; Basile, M.S.; Libra, M.; Falzone, L. Functional Roles of Matrix Metalloproteinases and Their Inhibitors in Melanoma. Cells 2020, 9, 1151. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Xu, J.; Wang, W.; Liang, C.; Hua, J.; Liu, J.; Zhang, B.; Meng, Q.; Yu, X.; Shi, S. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: New findings and future perspectives. Mol. Cancer 2021, 20, 131. [Google Scholar] [CrossRef]
- Jenkins, L.; Jungwirth, U.; Avgustinova, A.; Iravani, M.; Mills, A.; Haider, S.; Harper, J.; Isacke, C. Cancer-Associated Fibroblasts Suppress CD8+ T-cell Infiltration and Confer Resistance to Immune-Checkpoint Blockade. Cancer Res. 2022, 82, 2904–2917. [Google Scholar] [CrossRef] [PubMed]
- Hutchenreuther, J.; Nguyen, J.; Quesnel, K.; Vincent, K.M.; Petitjean, L.; Bourgeois, S.; Boyd, M.; Bou-Gharios, G.; Postovit, L.; Leask, A. Cancer-associated Fibroblast–specific Expression of the Matricellular Protein CCN1 Coordinates Neovascularization and Stroma Deposition in Melanoma Metastasis. Cancer Res. Commun. 2024, 4, 556–570. [Google Scholar] [CrossRef]
- Wu, Z.; Bian, Y.; Chu, T.; Wang, Y.; Man, S.; Song, Y.; Wang, Z. The role of angiogenesis in melanoma: Clinical treatments and future expectations. Front. Pharmacol. 2022, 13, 1028647. [Google Scholar] [CrossRef]
- Ghalehbandi, S.; Yuzugulen, J.; Pranjol, M.Z.I.; Pourgholami, M.H. The role of VEGF in cancer-induced angiogenesis and research progress of drugs targeting VEGF. Eur. J. Pharmacol. 2023, 949, 175586. [Google Scholar] [CrossRef]
- Ebeling, S.; Kowalczyk, A.; Perez-Vazquez, D.; Mattiola, I. Regulation of tumor angiogenesis by the crosstalk between innate immunity and endothelial cells. Front. Oncol. 2023, 13, 1171794. [Google Scholar] [CrossRef] [PubMed]
- Simiczyjew, A.; Wądzyńska, J.; Pietraszek-Gremplewicz, K.; Kot, M.; Ziętek, M.; Matkowski, R.; Nowak, D. Melanoma cells induce dedifferentiation and metabolic changes in adipocytes present in the tumor niche. Cell. Mol. Biol. Lett. 2023, 28, 58. [Google Scholar] [CrossRef] [PubMed]
- Olszańska, J.; Pietraszek-Gremplewicz, K.; Nowak, D. Melanoma Progression under Obesity: Focus on Adipokines. Cancers 2021, 13, 2281. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Di Martino, J.S.; Bowman, R.L.; Campbell, N.R.; Baksh, S.C.; Simon-Vermot, T.; Kim, I.S.; Haldeman, P.; Mondal, C.; Yong-Gonzales, V.; et al. Adipocyte-Derived Lipids Mediate Melanoma Progression via FATP Proteins. Cancer Discov. 2018, 8, 1006–1025. [Google Scholar] [CrossRef] [PubMed]
- Gam, D.-H.; Park, J.-H.; Kim, J.-H.; Beak, D.-H.; Kim, J.-W. Effects of Allium sativum Stem Extract on Growth and Migration in Melanoma Cells through Inhibition of VEGF, MMP-2, and MMP-9 Genes Expression. Molecules 2021, 27, 21. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Qin, J.; Li, Y.; Li, G.; Wang, Y.; Zhang, N.; Chen, P.; Li, C. Combination therapy of PKCζ and COX-2 inhibitors synergistically suppress melanoma metastasis. J. Exp. Clin. Cancer Res. 2017, 36, 115. [Google Scholar] [CrossRef] [PubMed]
- Aristorena, M.; Gallardo-Vara, E.; Vicen, M.; de Las Casas-Engel, M.; Ojeda-Fernandez, L.; Nieto, C.; Blanco, F.J.; Valbuena-Diez, A.C.; Botella, L.M.; Nachtigal, P. MMP-12, Secreted by Pro-Inflammatory Macrophages, Targets Endoglin in Human Macrophages and Endothelial Cells. Int. J. Mol. Sci. 2019, 20, 3107. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Kang, Q.; Chan, K.I.; Zhang, Y.; Zhong, Z.; Tan, W. The immunomodulatory role of matrix metalloproteinases in colitis-associated cancer. Front. Immunol. 2023, 13, 1093990. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, A.; Kawana, K.; Tomio, K.; Yamashita, A.; Isobe, Y.; Nagasaka, K.; Koga, K.; Inoue, T.; Nishida, H.; Kojima, S. Matrix Metalloproteinase (MMP)-9 in Cancer-Associated Fibroblasts (CAFs) Is Suppressed by Omega-3 Polyunsaturated Fatty Acids In Vitro and In Vivo. PLoS ONE 2014, 9, e89605. [Google Scholar] [CrossRef]
- Tanaka, N.; Sakamoto, T. MT1-MMP as a Key Regulator of Metastasis. Cells 2023, 12, 2187. [Google Scholar] [CrossRef]
- Bastian, A.; Nichita, L.; Zurac, S. Matrix Metalloproteinases in Melanoma with and without Regression. In The Role of Matrix Metalloproteinase in Human Body Pathologies; InTech: London, UK, 2017. [Google Scholar] [CrossRef]
- Nguyen, B.A.; Ho, J.; De La Cruz Diaz, J.S.; Nishimura, S.; Kaplan, D.H. TGFβ activating integrins β6 and β8 are dysregulated in inflammatory skin disease and cutaneous melanoma. J. Dermatol. Sci. 2022, 106, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Arias-Mejias, S.M.; Warda, K.Y.; Quattrocchi, E.; Alonso-Quinones, H.; Sominidi-Damodaran, S.; Meves, A. The role of integrins in melanoma: A review. Int. J. Dermatol. 2020, 59, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, H.; Leyton, L. CSK-mediated signalling by integrins in cancer. Front. Cell Dev. Biol. 2023, 11, 1214787. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Rofstad, E.K. Integrins as therapeutic targets in the organ-specific metastasis of human malignant melanoma. J. Exp. Clin. Cancer Res. 2018, 37, 92. [Google Scholar] [CrossRef] [PubMed]
- Lacy, P. Editorial: Secretion of Cytokines and Chemokines by Innate Immune Cells. Front. Immunol. 2015, 6, 190. [Google Scholar] [CrossRef]
- Paganelli, A.; Garbarino, F.; Toto, P.; Di Martino, G.; D’Urbano, M.; Auriemma, M.; Giovanni, P.D.; Panarese, F.; Staniscia, T.; Amerio, P.; et al. Serological landscape of cytokines in cutaneous melanoma. Cancer Biomark. 2019, 26, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Soler, M.F.; Abaurrea, A.; Azcoaga, P.; Araujo, A.M.; Caffarel, M.M. New perspectives in cancer immunotherapy: Targeting IL-6 cytokine family. J. Immunother. Cancer 2023, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Wang, W.; Wang, H.; Chen, L.; Liu, T. The expression pattern of immune-related genes and characterization of tumor immune microenvironment: Predicting prognosis and immunotherapeutic effects in cutaneous melanoma. World J. Surg. Oncol. 2022, 20, 303. [Google Scholar] [CrossRef]
- Niu, M.; Yi, M.; Wu, Y.; Lyu, L.; He, Q.; Yang, R.; Zeng, L.; Shi, J.; Zhang, J.; Zhou, P.; et al. Synergistic efficacy of simultaneous anti-TGF-β/VEGF bispecific antibody and PD-1 blockade in cancer therapy. J. Hematol. Oncol. 2023, 16, 94. [Google Scholar] [CrossRef]
- Courau, T.; Nehar-Belaid, D.; Florez, L.; Levacher, B.; Vazquez, T.; Brimaud, F.; Bellier, B.; Klatzmann, D. TGF-β and VEGF cooperatively control the immunotolerant tumor environment and the efficacy of cancer immunotherapies. JCI Insight 2016, 1, 2. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Turley, S.J.; Akhurst, R.J. TGFβ biology in cancer progression and immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 9–34. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Yang, J.; Deng, S.; Xu, H.; Wu, D.; Zeng, Q.; Wang, S.; Hu, T.; Wu, F.; Zhou, H. TGF-β signaling in the tumor metabolic microenvironment and targeted therapies. J. Hematol. Oncol. 2022, 15, 135. [Google Scholar] [CrossRef] [PubMed]
- Tasdogan, A.; Faubert, B.; Ramesh, V.; Ubellacker, J.M.; Shen, B.; Solmonson, A.; Murphy, M.M.; Gu, Z.; Gu, W.; Martin, M.; et al. Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 2020, 577, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Haffner, M.C.; Zwart, W.; Roudier, M.P.; True, L.D.; Nelson, W.G.; Epstein, J.I.; De Marzo, A.M.; Nelson, P.S.; Yegnasubramanian, S. Genomic and phenotypic heterogeneity in prostate cancer. Nat. Rev. Urol. 2021, 18, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Torborg, S.R.; Li, Z.; Chan, J.E.; Tammela, T. Cellular and molecular mechanisms of plasticity in cancer. Trends Cancer 2022, 8, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Grzywa, T.M.; Paskal, W.; Włodarski, P.K. Intratumor and Intertumor Heterogeneity in Melanoma. Transl. Oncol. 2017, 10, 956–975. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Tanaka, Y.; Murata, M.; Kaku-Ito, Y.; Furue, K.; Furue, M. BRAF Heterogeneity in Melanoma. Curr. Treat. Options Oncol. 2021, 22, 20. [Google Scholar] [CrossRef]
- Pellegrini, C.; Cardelli, L.; De Padova, M.; Di Nardo, L.; Ciciarelli, V.; Rocco, T.; Cipolloni, G.; Clementi, M.; Cortellini, A.; Ventura, A.; et al. Intra-patient Heterogeneity of BRAF and NRAS Molecular Alterations in Primary Melanoma and Metastases. Acta Derm. Venereol. 2020, 100, adv00040. [Google Scholar] [CrossRef]
- Seferbekova, Z.; Lomakin, A.; Yates, L.R.; Gerstung, M. Spatial biology of cancer evolution. Nat. Rev. Genet. 2023, 24, 295–313. [Google Scholar] [CrossRef]
- Anaka, M.; Hudson, C.; Lo, P.-H.; Do, H.; Caballero, O.L.; Davis, I.D.; Dobrovic, A.; Cebon, J.; Behren, A. Intratumoral genetic heterogeneity in metastatic melanoma is accompanied by variation in malignant behaviors. BMC Med. Genom. 2013, 6, 40. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Fung, C.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Hyman, J.; Shahheydari, H.; Tembe, V.; Thompson, J.F.; Saw, R.P.; et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat. Commun. 2014, 5, 5694. [Google Scholar] [CrossRef]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Kichina, J.V.; Maslov, A.; Kandel, E.S. PAK1 and Therapy Resistance in Melanoma. Cells 2023, 12, 2373. [Google Scholar] [CrossRef] [PubMed]
- Dharanipragada, P.; Zhang, X.; Liu, S.; Lomeli, S.H.; Hong, A.; Wang, Y.; Yang, Z.; Lo, K.Z.; Vega-Crespo, A.; Ribas, A.; et al. Blocking Genomic Instability Prevents Acquired Resistance to MAPK Inhibitor Therapy in Melanoma. Cancer Discov. 2023, 13, 880–909. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S.; Shaughnessy, M.; Tsao, H. Melanoma classification and management in the era of molecular medicine. Dermatol. Clin. 2023, 41, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Tangella, L.P.; Clark, M.E.; Gray, E.S. Resistance mechanisms to targeted therapy in BRAF-mutant melanoma—A mini review. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2021, 1865, 129736. [Google Scholar] [CrossRef] [PubMed]
- Strub, T.; Ballotti, R.; Bertolotto, C. The “ART” of Epigenetics in Melanoma: From histone “Alterations, to Resistance and Therapies”. Theranostics 2020, 10, 1777–1797. [Google Scholar] [CrossRef]
- Zhao, Y.; Murciano-Goroff, Y.R.; Xue, J.Y.; Ang, A.; Lucas, J.; Mai, T.T.; Da Cruz Paula, A.F.; Saiki, A.Y.; Mohn, D.; Achanta, P.; et al. Diverse alterations associated with resistance to KRAS(G12C) inhibition. Nature 2021, 599, 679–683. [Google Scholar] [CrossRef]
- Subhadarshini, S.; Sahoo, S.; Debnath, S.; Somarelli, J.A.; Jolly, M.K. Dynamical modeling of proliferative-invasive plasticity and IFNγ signaling in melanoma reveals mechanisms of PD-L1 expression heterogeneity. J. Immunother. Cancer 2023, 11, e006766. [Google Scholar] [CrossRef]
- Manfioletti, G.; Fedele, M. Epithelial-Mesenchymal Transition (EMT). Int. J. Mol. Sci. 2023, 24, 11386. [Google Scholar] [CrossRef] [PubMed]
- Pedri, D.; Karras, P.; Landeloos, E.; Marine, J.-C.; Rambow, F. Epithelial-to-mesenchymal-like transition events in melanoma. FEBS J. 2022, 289, 1352–1368. [Google Scholar] [CrossRef] [PubMed]
- Rapanotti, M.C.; Cugini, E.; Campione, E.; Di Raimondo, C.; Costanza, G.; Rossi, P.; Ferlosio, A.; Bernardini, S.; Orlandi, A.; De Luca, A.; et al. Epithelial-to-Mesenchymal Transition Gene Signature in Circulating Melanoma Cells: Biological and Clinical Relevance. Int. J. Mol. Sci. 2023, 24, 11792. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, M.P.; Marchbank, K.; Webster, M.R.; Valiga, A.A.; Kaur, A.; Vultur, A.; Li, L.; Herlyn, M.; Villanueva, J.; Liu, Q.; et al. Hypoxia Induces Phenotypic Plasticity and Therapy Resistance in Melanoma via the Tyrosine Kinase Receptors ROR1 and ROR2. Cancer Discov. 2013, 3, 1378–1393. [Google Scholar] [CrossRef]
- Wang, F.; Cheng, F.; Zheng, F. Stem cell like memory T cells: A new paradigm in cancer immunotherapy. Clin. Immunol. 2022, 241, 109078. [Google Scholar] [CrossRef] [PubMed]
- Magnoni, C.; Giudice, S.; Pellacani, G.; Bertazzoni, G.; Longo, C.; Veratti, E.; Morini, D.; Benassi, L.; Vaschieri, C.; Azzoni, P.; et al. Stem cell properties in cell cultures from different stage of melanoma progression. Appl. Immunohistochem. Mol. Morphol. 2014, 22, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.; Grulois, D.; Quadrana, L.; Chevin, L.-M. Phenotypic plasticity evolves at multiple biological levels in response to environmental predictability in a long-term experiment with a halotolerant microalga. PLoS Biol. 2023, 21, e3001895. [Google Scholar] [CrossRef] [PubMed]
- Comodo-Navarro, A.N.; Fernandes, M.; Barcelos, D.; Carapeto, F.C.L.; Guimarães, D.P.; de Sousa Moraes, L.; Cerutti, J.; Iwamura, E.S.M.; Landman, G. Intratumor Heterogeneity of KIT Gene Mutations in Acral Lentiginous Melanoma. Am. J. Dermatopathol. 2020, 42, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Castañeda, L.D.; Nova, J.A.; Tovar-Parra, J.D. Frequency of mutations in BRAF, NRAS, and KIT in different populations and histological subtypes of melanoma: A systemic review. Melanoma Res. 2020, 30, 62–70. [Google Scholar] [CrossRef]
- Minor, D.R.; Kashani-Sabet, M.; Garrido, M.; O’Day, S.J.; Hamid, O.; Bastian, B.C. Sunitinib Therapy for Melanoma Patients with KIT Mutations. Clin. Cancer Res. 2012, 18, 1457–1463. [Google Scholar] [CrossRef]
- Jung, S.; Armstrong, E.; Wei, A.Z.; Ye, F.; Lee, A.; Carlino, M.S.; Sullivan, R.J.; Carvajal, R.D.; Shoushtari, A.N.; Johnson, D.B. Clinical and genomic correlates of imatinib response in melanomas with KIT alterations. Br. J. Cancer 2022, 127, 1726–1732. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Ke, L.; Zhang, W.; Lu, J.; Chen, Y. Recurrent KRAS, KIT and SF3B1 mutations in melanoma of the female genital tract. BMC Cancer 2021, 21, 677. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Chen, S.; Sun, R.; Ashby, C.R.; Wei, L.; Huang, Z.; Chen, Z. Darovasertib, a novel treatment for metastatic uveal melanoma. Front. Pharmacol. 2023, 14, 1232787. [Google Scholar] [CrossRef] [PubMed]
- Silva-Rodríguez, P.; Fernández-Díaz, D.; Bande, M.; Pardo, M.; Loidi, L.; Blanco-Teijeiro, M.J. GNAQ and GNA11 Genes: A Comprehensive Review on Oncogenesis, Prognosis and Therapeutic Opportunities in Uveal Melanoma. Cancers 2022, 14, 3066. [Google Scholar] [CrossRef] [PubMed]
- Croce, M.; Ferrini, S.; Pfeffer, U.; Gangemi, R. Targeted Therapy of Uveal Melanoma: Recent Failures and New Perspectives. Cancers 2019, 11, 846. [Google Scholar] [CrossRef] [PubMed]
- Lietman, C.D.; McKean, M. Targeting GNAQ/11 through PKC inhibition in uveal melanoma. Cancer Gene Ther. 2022, 29, 1809–1813. [Google Scholar] [CrossRef] [PubMed]
- Toussi, A.; Mans, N.; Welborn, J.; Kiuru, M. Germline mutations predisposing to melanoma. J. Cutan. Pathol. 2020, 47, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting p53 pathways: Mechanisms, structures, and advances in therapy. Signal Transduct. Target Ther. 2023, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Sacconi, A.; Muti, P.; Pulito, C.; Urbani, G.; Allegretti, M.; Pellini, R.; Mehterov, N.; Ben-David, U.; Strano, S.; Bossi, P.; et al. Immunosignatures associated with TP53 status and co-mutations classify prognostically head and neck cancer patients. Mol. Cancer 2023, 22, 192. [Google Scholar] [CrossRef]
- Khan, R.; Pari, B.; Puszynski, K. Comprehensive Bioinformatic Investigation of TP53 Dysregulation in Diverse Cancer Landscapes. Genes 2024, 15, 577. [Google Scholar] [CrossRef]
- Álvarez-Garcia, V.; Tawil, Y.; Wise, H.M.; Leslie, N.R. Mechanisms of PTEN loss in cancer: It’s all about diversity. Semin. Cancer Biol. 2019, 59, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Ince, F.A.; Shariev, A.; Dixon, K. PTEN as a target in melanoma. J. Clin. Pathol. 2022, 75, 581–584. [Google Scholar] [CrossRef] [PubMed]
- Bazzichetto, C.; Conciatori, F.; Pallocca, M.; Falcone, I.; Fanciulli, M.; Cognetti, F.; Milella, M.; Ciuffreda, L. PTEN as a Prognostic/Predictive Biomarker in Cancer: An Unfulfilled Promise? Cancers 2019, 11, 435. [Google Scholar] [CrossRef]
- Cabrita, R.; Mitra, S.; Sanna, A.; Ekedahl, H.; Lövgren, K.; Olsson, H.; Ingvar, C.; Isaksson, K.; Lauss, M.; Carneiro, A.; et al. The Role of PTEN Loss in Immune Escape, Melanoma Prognosis and Therapy Response. Cancers 2020, 12, 742. [Google Scholar] [CrossRef]
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Teng, F.; Kong, L.; Yu, J. PD-L1 expression in human cancers and its association with clinical outcomes. Onco. Targets Ther. 2016, 9, 5023–5039. [Google Scholar] [CrossRef]
- Mariam, A.; Kamath, S.; Schveder, K.; McLeod, H.L.; Rotroff, D.M. Biomarkers for Response to Anti–PD-1/Anti–PD-L1 Immune Checkpoint Inhibitors: A Large Meta-Analysis. Oncology 2023, 37, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Sorroche, B.P.; Teixeira, R.d.J.; Pereira, C.A.D.; Santana, I.V.V.; Vujanovic, L.; Vazquez, V.d.L.; Arantes, L.M.R.B. PD-L1 Tumor Expression as a Predictive Biomarker of Immune Checkpoint Inhibitors’ Response and Survival in Advanced Melanoma Patients in Brazil. Diagnostics 2023, 13, 1041. [Google Scholar] [CrossRef]
- Davis, A.A.; Patel, V.G. The role of PD-L1 expression as a predictive biomarker: An analysis of all US Food and Drug Administration (FDA) approvals of immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 278. [Google Scholar] [CrossRef]
- Guterres, A.N.; Villanueva, J. Targeting telomerase for cancer therapy. Oncogene 2020, 39, 5811–5824. [Google Scholar] [CrossRef] [PubMed]
- Chun-On, P.; Hinchie, A.M.; Beale, H.C.; Gil Silva, A.A.; Rush, E.; Sander, C.; Connelly, C.J.; Seynnaeve, B.N.; Kirkwood, J.M.; Vaske, O.M.; et al. TPP1 promoter mutations cooperate with TERT promoter mutations to lengthen telomeres in melanoma. Science 2022, 378, 664–668. [Google Scholar] [CrossRef] [PubMed]
- Griewank, K.G.; Murali, R.; Puig-Butille, J.A.; Schilling, B.; Livingstone, E.; Potrony, M.; Carrera, C.; Schimming, T.; Möller, I.; Schwamborn, M.; et al. TERT Promoter Mutation Status as an Independent Prognostic Factor in Cutaneous Melanoma. JNCI J. Natl. Cancer Inst. 2014, 106, dju246b. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, Y.; Li, C.; Xiao, Z.; Yuan, H.; Zhang, Y.; Pang, D.; Tang, X.; Li, M.; Ouyang, H. TERT Promoter Revertant Mutation Inhibits Melanoma Growth through Intrinsic Apoptosis. Biology 2022, 11, 141. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Teng, M.W. 2018 Nobel Prize in physiology or medicine. Clin. Transl. Immunol. 2018, 7, e1041. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Chan, H.L.; Chen, P. Immune Checkpoint Inhibitors: Basics and Challenges. Curr. Med. Chem. 2019, 26, 3009–3025. [Google Scholar] [CrossRef]
- Chen, S.; Crabill, G.A.; Pritchard, T.S.; McMiller, T.L.; Wei, P.; Pardoll, D.M.; Pan, F.; Topalian, S.L. Mechanisms regulating PD-L1 expression on tumor and immune cells. J. Immunother. Cancer 2019, 7, 305. [Google Scholar] [CrossRef]
- Shiravand, Y.; Khodadadi, F.; Kashani, S.M.A.; Hosseini-Fard, S.R.; Hosseini, S.; Sadeghirad, H.; Ladwa, R.; O’Byrne, K.; Kulasinghe, A. Immune Checkpoint Inhibitors in Cancer Therapy. Curr. Oncol. 2022, 29, 3044–3060. [Google Scholar] [CrossRef]
- Madden, K.; Kasler, M.K. Immune Checkpoint Inhibitors in Lung Cancer and Melanoma. Semin. Oncol. Nurs. 2019, 35, 150932. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhang, J.; Xu, L.; Yang, H.; Liang, N.; Zhang, L.; Zhang, F.; Zhang, X. Safety and Efficacy of the Rechallenge of Immune Checkpoint Inhibitors after Immune-Related Adverse Events in Patients with Cancer: A Systemic Review and Meta-Analysis. Front. Immunol. 2021, 12, 730320. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Long-Term Outcomes with Nivolumab Plus Ipilimumab or Nivolumab Alone versus Ipilimumab in Patients with Advanced Melanoma. J. Clin. Oncol. 2022, 40, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Van Coillie, S.; Wiernicki, B.; Xu, J. Molecular and Cellular Functions of CTLA-4. Adv. Exp. Med. Biol. 2020, 1248, 7–32. [Google Scholar] [CrossRef]
- Nandi, D.; Pathak, S.; Verma, T.; Singh, M.; Chattopadhyay, A.; Thakur, S.; Raghavan, A.; Gokhroo, A.; Vijayamahantesh. T cell costimulation, checkpoint inhibitors and anti-tumor therapy. J. Biosci. 2020, 45, 50. [Google Scholar] [CrossRef]
- Weber, J.; Mandala, M.; Del Vecchio, M.; Gogas, H.J.; Arance, A.M.; Cowey, C.L.; Dalle, S.; Schenker, M.; Chiarion-Sileni, V.; Marquez-Rodas, I.; et al. Adjuvant Nivolumab versus Ipilimumab in Resected Stage III or IV Melanoma. N. Engl. J. Med. 2017, 377, 1824–1835. [Google Scholar] [CrossRef]
- Lorentzen, C.L.; Haanen, J.B.; Met, Ö.; Svane, I.M. Clinical advances and ongoing trials on mRNA vaccines for cancer treatment. Lancet Oncol. 2022, 23, e450–e458. [Google Scholar] [CrossRef]
- Bafaloukos, D.; Gazouli, I.; Koutserimpas, C.; Samonis, G. Evolution and Progress of mRNA Vaccines in the Treatment of Melanoma: Future Prospects. Vaccines 2023, 11, 636. [Google Scholar] [CrossRef]
- Peng, M.; Mo, Y.; Wang, Y.; Wu, P.; Zhang, Y.; Xiong, F.; Guo, C.; Wu, X.; Li, Y.; Li, X.; et al. Neoantigen vaccine: An emerging tumor immunotherapy. Mol. Cancer 2019, 18, 128. [Google Scholar] [CrossRef] [PubMed]
- Niemi, J.V.L.; Sokolov, A.V.; Schiöth, H.B. Neoantigen Vaccines; Clinical Trials, Classes, Indications, Adjuvants and Combinatorial Treatments. Cancers 2022, 14, 5163. [Google Scholar] [CrossRef] [PubMed]
- Gheena, S.; Ezhilarasan, D. Personalized mRNA cancer vaccines with immune checkpoint inhibitors: A promising therapeutic approach in oral cancer patients. Oral Oncol. 2023, 137, 106282. [Google Scholar] [CrossRef]
- Miao, L.; Zhang, Y.; Huang, L. mRNA vaccine for cancer immunotherapy. Mol. Cancer 2021, 20, 41. [Google Scholar] [CrossRef]
- Carvalho, T. Personalized anti-cancer vaccine combining mRNA and immunotherapy tested in melanoma trial. Nat. Med. 2023, 29, 2379–2380. [Google Scholar] [CrossRef] [PubMed]
- Cancer Discovery. mRNA Vaccine Slows Melanoma Recurrence. Cancer Discov 2023, 13, 1278. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Z.-X.; Chen, Y.-X.; Wu, H.-X.; Yin, L.; Zhao, Q.; Luo, H.; Zeng, Z.; Qiu, M.; Xu, R. Integrated analysis of single-cell and bulk RNA sequencing data reveals a pan-cancer stemness signature predicting immunotherapy response. Genome Med. 2022, 14, 45. [Google Scholar] [CrossRef] [PubMed]
- Schalck, A.; Sakellariou-Thompson, D.; Forget, M.-A.; Sei, E.; Hughes, T.G.; Reuben, A.; Bai, S.; Hu, M.; Kumar, T.; Hurd, M.W.; et al. Single-Cell Sequencing Reveals Trajectory of Tumor-Infiltrating Lymphocyte States in Pancreatic Cancer. Cancer Discov. 2022, 12, 2330–2349. [Google Scholar] [CrossRef] [PubMed]
- Huuhtanen, J.; Kasanen, H.; Peltola, K.; Lönnberg, T.; Glumoff, V.; Brück, O.; Dufva, O.; Peltonen, K.; Vikkula, J.; Jokinen, E.; et al. Single-cell characterization of anti-LAG-3 and anti-PD-1 combination treatment in patients with melanoma. J. Clin. Investig. 2023, 133, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Li, P.-H.; Kong, X.-Y.; He, Y.-Z.; Liu, Y.; Peng, X.; Li, Z.-H.; Xu, H.; Luo, H.; Park, J. Recent developments in application of single-cell RNA sequencing in the tumour immune microenvironment and cancer therapy. Mil. Med. Res. 2022, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Wang, F.; Hao, D.; Li, X.; Li, X.; Lei, T.; Yue, J.; Liu, C. Deciphering tumor-infiltrating dendritic cells in the single-cell era. Exp. Hematol. Oncol. 2023, 12, 97. [Google Scholar] [CrossRef]
- Khozyainova, A.A.; Valyaeva, A.A.; Arbatsky, M.S.; Isaev, S.V.; Iamshchikov, P.S.; Volchkov, E.V.; Sabirov, M.S.; Zainullina, V.R.; Chechekhin, V.I.; Vorobev, R.S.; et al. Complex Analysis of Single-Cell RNA Sequencing Data. Biochemistry 2023, 88, 231–252. [Google Scholar] [CrossRef]
- Kuksin, M.; Morel, D.; Aglave, M.; Danlos, F.-X.; Marabelle, A.; Zinovyev, A.; Gautheret, D.; Verlingue, L. Applications of single-cell and bulk RNA sequencing in onco-immunology. Eur. J. Cancer 2021, 149, 193–210. [Google Scholar] [CrossRef]
- Christodoulou, E.; Rashid, M.; Pacini, C.; Droop, A.; Robertson, H.; van Groningen, T.; Teunisse, A.F.A.S.; Iorio, F.; Jochemsen, A.G.; Adam, D.J.; et al. Analysis of CRISPR-Cas9 screens identifies genetic dependencies in melanoma. Pigment Cell Melanoma Res. 2021, 34, 122–131. [Google Scholar] [CrossRef]
- Wang, S.-W.; Gao, C.; Zheng, Y.-M.; Yi, L.; Lu, J.-C.; Huang, X.-Y.; Cai, J.-B.; Zhang, P.-F.; Cui, Y.-H.; Ke, A.-W. Current applications and future perspective of CRISPR/Cas9 gene editing in cancer. Mol. Cancer 2022, 21, 57. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y. Advances in CRISPR/Cas9. BioMed Res. Int. 2022, 2022, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Memi, F.; Ntokou, A.; Papangeli, I. CRISPR/Cas9 gene-editing: Research technologies, clinical applications and ethical considerations. Semin. Perinatol. 2018, 42, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Subica, A.M. CRISPR in Public Health: The Health Equity Implications and Role of Community in Gene-Editing Research and Applications. Am. J. Public Health 2023, 113, 874–882. [Google Scholar] [CrossRef] [PubMed]
- Bhinder, B.; Gilvary, C.; Madhukar, N.S.; Elemento, O. Artificial Intelligence in Cancer Research and Precision Medicine. Cancer Discov. 2021, 11, 900–915. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, Y.; Zheng, Q.; Li, J.; Huang, J.; Long, X. Artificial intelligence in melanoma: A systematic review. J. Cosmet. Dermatol. 2022, 21, 5993–6004. [Google Scholar] [CrossRef] [PubMed]
- Rajkomar, A.; Dean, J.; Kohane, I. Machine Learning in Medicine. N. Engl. J. Med. 2019, 380, 1347–1358. [Google Scholar] [CrossRef] [PubMed]
- Grossarth, S.; Mosley, D.; Madden, C.; Ike, J.; Smith, I.; Huo, Y.; Wheless, L. Recent Advances in Melanoma Diagnosis and Prognosis Using Machine Learning Methods. Curr. Oncol. Rep. 2023, 25, 635–645. [Google Scholar] [CrossRef]
- Tschandl, P.; Codella, N.; Akay, B.N.; Argenziano, G.; Braun, R.P.; Cabo, H.; Gutman, D.; Halpern, A.; Helba, B.; Hofmann-Wellenhof, R.; et al. Comparison of the accuracy of human readers versus machine-learning algorithms for pigmented skin lesion classification: An open, web-based, international, diagnostic study. Lancet Oncol. 2019, 20, 938–947. [Google Scholar] [CrossRef]
- Phillips, M.; Marsden, H.; Jaffe, W.; Matin, R.N.; Wali, G.N.; Greenhalgh, J.; McGrath, E.; James, R.; Ladoyanni, E.; Bewley, A.; et al. Assessment of Accuracy of an Artificial Intelligence Algorithm to Detect Melanoma in Images of Skin Lesions. JAMA Netw. Open 2019, 2, e1913436. [Google Scholar] [CrossRef]
- Prelaj, A.; Miskovic, V.; Zanitti, M.; Trovo, F.; Genova, C.; Viscardi, G.; Rebuzzi, S.E.; Mazzeo, L.; Provenzano, L.; Kosta, S.; et al. Artificial intelligence for predictive biomarker discovery in immuno-oncology: A systematic review. Ann. Oncol. 2024, 35, 29–65. [Google Scholar] [CrossRef]
- Kamińska, P.; Buszka, K.; Zabel, M.; Nowicki, M.; Alix-Panabières, C.; Budna-Tukan, J. Liquid Biopsy in Melanoma: Significance in Diagnostics, Prediction and Treatment Monitoring. Int. J. Mol. Sci. 2021, 22, 9714. [Google Scholar] [CrossRef]
- Wu, P.; Zhang, C.; Tang, X.; Li, D.; Zhang, G.; Zi, X.; Liu, J.; Yin, E.; Zhao, J.; Wang, P.; et al. Pan-cancer characterization of cell-free immune-related miRNA identified as a robust biomarker for cancer diagnosis. Mol. Cancer 2024, 23, 31. [Google Scholar] [CrossRef]
- Gaiser, M.R.; von Bubnoff, N.; Gebhardt, C.; Utikal, J.S. Liquid biopsy to monitor melanoma patients. JDDG J. Der Dtsch. Dermatol. Ges. 2018, 16, 405–414. [Google Scholar] [CrossRef]
- Boyer, M.; Cayrefourcq, L.; Dereure, O.; Meunier, L.; Becquart, O.; Alix-Panabières, C. Clinical Relevance of Liquid Biopsy in Melanoma and Merkel Cell Carcinoma. Cancers 2020, 12, 960. [Google Scholar] [CrossRef]
- Tivey, A.; Britton, F.; Scott, J.-A.; Rothwell, D.; Lorigan, P.; Lee, R. Circulating Tumour DNA in Melanoma—Clinic Ready? Curr. Oncol. Rep. 2022, 24, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Cassano, R.; Cuconato, M.; Calviello, G.; Serini, S.; Trombino, S. Recent Advances in Nanotechnology for the Treatment of Melanoma. Molecules 2021, 26, 785. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Gowda, B.H.J.; Ahmed, M.G.; Abourehab, M.A.S.; Chen, Z.-S.; Zhang, C.; Li, J.; Kesharwani, P. Advancements in nanoparticle-based treatment approaches for skin cancer therapy. Mol. Cancer 2023, 22, 10. [Google Scholar] [CrossRef] [PubMed]
- Adamus-Grabicka, A.A.; Hikisz, P.; Sikora, J. Nanotechnology as a Promising Method in the Treatment of Skin Cancer. Int. J. Mol. Sci. 2024, 25, 2165. [Google Scholar] [CrossRef]
- Volovat, S.; Negru, S.; Stolniceanu, C.; Volovat, C.; Lungulescu, C.; Scripcariu, D.; Cobzeanu, B.; Stefanescu, C.; Grigorescu, C.; Augustin, I.; et al. Nanomedicine to modulate immunotherapy in cutaneous melanoma (Review). Exp. Ther. Med. 2021, 21, 535. [Google Scholar] [CrossRef]
- Beiu, C.; Giurcaneanu, C.; Grumezescu, A.M.; Holban, A.M.; Popa, L.G.; Mihai, M.M. Nanosystems for Improved Targeted Therapies in Melanoma. J. Clin. Med. 2020, 9, 318. [Google Scholar] [CrossRef] [PubMed]
- Zahedipour, F.; Zamani, P.; Jamialahmadi, K.; Jaafari, M.R.; Sahebkar, A. Vaccines targeting angiogenesis in melanoma. Eur. J. Pharmacol. 2021, 912, 174565. [Google Scholar] [CrossRef] [PubMed]
- Haanen, J.; Los, C.; Phan, G.Q.; Betof Warner, A. Adoptive Cell Therapy for Solid Tumors: Current Status in Melanoma and Next-Generation Therapies. Am. Soc. Clin. Oncol. Educ. Book 2024, 44, e431608. [Google Scholar] [CrossRef] [PubMed]
- Soltantoyeh, T.; Akbari, B.; Karimi, A.; Mahmoodi Chalbatani, G.; Ghahri-Saremi, N.; Hadjati, J.; Hamblin, M.R.; Mirzaei, H.R. Chimeric Antigen Receptor (CAR) T Cell Therapy for Metastatic Melanoma: Challenges and Road Ahead. Cells 2021, 10, 1450. [Google Scholar] [CrossRef] [PubMed]
- Jilani, S.; Saco, J.D.; Mugarza, E.; Pujol-Morcillo, A.; Chokry, J.; Ng, C.; Abril-Rodriguez, G.; Berger-Manerio, D.; Pant, A.; Hu, J.; et al. CAR-T cell therapy targeting surface expression of TYRP1 to treat cutaneous and rare melanoma subtypes. Nat. Commun. 2024, 15, 1244. [Google Scholar] [CrossRef] [PubMed]
- Natarelli, N.; Aleman, S.J.; Mark, I.M.; Tran, J.T.; Kwak, S.; Botto, E.; Aflatooni, S.; Diaz, M.J.; Lipner, S.R. A Review of Current and Pipeline Drugs for Treatment of Melanoma. Pharmaceuticals 2024, 17, 214. [Google Scholar] [CrossRef] [PubMed]
- Chesney, J.; Lewis, K.D.; Kluger, H.; Hamid, O.; Whitman, E.; Thomas, S.; Wermke, M.; Cusnir, M.; Domingo-Musibay, E.; Phan, G.Q.; et al. Efficacy and safety of lifileucel, a one-time autologous tumor-infiltrating lymphocyte (TIL) cell therapy, in patients with advanced melanoma after progression on immune checkpoint inhibitors and targeted therapies: Pooled analysis of consecutive cohorts of the C-144-01 study. J. Immunother. Cancer 2022, 10, e005755. [Google Scholar] [CrossRef]
- Zhao, Z.; Chen, Y.; Francisco, N.M.; Zhang, Y.; Wu, M. The application of CAR-T cell therapy in hematological malignancies: Advantages and challenges. Acta Pharm. Sin. B 2018, 8, 539–551. [Google Scholar] [CrossRef]
- Sivori, S.; Pende, D.; Quatrini, L.; Pietra, G.; Della Chiesa, M.; Vacca, P.; Tumino, N.; Moretta, F.; Mingari, M.C.; Locatelli, F.; et al. NK cells and ILCs in tumor immunotherapy. Mol. Aspects Med. 2021, 80, 100870. [Google Scholar] [CrossRef]
- Chen, J.; López-Moyado, I.F.; Seo, H.; Lio, C.-W.J.; Hempleman, L.J.; Sekiya, T.; Yoshimura, A.; Scott-Browne, J.P.; Rao, A. NR4A transcription factors limit CAR T cell function in solid tumours. Nature 2019, 567, 530–534. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valdez-Salazar, F.; Jiménez-Del Rio, L.A.; Padilla-Gutiérrez, J.R.; Valle, Y.; Muñoz-Valle, J.F.; Valdés-Alvarado, E. Advances in Melanoma: From Genetic Insights to Therapeutic Innovations. Biomedicines 2024, 12, 1851. https://doi.org/10.3390/biomedicines12081851
Valdez-Salazar F, Jiménez-Del Rio LA, Padilla-Gutiérrez JR, Valle Y, Muñoz-Valle JF, Valdés-Alvarado E. Advances in Melanoma: From Genetic Insights to Therapeutic Innovations. Biomedicines. 2024; 12(8):1851. https://doi.org/10.3390/biomedicines12081851
Chicago/Turabian StyleValdez-Salazar, Fernando, Luis A. Jiménez-Del Rio, Jorge R. Padilla-Gutiérrez, Yeminia Valle, José F. Muñoz-Valle, and Emmanuel Valdés-Alvarado. 2024. "Advances in Melanoma: From Genetic Insights to Therapeutic Innovations" Biomedicines 12, no. 8: 1851. https://doi.org/10.3390/biomedicines12081851