
Dysregulated Non-Coding RNA Expression in T Cells from Patients with Ankylosing Spondylitis Contributes to Its Immunopathogenesis


Abstract
:1. Introduction
2. Pathogenesis of AS
2.1. Genetic Inheritance
2.2. Environmental Factors
2.3. T Cells
2.3.1. CD4+ T Cells
2.3.2. Formation of Autoantibodies
2.3.3. CD8+ T Cells
3. NcRNAs
3.1. Overview of Abnormal Expression of ncRNA in Patients with AS
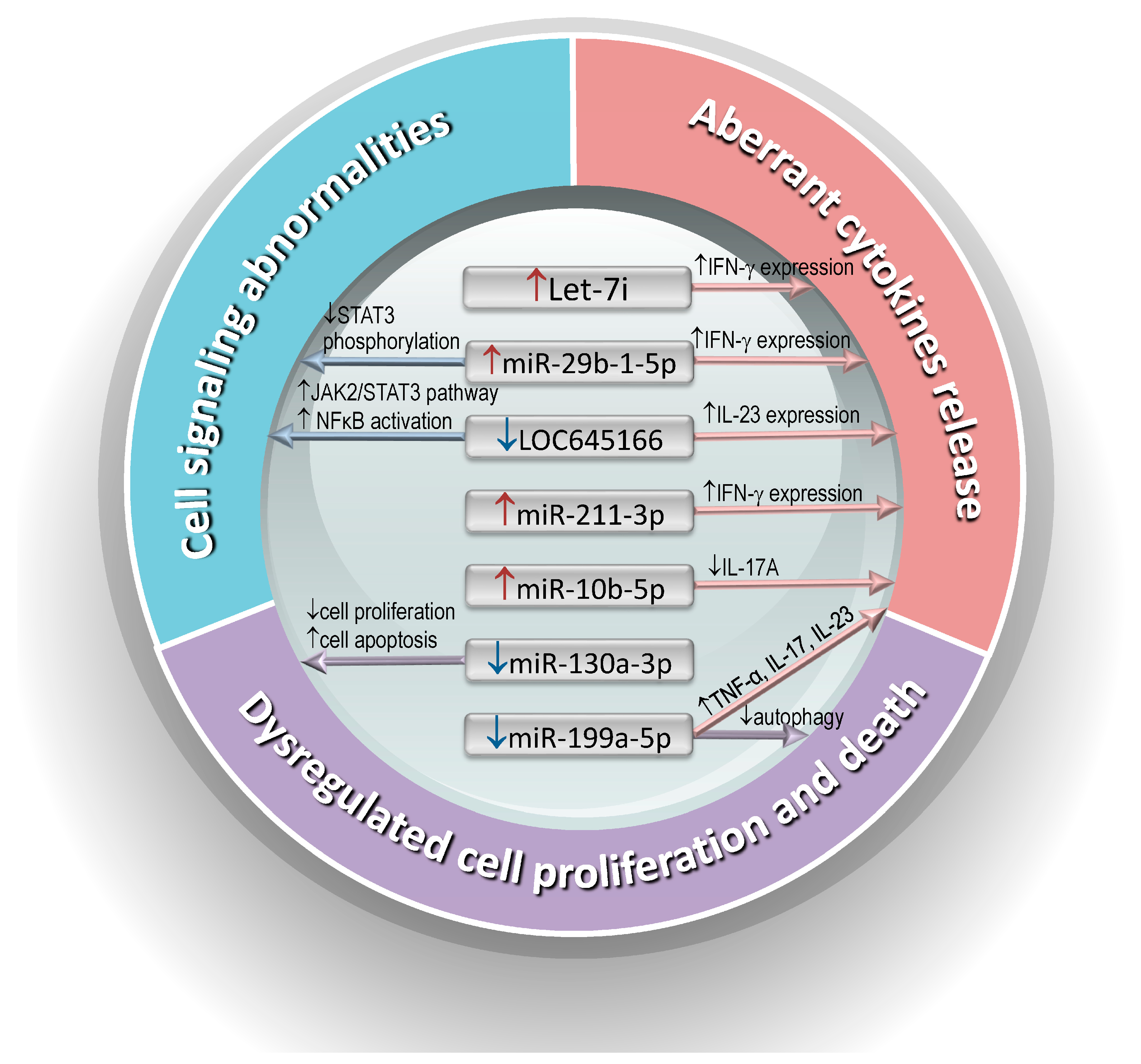
3.2. Abnormal Expression of ncRNAs in AS T Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- van der Heijde, D.; Molto, A.; Ramiro, S.; Braun, J.; Dougados, M.; van Gaalen, F.A.; Gensler, L.S.; Inman, R.D.; Landewé, R.B.M.; Marzo-Ortega, H.; et al. Goodbye to the term ‘ankylosing spondylitis’, hello ‘axial spondyloarthritis’: Time to embrace the ASAS-defined nomenclature. Ann. Rheum. Dis. 2024, 83, 547–549. [Google Scholar] [CrossRef] [PubMed]
- Taurog, J.D.; Chhabra, A.; Colbert, R.A. Ankylosing Spondylitis and Axial Spondyloarthritis. N. Engl. J. Med. 2016, 374, 2563–2574. [Google Scholar] [CrossRef]
- Sieper, J.; Poddubnyy, D. Axial spondyloarthritis. Lancet 2017, 390, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, H.; Bohra, N.; Syed, K.; Donato, A.; Murad, M.H.; Karmacharya, P. All-cause and cause-specific mortality in psoriatic arthritis and ankylosing spondylitis: A systematic review and meta-analysis. Arthritis Care Res. 2023, 75, 1052–1065. [Google Scholar] [CrossRef] [PubMed]
- Schlosstein, L.; Terasaki, P.I.; Bluestone, R.; Pearson, C.M. High association of an HL-A antigen, W27, with ankylosing spondylitis. N. Engl. J. Med. 1973, 288, 704–706. [Google Scholar] [CrossRef] [PubMed]
- Ganau, L.; Prisco, L.; Ligarotti, G.K.I.; Ambu, R.; Ganau, M. Understanding the pathological basis of neurological diseases through diagnostic platforms based on innovations in biomedical engineering: New concepts and theranostics perspectives. Medicines 2018, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Rosine, N.; Fogel, O.; Koturan, S.; Rogge, L.; Bianchi, E.; Miceli-Richard, C. T cells in the pathogenesis of axial spondyloarthritis. Jt. Bone Spine 2023, 90, 105619. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chang, C.; Lu, Q. Epigenetics of CD4+ T cells in autoimmune diseases. Curr. Opin. Rheumatol. 2017, 29, 361–368. [Google Scholar] [CrossRef] [PubMed]
- van der Linden, S.; Valkenburg, H.A.; Cats, A. Evaluation of diagnostic criteria for ankylosing spondylitis. Arthritis Rheum. 1984, 27, 361–368. [Google Scholar] [CrossRef]
- Rudwaleit, M.; van der Heijde, D.; Landewé, R.; Akkoc, N.; Brandt, J.; Chou, C.T.; Dougados, M.; Huang, F.; Gu, J.; Kirazli, Y.; et al. The Assessment of SpondyloArthritis International Society classification criteria for peripheral spondyloarthritis and for spondyloarthritis in general. Ann. Rheum. Dis. 2011, 70, 25–31. [Google Scholar] [CrossRef]
- Garrett, S.; Jenkinson, T.; Kennedy, L.G.; Whitelock, H.; Gaisford, P.; Calin, A. A new approach to defining disease status in ankylosing spondylitis: The Bath Ankylosing Spondylitis Disease Activity Index. J. Rheumatol. 1994, 21, 2286–2291. [Google Scholar]
- Machado, P.; Landewé, R.; Lie, E.; Kvien, T.K.; Braun, J.; Baker, D.; van der Heijde, D. Ankylosing Spondylitis Disease Activity Score (ASDAS): Defining cut-off values for disease activity states and improvement scores. Ann. Rheum. Dis. 2011, 70, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Ambarus, C.; Yeremenko, N.; Tak, P.P.; Baeten, D. Pathogenesis of spondyloarthritis: Autoimmune or autoinflammatory? Curr. Opin. Rheumatol. 2012, 24, 351–358. [Google Scholar] [CrossRef]
- McGonagle, D.; McDermott, M.F. A proposed classification of the immunological diseases. PLoS Med. 2006, 3, e297. [Google Scholar] [CrossRef]
- Braun, J.; Bollow, M.; Remlinger, G.; Eggens, U.; Rudwaleit, M.; Distler, A.; Sieper, J. Prevalence of spondylarthropathies in HLA-B27 positive and negative blood donors. Arthritis Rheum. 1998, 41, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.; Sieper, J. Fifty years after the discovery of the association of HLA B27 with ankylosing spondylitis. RMD Open 2023, 9, e003102. [Google Scholar] [CrossRef]
- DeLay, M.L.; Turner, M.J.; Klenk, E.I.; Smith, J.A.; Sowders, D.P.; Colbert, R.A. HLA-B27 misfolding and the unfolded protein response augment interleukin-23 production and are associated with Th17 activation in transgenic rats. Arthritis Rheum. 2009, 60, 2633–2643. [Google Scholar] [CrossRef]
- Yu, H.C.; Lu, M.C.; Li, C.; Huang, H.L.; Huang, K.Y.; Liu, S.Q.; Lai, N.S.; Huang, H.B. Targeted delivery of an antigenic peptide to the endoplasmic reticulum: Application for development of a peptide therapy for ankylosing spondylitis. PLoS ONE 2013, 8, e77451. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.C.; Huang, K.Y.; Lu, M.C.; Huang, H.L.; Liu, S.Q.; Lai, N.S.; Huang, H.B. Targeted Delivery of the HLA-B∗27-Binding Peptide into the Endoplasmic Reticulum Suppresses the IL-23/IL-17 Axis of Immune Cells in Spondylarthritis. Mediat. Inflamm. 2017, 2017, 4016802. [Google Scholar] [CrossRef]
- Bowness, P.; Ridley, A.; Shaw, J.; Chan, A.T.; Wong-Baeza, I.; Fleming, M.; Cummings, F.; McMichael, A.; Kollnberger, S. Th17 cells expressing KIR3DL2+ and responsive to HLA-B27 homodimers are increased in ankylosing spondylitis. J. Immunol. 2011, 186, 2672–2680. [Google Scholar] [CrossRef]
- Reveille, J.D. The genetic basis of spondyloarthritis. Ann. Rheum. Dis. 2011, 70 (Suppl. S1), i44–i50. [Google Scholar] [CrossRef] [PubMed]
- Simone, D.; Al Mossawi, M.H.; Bowness, P. Progress in our understanding of the pathogenesis of ankylosing spondylitis. Rheumatology 2018, 57 (Suppl. S6), vi4–vi9. [Google Scholar] [CrossRef] [PubMed]
- Rudwaleit, M.; Baeten, D. Ankylosing spondylitis and bowel disease. Best Pract. Research. Clin. Rheumatol. 2006, 20, 451–471. [Google Scholar] [CrossRef]
- Song, Z.Y.; Yuan, D.; Zhang, S.X. Role of the microbiome and its metabolites in ankylosing spondylitis. Front. Immunol. 2022, 13, 1010572. [Google Scholar] [CrossRef] [PubMed]
- Costello, M.E.; Ciccia, F.; Willner, D.; Warrington, N.; Robinson, P.C.; Gardiner, B.; Marshall, M.; Kenna, T.J.; Triolo, G.; Brown, M.A. Brief Report: Intestinal Dysbiosis in Ankylosing Spondylitis. Arthritis Rheumatol. 2015, 67, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Lee, S.H. Updates on ankylosing spondylitis: Pathogenesis and therapeutic agents. J. Rheum. Dis. 2023, 30, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.; Yen-Ting Chen, T.; Wang, S.I.; Hung, Y.M.; Chen, H.Y.; Wei, C.J. Risk of autoimmune diseases in patients with COVID-19: A retrospective cohort study. EClinicalMedicine 2023, 56, 101783. [Google Scholar] [CrossRef] [PubMed]
- Jacques, P.; Lambrecht, S.; Verheugen, E.; Pauwels, E.; Kollias, G.; Armaka, M.; Verhoye, M.; Van der Linden, A.; Achten, R.; Lories, R.J.; et al. Proof of concept: Enthesitis and new bone formation in spondyloarthritis are driven by mechanical strain and stromal cells. Ann. Rheum. Dis. 2014, 73, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Baeten, D.; Kruithof, E.; Van den Bosch, F.; Demetter, P.; Van Damme, N.; Cuvelier, C.; De Vos, M.; Mielants, H.; Veys, E.M.; De Keyser, F. Immunomodulatory effects of anti-tumor necrosis factor alpha therapy on synovium in spondylarthropathy: Histologic findings in eight patients from an open-label pilot study. Arthritis Rheum. 2001, 44, 186–195. [Google Scholar] [CrossRef]
- Bollow, M.; Fischer, T.; Reisshauer, H.; Backhaus, M.; Sieper, J.; Hamm, B.; Braun, J. Quantitative analyses of sacroiliac biopsies in spondyloarthropathies: T cells and macrophages predominate in early and active sacroiliitis—Cellularity correlates with the degree of enhancement detected by magnetic resonance imaging. Ann. Rheum. Dis. 2000, 59, 135–140. [Google Scholar] [CrossRef]
- Wen, J.T.; Zhang, D.H.; Fang, P.F.; Li, M.H.; Wang, R.J.; Li, S.H. Role of Th1/Th2 cytokines in the diagnosis and prognostic evaluation of ankylosing spondylitis. Genet. Mol. Res. 2017, 16, 10–4238. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.R.; Vecellio, M.; Chen, L.; Ridley, A.; Cortes, A.; Knight, J.C.; Bowness, P.; Cohen, C.J.; Wordsworth, B.P. An ankylosing spondylitis-associated genetic variant in the IL23R-IL12RB2 intergenic region modulates enhancer activity and is associated with increased Th1-cell differentiation. Ann. Rheum. Dis. 2016, 75, 2150–2156. [Google Scholar] [CrossRef]
- Shesternya, P.A.; Savchenko, A.A.; Gritsenko, O.D.; Vasileva, A.O.; Kudryavtsev, I.V.; Masterova, A.A.; Isakov, D.V.; Borisov, A.G. Features of peripheral blood th-cell subset composition and serum cytokine level in patients with activity-driven ankylosing spondylitis. Pharmaceuticals 2022, 15, 1370. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.S.; Chang, Y.S.; Lin, K.C.; Lai, C.C.; Wang, S.H.; Hsiao, K.H.; Lee, H.T.; Chen, M.H.; Tsai, C.Y.; Chou, C.T. Association of serum interleukin-17 and interleukin-23 levels with disease activity in Chinese patients with ankylosing spondylitis. J. Chin. Med. Assoc. 2012, 75, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.Y.; Zheng, J.W.; Yang, J.Y.; Zhang, T.Y.; Song, S.; Zhao, R.; Di, J.K.; Zhang, S.X.; Wang, C.H.; Gao, H.Y. Levels of peripheral th17 cells and th17-related cytokines in patients with ankylosing spondylitis: A meta-analysis. Adv. Ther. 2022, 39, 4423–4439. [Google Scholar] [CrossRef] [PubMed]
- van der Heijde, D.; Cheng-Chung Wei, J.; Dougados, M.; Mease, P.; Deodhar, A.; Maksymowych, W.P.; Van den Bosch, F.; Sieper, J.; Tomita, T.; Landewé, R.; et al. Ixekizumab, an interleukin-17A antagonist in the treatment of ankylosing spondylitis or radiographic axial spondyloarthritis in patients previously untreated with biological disease-modifying anti-rheumatic drugs (COAST-V): 16 week results of a phase 3 randomised, double-blind, active-controlled and placebo-controlled trial. Lancet 2018, 392, 2441–2451. [Google Scholar] [CrossRef] [PubMed]
- Lai, N.L.; Zhang, S.X.; Wang, J.; Zhang, J.Q.; Wang, C.H.; Gao, C.; Li, X.F. The Proportion of Regulatory T Cells in Patients with Ankylosing Spondylitis: A Meta-Analysis. J. Immunol. Res. 2019, 2019, 1058738. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.T.; Tsai, C.Y. Cytokines and regulatory T cells in ankylosing spondylitis. Bone Jt. Res. 2023, 12, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Rodolfi, S.; Davidson, C.; Vecellio, M. Regulatory T cells in spondyloarthropathies: Genetic evidence, functional role, and therapeutic possibilities. Front. Immunol. 2023, 14, 1303640. [Google Scholar] [CrossRef] [PubMed]
- Lejon, K.; Hellman, U.; Do, L.; Kumar, A.; Forsblad-d’Elia, H. Increased proportions of inflammatory T cells and their correlations with cytokines and clinical parameters in patients with ankylosing spondylitis from northern Sweden. Scand. J. Immunol. 2022, 96, e13190. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Y.G.; Li, Y.H.; Qi, L.; Liu, X.G.; Yuan, C.Z.; Hu, N.W.; Ma, D.X.; Li, Z.F.; Yang, Q.; et al. Increased frequencies of Th22 cells as well as Th17 cells in the peripheral blood of patients with ankylosing spondylitis and rheumatoid arthritis. PLoS ONE 2012, 7, e31000. [Google Scholar] [CrossRef]
- Xiao, F.; Zhang, H.Y.; Liu, Y.J.; Zhao, D.; Shan, Y.X.; Jiang, Y.F. Higher frequency of peripheral blood interleukin 21 positive follicular helper T cells in patients with ankylosing spondylitis. J. Rheumatol. 2013, 40, 2029–2037. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Chen, L.; Zhao, Q.; Zheng, Z.H.; Chen, Z.N.; Bian, H.; Yang, X.; Lu, H.Y.; Lin, P.; Chen, X.; et al. Cysteine carboxyethylation generates neoantigens to induce HLA-restricted autoimmunity. Science 2023, 379, eabg2482. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jarvis, L.B.; Baek, H.J.; Gaston, J.S. Regulatory IL4+CD8+ T cells in patients with ankylosing spondylitis and healthy controls. Ann. Rheum. Dis. 2009, 68, 1345–1351. [Google Scholar] [CrossRef]
- Baek, H.J.; Zhang, L.; Jarvis, L.B.; Gaston, J.S. Increased IL-4+ CD8+ T cells in peripheral blood and autoreactive CD8+ T cell lines of patients with inflammatory arthritis. Rheumatology 2008, 47, 795–803. [Google Scholar] [CrossRef]
- Gracey, E.; Yao, Y.; Qaiyum, Z.; Lim, M.; Tang, M.; Inman, R.D. Altered Cytotoxicity Profile of CD8+ T Cells in Ankylosing Spondylitis. Arthritis Rheumatol. 2020, 72, 428–434. [Google Scholar] [CrossRef]
- Perl, A.; Morel, L. Expanding scope of TEMRA in autoimmunity. EBioMedicine 2023, 90, 104520. [Google Scholar] [CrossRef] [PubMed]
- Martini, V.; Silvestri, Y.; Ciurea, A.; Möller, B.; Danelon, G.; Flamigni, F.; Jarrossay, D.; Kwee, I.; Foglierini, M.; Rinaldi, A.; et al. Patients with ankylosing spondylitis present a distinct CD8 T cell subset with osteogenic and cytotoxic potential. RMD Open 2024, 10, e003926. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.C. Regulatory RNAs in rheumatology: From pathogenesis to potential therapy. Int. J. Rheum. Dis. 2023, 26, 605–606. [Google Scholar] [CrossRef]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef]
- Lai, N.S.; Koo, M.; Yu, C.L.; Lu, M.C. Immunopathogenesis of systemic lupus erythematosus and rheumatoid arthritis: The role of aberrant expression of non-coding RNAs in T cells. Clin. Exp. Immunol. 2017, 187, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Chen, Y.; Xu, W.; Shao, M.; Deng, J.; Xu, S.; Gao, X.; Guan, S.; Wang, J.; Xu, S.; et al. Epigenetics of ankylosing spondylitis: Recent developments. Int. J. Rheum. Dis. 2021, 24, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Ren, R.; Zhang, J.; Huang, Z.; Niu, Q.; Yang, B. Analysis of inflammation-related microRNA expression in patients with ankylosing spondylitis. Immunol. Res. 2022, 70, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.P.; Zhang, Q.B.; Dai, F.; Liao, X.; Dong, Z.R.; Yi, T.; Qing, Y.F. Circular RNAs in peripheral blood mononuclear cells from ankylosing spondylitis. Chin. Med. J. 2021, 134, 2573–2582. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhou, X. Long noncoding RNA intersectin 1-2 gradually declines during adalimumab treatment, and its reduction correlates with treatment efficacy in patients with ankylosing spondylitis. Inflammopharmacology 2021, 29, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Wang, X.; Sun, X.; Zhao, B.; Zhang, X.; Gong, X.; Wong, S.H.; Chan, M.T.V.; Wu, W.K.K. emerging roles of long non-coding RNAS in ankylosing spondylitis. Front. Immunol. 2022, 13, 790924. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Liu, J. Novel regulatory role of non-coding RNAs in ankylosing spondylitis. Front. Immunol. 2023, 14, 1131355. [Google Scholar] [CrossRef]
- Lai, N.S.; Yu, H.C.; Chen, H.C.; Yu, C.L.; Huang, H.B.; Lu, M.C. Aberrant expression of microRNAs in T cells from patients with ankylosing spondylitis contributes to the immunopathogenesis. Clin. Exp. Immunol. 2013, 173, 47–57. [Google Scholar] [CrossRef]
- Reyes-Loyola, P.; Rodríguez-Henríquez, P.; Ballinas-Verdugo, M.A.; Amezcua-Castillo, L.M.; Juárez-Vicuña, Y.; Jiménez-Rojas, V.; Márquez-Velasco, R.; Sánchez-Muñoz, F.; Amezcua-Guerra, L.M. Plasma let-7i, miR-16, and miR-221 levels as candidate biomarkers for the assessment of ankylosing spondylitis in Mexican patients naïve to anti-TNF therapy. Clin. Rheumatol. 2019, 38, 1367–1373. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Hou, C.; Zhu, M.; Sun, M.; Lin, Y. MicroRNA let-7i induced autophagy to protect T cell from apoptosis by targeting IGF1R. Biochem. Biophys. Res. Commun. 2014, 453, 728–734. [Google Scholar] [CrossRef]
- Lu, L.; Fang, H.; Gu, M.; Wang, H.; Yu, Q.; Chen, A.; Gan, K.F. MicroRNA Let-7i Regulates Innate TLR4 Pathways in Peripheral Blood Mononuclear Cells of Patients with Ankylosing Spondylitis. Int. J. Gen. Med. 2023, 16, 1393–1401. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, J.; Wang, X.; Yang, B.; Cui, L. MicroRNA-199a-5p induced autophagy and inhibits the pathogenesis of ankylosing spondylitis by modulating the mTOR signaling via directly targeting Ras homolog enriched in brain (Rheb). Cell. Physiol. Biochem. 2017, 42, 2481–2491. [Google Scholar] [CrossRef]
- Tan, M.; Zhang, Q.B.; Liu, T.H.; Yang, Y.Y.; Zheng, J.X.; Zhou, W.J.; Xiong, Q.; Qing, Y.F. Autophagy dysfunction may be involved in the pathogenesis of ankylosing spondylitis. Exp. Ther. Med. 2020, 20, 3578–3586. [Google Scholar] [CrossRef]
- Chen, L.; Al-Mossawi, M.H.; Ridley, A.; Sekine, T.; Hammitzsch, A.; de Wit, J.; Simone, D.; Shi, H.; Penkava, F.; Kurowska-Stolarska, M.; et al. miR-10b-5p is a novel Th17 regulator present in Th17 cells from ankylosing spondylitis. Ann. Rheum. Dis. 2017, 76, 620–625. [Google Scholar] [CrossRef]
- Schinocca, C.; Rizzo, C.; Fasano, S.; Grasso, G.; La Barbera, L.; Ciccia, F.; Guggino, G. Role of the IL-23/IL-17 pathway in rheumatic diseases: An overview. Front. Immunol. 2021, 12, 637829. [Google Scholar] [CrossRef] [PubMed]
- Lai, N.S.; Yu, H.C.; Tung, C.H.; Huang, K.Y.; Huang, H.B.; Lu, M.C. Aberrant expression of interleukin-23-regulated miRNAs in T cells from patients with ankylosing spondylitis. Arthritis Res. Ther. 2018, 20, 259. [Google Scholar] [CrossRef] [PubMed]
- Baeten, D.; Østergaard, M.; Wei, J.C.; Sieper, J.; Järvinen, P.; Tam, L.S.; Salvarani, C.; Kim, T.H.; Solinger, A.; Datsenko, Y.; et al. Risankizumab, an IL-23 inhibitor, for ankylosing spondylitis: Results of a randomised, double-blind, placebo-controlled, proof-of-concept, dose-finding phase 2 study. Ann. Rheum. Dis. 2018, 77, 1295–1302. [Google Scholar] [CrossRef]
- Baeten, D.; Adamopoulos, I.E. IL-23 Inhibition in ankylosing spondylitis: Where did it go wrong? Front. Immunol. 2020, 11, 623874. [Google Scholar] [CrossRef] [PubMed]
- Fogel, O.; Bugge Tinggaard, A.; Fagny, M.; Sigrist, N.; Roche, E.; Leclere, L.; Deleuze, J.F.; Batteux, F.; Dougados, M.; Miceli-Richard, C.; et al. Deregulation of microRNA expression in monocytes and CD4+ T lymphocytes from patients with axial spondyloarthritis. Arthritis Res. Ther. 2019, 21, 51. [Google Scholar] [CrossRef]
- Li, F.; Si, D.; Guo, X.; Guo, N.; Li, D.; Zhang, L.; Jian, X.; Ma, J. Aberrant expression of miR-130a-3p in ankylosing spondylitis and its role in regulating T-cell survival. Mol. Med. Rep. 2019, 20, 3388–3394. [Google Scholar] [CrossRef]
- Yu, H.C.; Huang, K.Y.; Lu, M.C.; Huang Tseng, H.Y.; Liu, S.Q.; Lai, N.S.; Huang, H.B. Down-regulation of LOC645166 in T cells of ankylosing spondylitis patients promotes the NF-κB signaling via decreasingly blocking recruitment of the IKK complex to K63-linked polyubiquitin chains. Front. Immunol. 2021, 12, 591706. [Google Scholar] [CrossRef]
- Raychaudhuri, S.P.; Shah, R.J.; Banerjee, S.; Raychaudhuri, S.K. JAK-STAT signaling and beyond in the pathogenesis of spondyloarthritis and their clinical significance. Curr. Rheumatol. Rep. 2024, 26, 204–213. [Google Scholar] [CrossRef]
- Deodhar, A.; Sliwinska-Stanczyk, P.; Xu, H.; Baraliakos, X.; Gensler, L.S.; Fleishaker, D.; Wang, L.; Wu, J.; Menon, S.; Wang, C.; et al. Tofacitinib for the treatment of ankylosing spondylitis: A phase III, randomised, double-blind, placebo-controlled study. Ann. Rheum. Dis. 2021, 80, 1004–1013. [Google Scholar] [CrossRef]
- Tavasolian, F.; Lively, S.; Pastrello, C.; Tang, M.; Lim, M.; Pacheco, A.; Qaiyum, Z.; Yau, E.; Baskurt, Z.; Jurisica, I.; et al. Proteomic and genomic profiling of plasma exosomes from patients with ankylosing spondylitis. Ann. Rheum. Dis. 2023, 82, 1429–1443. [Google Scholar] [CrossRef]
- Liu, C.; Yang, H.; Shi, W.; Wang, T.; Ruan, Q. MicroRNA-mediated regulation of T helper type 17/regulatory T-cell balance in autoimmune disease. Immunology 2018, 155, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Bi, K.; Zhang, X.; Chen, W.; Diao, H. MicroRNAs regulate intestinal immunity and gut microbiota for gastrointestinal health: A comprehensive review. Genes 2020, 11, 1075. [Google Scholar] [CrossRef] [PubMed]
- Cuesta, C.M.; Guerri, C.; Ureña, J.; Pascual, M. Role of microbiota-derived extracellular vesicles in gut-brain communication. Int. J. Mol. Sci. 2021, 22, 4235. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| ncRNAs | Validated Targets | Signaling Pathway | Function | Patient Number | Reference and Study Year |
|---|---|---|---|---|---|
| miR-16↑, miR-221↑and let-7i↑ | TLR-4 | LPS/TLR-4 | Enhanced IFN-γ expression | 22 | 2013 [58] |
| let-7i | IGF-1R | mTOR/AKT | Induced autophagy | - | 2014 [61] |
| miR-199a-5p↓ | Rheb | mTOR | Inhibited autophagy | 41 | 2017 [63] |
| miR-10b-5p↑, miR-155-5p↑, and miR-210-3p↑ | MAP3K7 | MAPKs | Reduced Th17 differentiation and IL-17A production | 15 | 2017 [65] |
| miR-16-1-3p↑, miR-28-5p↑, miR-199a-5p↑, and miR-126-3p↑ | - | - | - | 81 | 2019 [70] |
| miR-130a-3p↓ | Homeobox B1 | Bcl-2/Bax pathway | Inhibited T-cell proliferation and induced T-cell apoptosis | 30 | 2019 [71] |
| miR-29b-1-5p↑, miR-4449↑, miR-211-3p↑, miR-1914-3p↑, and miR-7114-5p↑ | Angiogenin | JAK/STAT pathway | Enhanced IFN-γ expression | 24 | 2018 [67] |
| LOC100506014↓, LOC645166↓, lncEXD2-1↓, and LINC00282↓ | K63-linked polyubiquitin chains | JAK/STAT and NFkB pathway | Enhanced IL-23 expression | 30 | 2021 [72] |
| * let-7b-5p↓ and let-7c-5p↓ miR-30d-5p↑, miR-93-5p↑, miR-23b-3p↑, miR-191–5p↑, miR-148b-3p↑,miR-146b-5p↑,miR-1260b↑, miR-130a-5p↑, miR-140-3p↑, miR-145-3p↑, miR-22-5p↑, miR-2277-5p↑, miR-27a-3p↑, miR-29a-3p↑, miR-30c-5p↑, miR-330-5p↑, miR-345-5p↑, miR-4717-3p↑, miR-500a-3p↑, miR-502-3p↑, miR-598-3p↑, miR-6810-3p↑ | - | Covalent chromatin modification, chromatin modifying enzymes, nuclear chromatin, chromatin DNA binding | Inhibited the proliferation of regulatory T cells | 17 | 2023 [75] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, H.-C.; Wang, S.-T.; Lu, M.-C. Dysregulated Non-Coding RNA Expression in T Cells from Patients with Ankylosing Spondylitis Contributes to Its Immunopathogenesis. Biomedicines 2024, 12, 1873. https://doi.org/10.3390/biomedicines12081873
Yu H-C, Wang S-T, Lu M-C. Dysregulated Non-Coding RNA Expression in T Cells from Patients with Ankylosing Spondylitis Contributes to Its Immunopathogenesis. Biomedicines. 2024; 12(8):1873. https://doi.org/10.3390/biomedicines12081873
Chicago/Turabian StyleYu, Hui-Chun, Sz-Tsan Wang, and Ming-Chi Lu. 2024. "Dysregulated Non-Coding RNA Expression in T Cells from Patients with Ankylosing Spondylitis Contributes to Its Immunopathogenesis" Biomedicines 12, no. 8: 1873. https://doi.org/10.3390/biomedicines12081873