
Application of PPAR Ligands and Nanoparticle Technology in Metabolic Steatohepatitis Treatment


Abstract
:1. Introduction
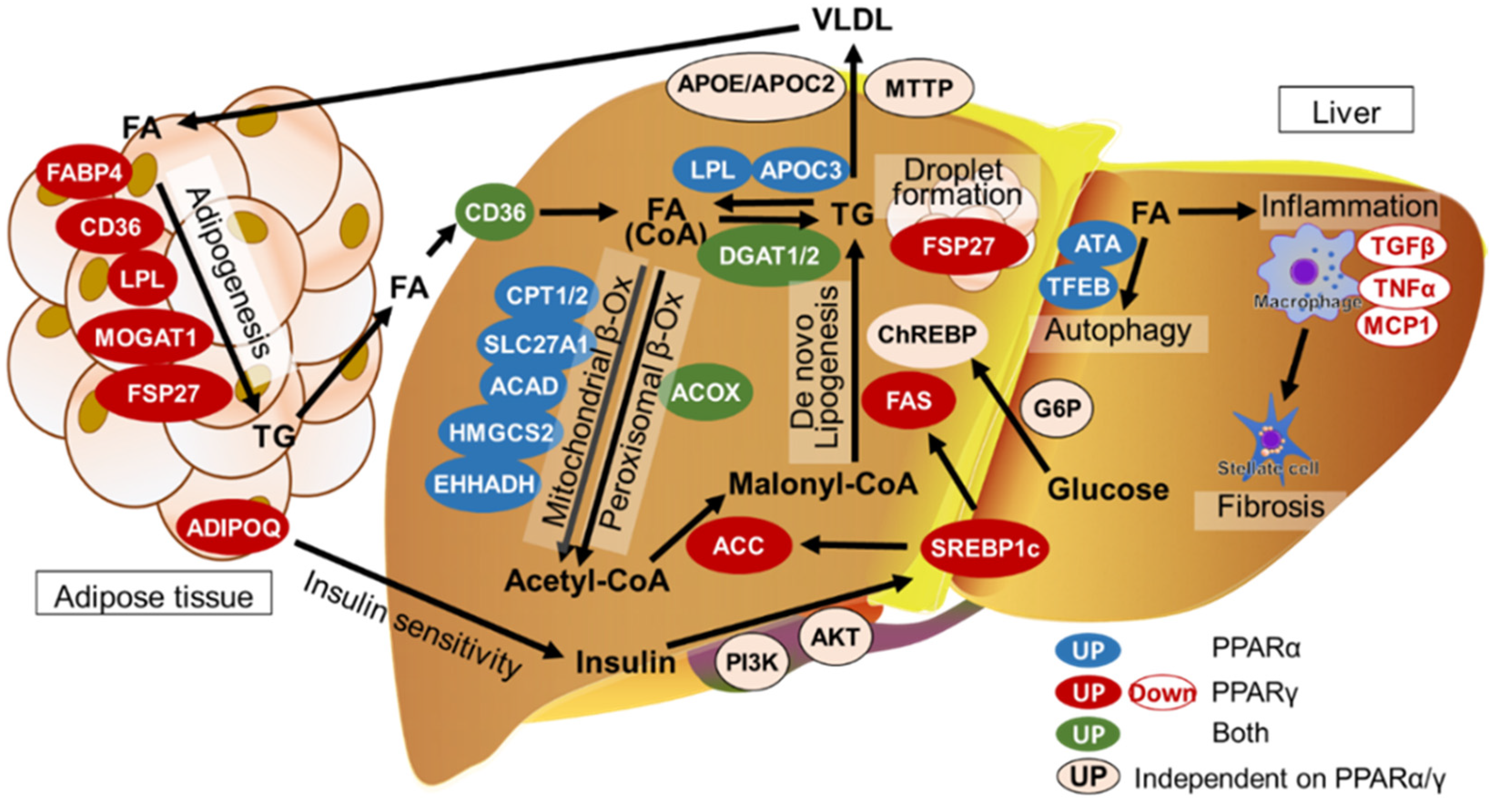
2. Lipid Homeostasis in the Liver
3. Pathogenesis of MASLD/MASH
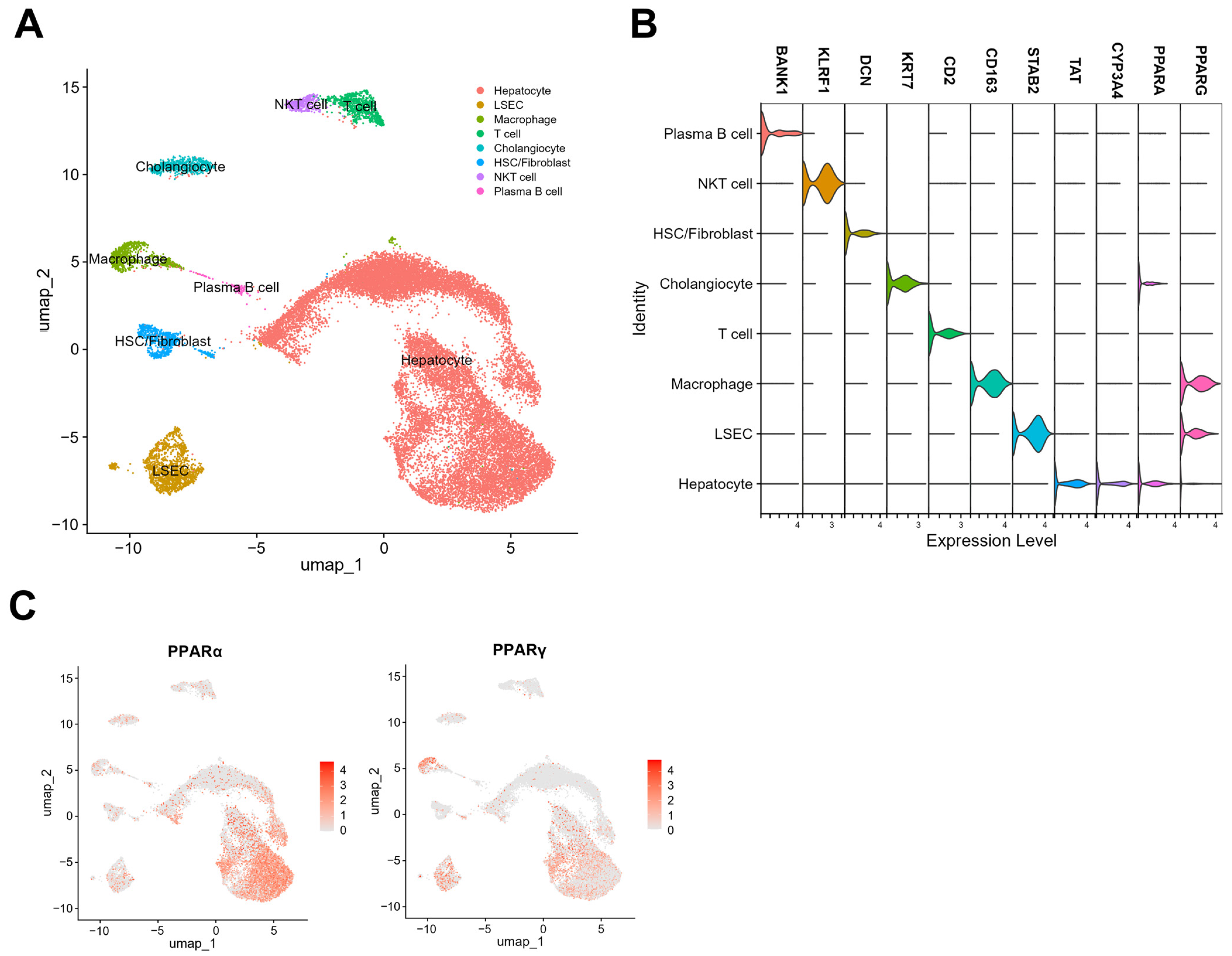
4. Single-Nuclei RNA Sequencing of PPARα and γ Expression Levels in Hepatic Cells
5. PPARα Functions in Hepatocytes, Cholangiocytes, and Macrophages
6. PPARγ Functions in Macrophages, LSECs, and Hepatocytes
7. PPARγ Functions in HSCs
8. PPARα and PPARγ Functions in Adipose Tissues
9. Clinical Trials and Drugs Used for MASH: Potency of PPAR Ligands as Therapeutic Agents
9.1. PPARα Agonists
9.2. PPARγ Agonists
9.3. PPARα/γ Agonists

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PPAR Ligands | Isotype | Status | Source | Reference |
|---|---|---|---|---|
| Arachidonic acid | α | Nature | [117] | |
| Leukotriene B4 | α | Nature | [118,119] | |
| Phosphatidylcholine | α | Nature | [120,121] | |
| Resveratrol | α | Nature | [122,123] | |
| Linoleic acid | γ | Nature | [124] | |
| Prostaglandin D2 | γ | Nature | [125] | |
| 15-deoxy-delta 12,14-prostaglandin J2 | γ | Nature | [126] | |
| Lysophosphatidic acid | γ | Nature | [127] | |
| Clofibrate | α | Clinical | Synthesis | [128] |
| Fenofibrate | α | Clinical | Synthesis | [129,130] |
| Bezafibrate | α | Clinical | Synthesis | [131] |
| Gemfibrozil | α | Clinical | Synthesis | [132] |
| Pemafibrate | α | Clinical | Synthesis | NCT03350165 [110,133,134,135,136,137,138] |
| WY14643 | α | Basic | Synthesis | [139] |
| GW9578 | α | Basic | Synthesis | [140] |
| GW7647 | α | Basic | Synthesis | [141,142] |
| Pioglitazone | γ | Clinical | Synthesis | NCT00063622 NCT00062764 NCT00013598 NCT03646292 NCT04976283 NCT04501406 |
| Ciglitazone | γ | Clinical | Synthesis | [143] |
| Troglitazone | γ | Clinical | Synthesis | [144,145] |
| Rosiglitazone | γ | Clinical | Synthesis | [146,147,148] |
| S26948 | γ | Clinical | Synthesis | [149] |
| INT131 | γ | Clinical | Synthesis | [150,151] |
| Saroglitazar | α/γ | Clinical | Synthesis | NCT05011305 NCT03639623 NCT03061721 NCT02265276 NCT03863574 NCT03617263 NCT03112681 NCT04193982 |
| Lobeglitazone | α/γ | Clinical | Synthesis | [152,153] |
| Lanifibranor (IVA-337) | pan | Clinical | Synthesis | NCT03008070 NCT02503644 NCT05232071 NCT04849728 |
10. NP-Mediated Drug Delivery Systems (NDDSs) as Potential Therapeutic Agents
10.1. Nanoparticles System
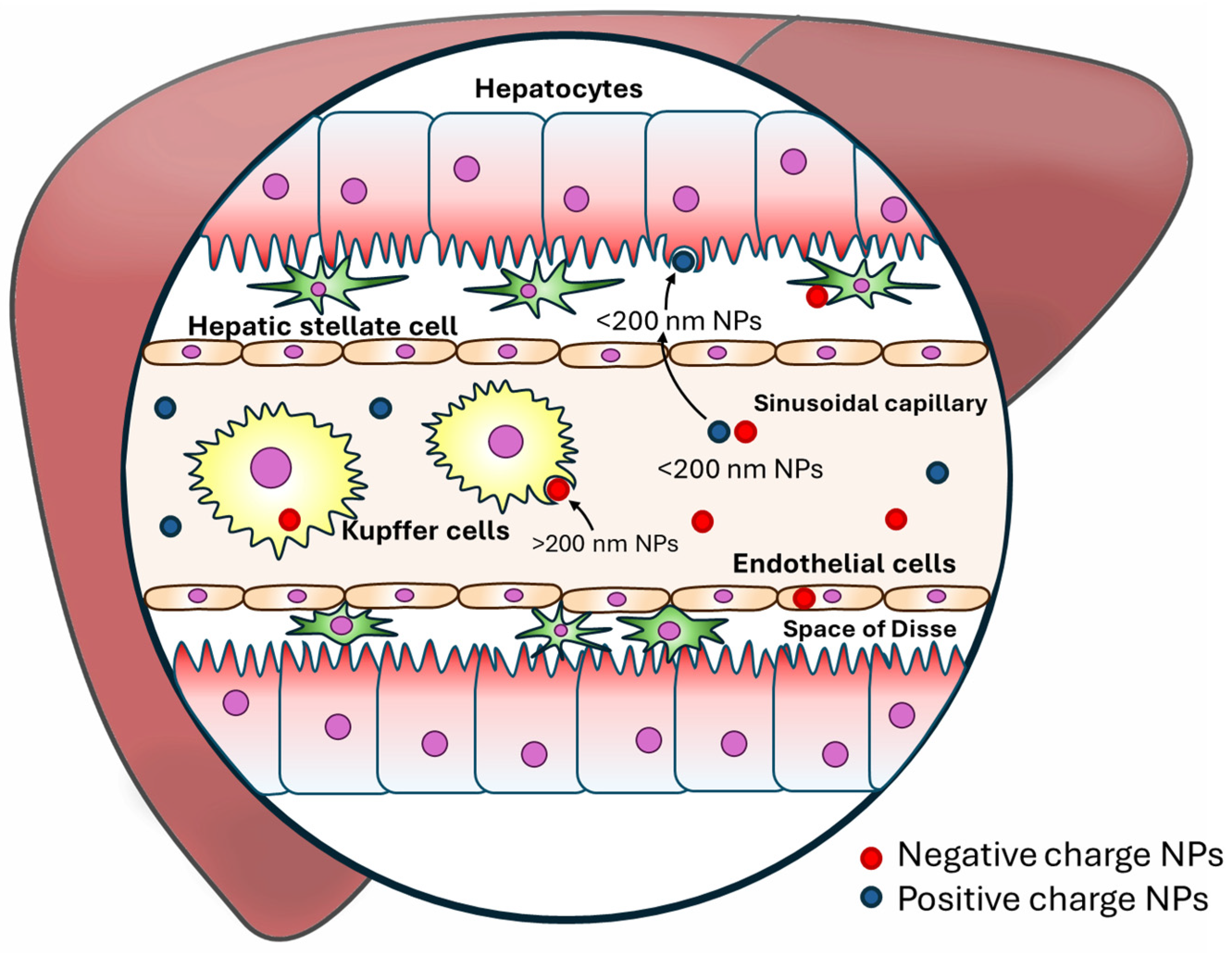
10.2. Mechanisms of NPs in Targeting Liver Disease
10.2.1. Passive Targeting
10.2.2. Active Targeting
| Types of Nanoparticles | Nanoparticle Formulation | Drug | Target Cell | Impact on Liver Diseases | Reference |
|---|---|---|---|---|---|
| Polymeric NPs | PLGA-PEG-Mal | Nilotinib | HSC | Efficiently degrading pericellular collagen I showed optimal antifibrotic activity, liver fibrosis therapy | [188] |
| Diblock copolymers poly[oligo(ethylene glycol) methyl ether methacrylate]-block-VDM | S-nitrosoglutathione | HSC | Alleviating both liver fibrosis and portal hypertension | [189] | |
| PEG-PLGA | Sorafenib | HSC | Efficiently ameliorated liver fibrosis by decreased alpha-smooth muscle actin (α-SMA) content and collagen production in the livers of CCl4-treated mice, shrank the abnormal blood vessels, and decreased microvascular density | [190] | |
| Retinol–chitosan NPs | JQ1- and atorvastatin | HSC | Preventing HSC activation showed reduction in α-SMA expression | [191] | |
| Monomethoxy-PEG-PLGA | Rapamycin | Hepatocyte | Improve hepatic steatosis and liver injury in MASLD | [192] | |
| Polyurethane NPs | Fenofibrate | Hepatocyte | Inhibitory effects on MASLD | [193] | |
| Chondroitin sulfate nanomicelles codelivery system | Retinoic acid and doxorubicin | HSC, Hepatocyte | Destroyed the Golgi structure and ultimately downregulated COL1 production | [194] | |
| Cyclic RGD peptides-PEG-PLGA | miR-29b and germacrone | HSC | Co-delivery of miR-29b and germacrone based on Cyclic RGD peptides-modified NPs for liver fibrosis therapy | [195] | |
| Poly(beta-amino-ester) (PBAE) 536 | DNA labeled with a fluorescent marker | Hep3b | Theranostic gene delivery to HCC | [196] | |
| Lipid-based NPs | pRelaxin-Lipid protamine DNA | Aminoethyl anisamide | HSC | Deactivated HSC, macrophage phenotype switch, the relaxin-primed alleviation of liver fibrosis | [197] |
| cRGDyK-guided liposomes | The hedgehog inhibitor Vismodegib | HSC | Preferentially internalized by activated HSCs in vitro and in vivo, deliver therapeutic agents to activated HSC to treat liver fibrosis | [198] | |
| Asialoglycoprote-liposomes loaded | Norcantharidin | HepG2 | Effectively reduce renal and hepatocellular toxicity | [199] | |
| Vitamin A-coupled liposomes | siRNA against a collagen-specific chaperone | HSC | Effective in suppressing collagen secretion and treating fibrosis induced by CCl4 or bile duct ligation | [176] | |
| Inorganic NPs | Gold nanorods-PEG-PDGFRβ | HSC | Decreased fibrosis, hepatic inflammation, and hepatocyte injury | [200] | |
| Calcium phosphate NPs | Tumor necrosis factor-stimulated gene 6 | Macrophages | Tumor necrosis factor-stimulated gene 6 exerts an antifibrotic effect by facilitating the transition of macrophages to the M2 phenotype and by enhancing the expression of matrix metalloproteinase-12 | [201] |
11. Discussion and Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef]
- Estes, C.; Anstee, Q.M.; Arias-Loste, M.T.; Bantel, H.; Bellentani, S.; Caballeria, J.; Colombo, M.; Craxi, A.; Crespo, J.; Day, C.P.; et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016–2030. J. Hepatol. 2018, 69, 896–904. [Google Scholar] [CrossRef]
- Mitra, S.; De, A.; Chowdhury, A. Epidemiology of non-alcoholic and alcoholic fatty liver diseases. Transl. Gastroenterol. Hepatol. 2020, 5, 16. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef]
- Devarbhavi, H.; Asrani, S.K.; Arab, J.P.; Nartey, Y.A.; Pose, E.; Kamath, P.S. Global burden of liver disease: 2023 update. J. Hepatol. 2023, 79, 516–537. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Golabi, P.; de Avila, L.; Paik, J.M.; Srishord, M.; Fukui, N.; Qiu, Y.; Burns, L.; Afendy, A.; Nader, F. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J. Hepatol. 2019, 71, 793–801. [Google Scholar] [CrossRef]
- Le, M.H.; Yeo, Y.H.; Li, X.; Li, J.; Zou, B.; Wu, Y.; Ye, Q.; Huang, D.Q.; Zhao, C.; Zhang, J.; et al. 2019 Global NAFLD Prevalence: A Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol. 2022, 20, 2809–2817.e28. [Google Scholar] [CrossRef]
- Lazarus, J.V.; Mark, H.E.; Villota-Rivas, M.; Palayew, A.; Carrieri, P.; Colombo, M.; Ekstedt, M.; Esmat, G.; George, J.; Marchesini, G.; et al. The global NAFLD policy review and preparedness index: Are countries ready to address this silent public health challenge? J. Hepatol. 2022, 76, 771–780. [Google Scholar] [CrossRef]
- Boyle, M.; Masson, S.; Anstee, Q.M. The bidirectional impacts of alcohol consumption and the metabolic syndrome: Cofactors for progressive fatty liver disease. J. Hepatol. 2018, 68, 251–267. [Google Scholar] [CrossRef]
- Inan-Eroglu, E.; Huang, B.H.; Ahmadi, M.N.; Johnson, N.; El-Omar, E.M.; Stamatakis, E. Joint associations of adiposity and alcohol consumption with liver disease-related morbidity and mortality risk: Findings from the UK Biobank. Eur. J. Clin. Nutr. 2022, 76, 74–83. [Google Scholar] [CrossRef]
- Paik, J.M.; Golabi, P.; Biswas, R.; Alqahtani, S.; Venkatesan, C.; Younossi, Z.M. Nonalcoholic Fatty Liver Disease and Alcoholic Liver Disease are Major Drivers of Liver Mortality in the United States. Hepatol. Commun. 2020, 4, 890–903. [Google Scholar] [CrossRef]
- Singal, A.K.; Bataller, R.; Ahn, J.; Kamath, P.S.; Shah, V.H. ACG Clinical Guideline: Alcoholic Liver Disease. Am. J. Gastroenterol. 2018, 113, 175–194. [Google Scholar] [CrossRef]
- Kupcova, V.; Fedelesova, M.; Bulas, J.; Kozmonova, P.; Turecky, L. Overview of the Pathogenesis, Genetic, and Non-Invasive Clinical, Biochemical, and Scoring Methods in the Assessment of NAFLD. Int. J. Environ. Res. Public Health 2019, 16, 3570. [Google Scholar] [CrossRef]
- Geier, A.; Tiniakos, D.; Denk, H.; Trauner, M. From the origin of NASH to the future of metabolic fatty liver disease. Gut 2021, 70, 1570–1579. [Google Scholar] [CrossRef]
- Wang, X.; Moore, M.P.; Shi, H.; Miyata, Y.; Donnelly, S.K.; Radiloff, D.R.; Tabas, I. Hepatocyte-targeted siTAZ therapy lowers liver fibrosis in NASH diet-fed chimeric mice with hepatocyte-humanized livers. Mol. Ther. Methods Clin. Dev. 2023, 31, 101165. [Google Scholar] [CrossRef] [PubMed]
- Sheka, A.C.; Adeyi, O.; Thompson, J.; Hameed, B.; Crawford, P.A.; Ikramuddin, S. Nonalcoholic Steatohepatitis: A Review. JAMA 2020, 323, 1175–1183. [Google Scholar] [CrossRef]
- Jarvis, H.; Craig, D.; Barker, R.; Spiers, G.; Stow, D.; Anstee, Q.M.; Hanratty, B. Metabolic risk factors and incident advanced liver disease in non-alcoholic fatty liver disease (NAFLD): A systematic review and meta-analysis of population-based observational studies. PLoS Med. 2020, 17, e1003100. [Google Scholar] [CrossRef]
- Shi, K.; Li, R.; Xu, Z.; Zhang, Q. Identification of Crucial Genetic Factors, Such as PPARγ, that Regulate the Pathogenesis of Fatty Liver Disease in Dairy Cows Is Imperative for the Sustainable Development of Dairy Industry. Animals 2020, 10, 639. [Google Scholar] [CrossRef]
- De Duve, C.; Baudhuin, P. Peroxisomes (microbodies and related particles). Physiol. Rev. 1966, 46, 323–357. [Google Scholar] [CrossRef]
- Imanaka, T. Biogenesis and Function of Peroxisomes in Human Disease with a Focus on the ABC Transporter. Biol. Pharm. Bull. 2019, 42, 649–665. [Google Scholar] [CrossRef]
- Saito, K.; Sekiya, M.; Kainoh, K.; Yoshino, R.; Hayashi, A.; Han, S.I.; Araki, M.; Ohno, H.; Takeuchi, Y.; Tsuyuzaki, T.; et al. Obesity-induced metabolic imbalance allosterically modulates CtBP2 to inhibit PPAR-α transcriptional activity. J. Biol. Chem. 2023, 299, 104890. [Google Scholar] [CrossRef]
- Duszka, K.; Gregor, A.; Guillou, H.; Konig, J.; Wahli, W. Peroxisome Proliferator-Activated Receptors and Caloric Restriction-Common Pathways Affecting Metabolism, Health, and Longevity. Cells 2020, 9, 1708. [Google Scholar] [CrossRef]
- Christofides, A.; Konstantinidou, E.; Jani, C.; Boussiotis, V.A. The role of peroxisome proliferator-activated receptors (PPAR) in immune responses. Metab. Clin. Exp. 2021, 114, 154338. [Google Scholar] [CrossRef]
- Machado, M.V. MASLD treatment-a shift in the paradigm is imminent. Front. Med. 2023, 10, 1316284. [Google Scholar] [CrossRef]
- Ferguson, D.; Finck, B.N. Emerging therapeutic approaches for the treatment of NAFLD and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2021, 17, 484–495. [Google Scholar] [CrossRef]
- Kang, J.H.; Toita, R.; Murata, M. Liver cell-targeted delivery of therapeutic molecules. Crit. Rev. Biotechnol. 2016, 36, 132–143. [Google Scholar] [CrossRef]
- Freitas, R.A., Jr. What is nanomedicine? Nanomedicine 2005, 1, 2–9. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, M.; Xu, X.; Liu, Y.; Chen, Q.; Wu, B.; Zhang, Y. PPARs/macrophages: A bridge between the inflammatory response and lipid metabolism in autoimmune diseases. Biochem. Biophys. Res. Commun. 2023, 684, 149128. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef]
- Linden, A.G.; Li, S.; Choi, H.Y.; Fang, F.; Fukasawa, M.; Uyeda, K.; Hammer, R.E.; Horton, J.D.; Engelking, L.J.; Liang, G. Interplay between ChREBP and SREBP-1c coordinates postprandial glycolysis and lipogenesis in livers of mice [S]. J. Lipid Res. 2018, 59, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K. The Roles of Carbohydrate Response Element Binding Protein in the Relationship between Carbohydrate Intake and Diseases. Int. J. Mol. Sci. 2021, 22, 12058. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Lin, X.; Wang, G. Targeting SREBP-1-Mediated Lipogenesis as Potential Strategies for Cancer. Front. Oncol. 2022, 12, 952371. [Google Scholar] [CrossRef]
- Sakurai, Y.; Kubota, N.; Yamauchi, T.; Kadowaki, T. Role of Insulin Resistance in MAFLD. Int. J. Mol. Sci. 2021, 22, 4156. [Google Scholar] [CrossRef]
- Daniel, P.V.; Mondal, P. Causative and Sanative dynamicity of ChREBP in Hepato-Metabolic disorders. Eur. J. Cell Biol. 2020, 99, 151128. [Google Scholar] [CrossRef]
- Geng, Y.; Faber, K.N.; de Meijer, V.E.; Blokzijl, H.; Moshage, H. How does hepatic lipid accumulation lead to lipotoxicity in non-alcoholic fatty liver disease? Hepatol. Int. 2021, 15, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Todisco, S.; Santarsiero, A.; Convertini, P.; De Stefano, G.; Gilio, M.; Iacobazzi, V.; Infantino, V. PPAR Alpha as a Metabolic Modulator of the Liver: Role in the Pathogenesis of Nonalcoholic Steatohepatitis (NASH). Biology 2022, 11, 792. [Google Scholar] [CrossRef] [PubMed]
- Judyta, Z.; Tomasz, S.; Ewa, S. Acyl-Coenzyme A: Cholesterol Acyltransferase Inhibition in Cancer Treatment. Anticancer. Res. 2019, 39, 3385–3394. [Google Scholar] [CrossRef]
- Li, S.; Wu, T.; Lu, Y.-X.; Wang, J.-X.; Yu, F.-H.; Yang, M.-Z.; Huang, Y.-J.; Li, Z.-J.; Wang, S.-L.; Huang, L.; et al. Obesity promotes gastric cancer metastasis via diacylglycerol acyltransferase 2-dependent lipid droplets accumulation and redox homeostasis. Redox Biol. 2020, 36, 101596. [Google Scholar] [CrossRef]
- Løvsletten, N.G.; Vu, H.; Skagen, C.; Lund, J.; Kase, E.T.; Thoresen, G.H.; Zammit, V.A.; Rustan, A.C. Treatment of human skeletal muscle cells with inhibitors of diacylglycerol acyltransferases 1 and 2 to explore isozyme-specific roles on lipid metabolism. Sci. Rep. 2020, 10, 238. [Google Scholar] [CrossRef]
- Heeren, J.; Scheja, L. Metabolic-associated fatty liver disease and lipoprotein metabolism. Mol. Metab. 2021, 50, 101238. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, Q.; Pan, Y.; Chen, S.; Zhao, Y.; Hu, Y. New insights into the role of dietary triglyceride absorption in obesity and metabolic diseases. Front. Pharmacol. 2023, 14, 1097835. [Google Scholar] [CrossRef]
- Gandhi, A.Y.; Yu, J.; Gupta, A.; Guo, T.; Iyengar, P.; Infante, R.E. Cytokine-Mediated STAT3 Transcription Supports ATGL/CGI-58-Dependent Adipocyte Lipolysis in Cancer Cachexia. Front. Oncol. 2022, 12, 841758. [Google Scholar] [CrossRef]
- Haemmerle, G.; Lass, A. Genetically modified mouse models to study hepatic neutral lipid mobilization. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 879–894. [Google Scholar] [CrossRef]
- Tsamos, G.; Vasdeki, D.; Koufakis, T.; Michou, V.; Makedou, K.; Tzimagiorgis, G. Therapeutic Potentials of Reducing Liver Fat in Non-Alcoholic Fatty Liver Disease: Close Association with Type 2 Diabetes. Metabolites 2023, 13, 517. [Google Scholar] [CrossRef] [PubMed]
- Su, L.-J.; Zhang, J.-H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.-Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxidative Med. Cell. Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Lee, G.; Heo, S.-Y.; Roh, Y.-S. Oxidative Stress Is a Key Modulator in the Development of Nonalcoholic Fatty Liver Disease. Antioxidants 2022, 11, 91. [Google Scholar] [CrossRef]
- Koo, J.H.; Han, C.Y. Signaling Nodes Associated with Endoplasmic Reticulum Stress during NAFLD Progression. Biomolecules 2021, 11, 242. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Shen, H.; Li, X.; Wang, H. Endoplasmic reticulum stress in innate immune cells—A significant contribution to non-alcoholic fatty liver disease. Front. Immunol. 2022, 13, 951406. [Google Scholar] [CrossRef]
- Amen, O.M.; Sarker, S.D.; Ghildyal, R.; Arya, A. Endoplasmic Reticulum Stress Activates Unfolded Protein Response Signaling and Mediates Inflammation, Obesity, and Cardiac Dysfunction: Therapeutic and Molecular Approach. Front. Pharmacol. 2019, 10, 977. [Google Scholar] [CrossRef]
- Batchuluun, B.; Pinkosky, S.L.; Steinberg, G.R. Lipogenesis inhibitors: Therapeutic opportunities and challenges. Nat. Rev. Drug Discov. 2022, 21, 283–305. [Google Scholar] [CrossRef]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants 2021, 10, 174. [Google Scholar] [CrossRef]
- Bruneau, A.; Hundertmark, J.; Guillot, A.; Tacke, F. Molecular and Cellular Mediators of the Gut-Liver Axis in the Progression of Liver Diseases. Front. Med. 2021, 8, 725390. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Zou, X.B.; Chai, Y.F.; Yao, Y.M. Macrophage polarization in inflammatory diseases. Int. J. Biol. Sci. 2014, 10, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Feng, D.; Guillot, A.; Dai, S.; Liu, F.; Hwang, S.; Parker, R.; Seo, W.; He, Y.; Godlewski, G.; et al. Adipocyte Death Preferentially Induces Liver Injury and Inflammation Through the Activation of Chemokine (C-C Motif) Receptor 2-Positive Macrophages and Lipolysis. Hepatology 2019, 69, 1965–1982. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef]
- Sun, H.; Feng, J.; Tang, L. Function of TREM1 and TREM2 in Liver-Related Diseases. Cells 2020, 9, 2626. [Google Scholar] [CrossRef]
- Yu, X.; Lan, P.; Hou, X.; Han, Q.; Lu, N.; Li, T.; Jiao, C.; Zhang, J.; Zhang, C.; Tian, Z. HBV inhibits LPS-induced NLRP3 inflammasome activation and IL-1β production via suppressing the NF-κB pathway and ROS production. J. Hepatol. 2017, 66, 693–702. [Google Scholar] [CrossRef]
- Ferro, D.; Baratta, F.; Pastori, D.; Cocomello, N.; Colantoni, A.; Angelico, F.; Del Ben, M. New Insights into the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Gut-Derived Lipopolysaccharides and Oxidative Stress. Nutrients 2020, 12, 2762. [Google Scholar] [CrossRef]
- Elchaninov, A.V.; Fatkhudinov, T.K.; Vishnyakova, P.A.; Lokhonina, A.V.; Sukhikh, G.T. Phenotypical and Functional Polymorphism of Liver Resident Macrophages. Cells 2019, 8, 1032. [Google Scholar] [CrossRef]
- Lee, Y.K.; Park, J.E.; Lee, M.; Hardwick, J.P. Hepatic lipid homeostasis by peroxisome proliferator-activated receptor gamma 2. Liver Res. 2018, 2, 209–215. [Google Scholar] [CrossRef]
- Filliol, A.; Saito, Y.; Nair, A.; Dapito, D.H.; Yu, L.-X.; Ravichandra, A.; Bhattacharjee, S.; Affo, S.; Fujiwara, N.; Su, H.; et al. Opposing roles of hepatic stellate cell subpopulations in hepatocarcinogenesis. Nature 2022, 610, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.-R.; Raychaudhuri, S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- Bougarne, N.; Weyers, B.; Desmet, S.J.; Deckers, J.; Ray, D.W.; Staels, B.; De Bosscher, K. Molecular Actions of PPARα in Lipid Metabolism and Inflammation. Endocr. Rev. 2018, 39, 760–802. [Google Scholar] [CrossRef]
- Dorotea, D.; Koya, D.; Ha, H. Recent Insights Into SREBP as a Direct Mediator of Kidney Fibrosis via Lipid-Independent Pathways. Front. Pharmacol. 2020, 11, 265. [Google Scholar] [CrossRef]
- Yamashita, S.; Masuda, D.; Matsuzawa, Y. Pemafibrate, a New Selective PPARα Modulator: Drug Concept and Its Clinical Applications for Dyslipidemia and Metabolic Diseases. Curr. Atheroscler. Rep. 2020, 22, 5. [Google Scholar] [CrossRef]
- Zhao, Q.; Yang, R.; Wang, J.; Hu, D.D.; Li, F. PPARα activation protects against cholestatic liver injury. Sci. Rep. 2017, 7, 9967. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Qiu, F.; Zhou, K.; Matlock, H.G.; Takahashi, Y.; Rajala, R.V.S.; Yang, Y.; Moran, E.; Ma, J.X. Pathogenic Role of microRNA-21 in Diabetic Retinopathy Through Downregulation of PPARα. Diabetes 2017, 66, 1671–1682. [Google Scholar] [CrossRef]
- Adamowicz, M.; Kempinska-Podhorodecka, A.; Abramczyk, J.; Banales, J.M.; Milkiewicz, P.; Milkiewicz, M. Suppression of Hepatic PPARα in Primary Biliary Cholangitis Is Modulated by miR-155. Cells 2022, 11, 2880. [Google Scholar] [CrossRef]
- Zimmermann, R.; Panzenbock, U.; Wintersperger, A.; Levak-Frank, S.; Graier, W.; Glatter, O.; Fritz, G.; Kostner, G.M.; Zechner, R. Lipoprotein lipase mediates the uptake of glycated LDL in fibroblasts, endothelial cells, and macrophages. Diabetes 2001, 50, 1643–1653. [Google Scholar] [CrossRef] [PubMed]
- Gbaguidi, F.G.; Chinetti, G.; Milosavljevic, D.; Teissier, E.; Chapman, J.; Olivecrona, G.; Fruchart, J.C.; Griglio, S.; Fruchart-Najib, J.; Staels, B. Peroxisome proliferator-activated receptor (PPAR) agonists decrease lipoprotein lipase secretion and glycated LDL uptake by human macrophages. FEBS Lett. 2002, 512, 85–90. [Google Scholar] [CrossRef]
- Chinetti-Gbaguidi, G.; Rigamonti, E.; Helin, L.; Mutka, A.L.; Lepore, M.; Fruchart, J.C.; Clavey, V.; Ikonen, E.; Lestavel, S.; Staels, B. Peroxisome proliferator-activated receptor alpha controls cellular cholesterol trafficking in macrophages. J. Lipid Res. 2005, 46, 2717–2725. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Natarajan, R. Cloning of the human cholesteryl ester hydrolase promoter: Identification of functional peroxisomal proliferator-activated receptor responsive elements. Biochem. Biophys. Res. Commun. 2001, 284, 1065–1070. [Google Scholar] [CrossRef]
- Argmann, C.A.; Sawyez, C.G.; McNeil, C.J.; Hegele, R.A.; Huff, M.W. Activation of peroxisome proliferator-activated receptor gamma and retinoid X receptor results in net depletion of cellular cholesteryl esters in macrophages exposed to oxidized lipoproteins. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 475–482. [Google Scholar] [CrossRef]
- Chinetti, G.; Lestavel, S.; Fruchart, J.C.; Clavey, V.; Staels, B. Peroxisome proliferator-activated receptor alpha reduces cholesterol esterification in macrophages. Circ. Res. 2003, 92, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Rigamonti, E.; Chinetti-Gbaguidi, G.; Staels, B. Regulation of macrophage functions by PPAR-α, PPAR-γ, and LXRs in mice and men. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1050–1059. [Google Scholar] [CrossRef]
- Su, M.; Cao, J.; Huang, J.; Liu, S.; Im, D.S.; Yoo, J.W.; Jung, J.H. The In Vitro and In Vivo Anti-Inflammatory Effects of a Phthalimide PPAR-γ Agonist. Mar. Drugs 2017, 15, 7. [Google Scholar] [CrossRef] [PubMed]
- Linares, I.; Farrokhi, K.; Echeverri, J.; Kaths, J.M.; Kollmann, D.; Hamar, M.; Urbanellis, P.; Ganesh, S.; Adeyi, O.A.; Yip, P.; et al. PPAR-γ activation is associated with reduced liver ischemia-reperfusion injury and altered tissue-resident macrophages polarization in a mouse model. PLoS ONE 2018, 13, e0195212. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Jiang, Q.; Wang, Z.; Li, M.; Zhang, Q.; Lu, W.; Wang, J. Mutual inhibitory mechanisms between PPARγ and Hif-1α: Implication in pulmonary hypertension. Recept. Clin. Investig. 2015, 2, e626. [Google Scholar]
- Heming, M.; Gran, S.; Jauch, S.L.; Fischer-Riepe, L.; Russo, A.; Klotz, L.; Hermann, S.; Schafers, M.; Roth, J.; Barczyk-Kahlert, K. Peroxisome Proliferator-Activated Receptor-gamma Modulates the Response of Macrophages to Lipopolysaccharide and Glucocorticoids. Front. Immunol. 2018, 9, 893. [Google Scholar] [CrossRef]
- Yu, T.; Gao, M.; Yang, P.; Liu, D.; Wang, D.; Song, F.; Zhang, X.; Liu, Y. Insulin promotes macrophage phenotype transition through PI3K/Akt and PPAR-γ signaling during diabetic wound healing. J. Cell. Physiol. 2019, 234, 4217–4231. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.X.; Ji, P.X.; Chen, Y.X.; Li, X.Y.; Sheng, L.; Lian, M.; Guo, C.J.; Hua, J. Regulation of the macrophage-hepatic stellate cell interaction by targeting macrophage peroxisome proliferator-activated receptor gamma to prevent non-alcoholic steatohepatitis progression in mice. Liver Int. 2022, 42, 2696–2712. [Google Scholar] [CrossRef]
- Li, X.Y.; Ji, P.X.; Ni, X.X.; Chen, Y.X.; Sheng, L.; Lian, M.; Guo, C.J.; Hua, J. Regulation of PPAR-γ activity in lipid-laden hepatocytes affects macrophage polarization and inflammation in nonalcoholic fatty liver disease. World J. Hepatol. 2022, 14, 1365–1381. [Google Scholar] [CrossRef] [PubMed]
- Monroy-Ramirez, H.C.; Galicia-Moreno, M.; Sandoval-Rodriguez, A.; Meza-Rios, A.; Santos, A.; Armendariz-Borunda, J. PPARs as Metabolic Sensors and Therapeutic Targets in Liver Diseases. Int. J. Mol. Sci. 2021, 22, 8298. [Google Scholar] [CrossRef]
- Schulte, R.; Wohlleber, D.; Unrau, L.; Geers, B.; Metzger, C.; Erhardt, A.; Tiegs, G.; van Rooijen, N.; Heukamp, L.C.; Klotz, L.; et al. Pioglitazone-Mediated Peroxisome Proliferator-Activated Receptor gamma Activation Aggravates Murine Immune-Mediated Hepatitis. Int. J. Mol. Sci. 2020, 21, 2523. [Google Scholar] [CrossRef]
- Fougerat, A.; Montagner, A.; Loiseau, N.; Guillou, H.; Wahli, W. Peroxisome Proliferator-Activated Receptors and Their Novel Ligands as Candidates for the Treatment of Non-Alcoholic Fatty Liver Disease. Cells 2020, 9, 1638. [Google Scholar] [CrossRef]
- Decara, J.; Rivera, P.; López-Gambero, A.J.; Serrano, A.; Pavón, F.J.; Baixeras, E.; Rodríguez de Fonseca, F.; Suárez, J. Peroxisome Proliferator-Activated Receptors: Experimental Targeting for the Treatment of Inflammatory Bowel Diseases. Front. Pharmacol. 2020, 11, 730. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.-Q.; Feng, Z.-C.; Zhang, X.-L.; Hu, L.-Q.; Wang, M.; Zhang, H.-F.; Li, S.-M. PPAR-γ Activation Exerts an Anti-inflammatory Effect by Suppressing the NLRP3 Inflammasome in Spinal Cord-Derived Neurons. Mediat. Inflamm. 2019, 2019, 6386729. [Google Scholar] [CrossRef]
- Li, L.; Fu, J.; Liu, D.; Sun, J.; Hou, Y.; Chen, C.; Shao, J.; Wang, L.; Wang, X.; Zhao, R.; et al. Hepatocyte-specific Nrf2 deficiency mitigates high-fat diet-induced hepatic steatosis: Involvement of reduced PPARγ expression. Redox Biol. 2020, 30, 101412. [Google Scholar] [CrossRef]
- Ni, X.X.; Li, X.Y.; Wang, Q.; Hua, J. Regulation of peroxisome proliferator-activated receptor-gamma activity affects the hepatic stellate cell activation and the progression of NASH via TGF-β1/Smad signaling pathway. J. Physiol. Biochem. 2021, 77, 35–45. [Google Scholar] [CrossRef]
- He, J.; Hong, B.; Bian, M.; Jin, H.; Chen, J.; Shao, J.; Zhang, F.; Zheng, S. Docosahexaenoic acid inhibits hepatic stellate cell activation to attenuate liver fibrosis in a PPARγ-dependent manner. Int. Immunopharmacol. 2019, 75, 105816. [Google Scholar] [CrossRef]
- de Souza Basso, B.; Haute, G.V.; Ortega-Ribera, M.; Luft, C.; Antunes, G.L.; Bastos, M.S.; Carlessi, L.P.; Levorse, V.G.; Cassel, E.; Donadio, M.V.F.; et al. Methoxyeugenol deactivates hepatic stellate cells and attenuates liver fibrosis and inflammation through a PPAR-ɣ and NF-kB mechanism. J. Ethnopharmacol. 2021, 280, 114433. [Google Scholar] [CrossRef]
- Yu, J.; Zhang, S.; Chu, E.S.; Go, M.Y.; Lau, R.H.; Zhao, J.; Wu, C.W.; Tong, L.; Zhao, J.; Poon, T.C.; et al. Peroxisome proliferator-activated receptors gamma reverses hepatic nutritional fibrosis in mice and suppresses activation of hepatic stellate cells in vitro. Int. J. Biochem. Cell Biol. 2010, 42, 948–957. [Google Scholar] [CrossRef]
- Liu, X.; Xu, J.; Rosenthal, S.; Zhang, L.J.; McCubbin, R.; Meshgin, N.; Shang, L.; Koyama, Y.; Ma, H.Y.; Sharma, S.; et al. Identification of Lineage-Specific Transcription Factors That Prevent Activation of Hepatic Stellate Cells and Promote Fibrosis Resolution. Gastroenterology 2020, 158, 1728–1744.e14. [Google Scholar] [CrossRef] [PubMed]
- Yum, Y.J.; Yoo, J.; Bang, K.; Jun, J.E.; Jeong, I.K.; Ahn, K.J.; Chung, H.Y.; Hwang, Y.C. Peroxisome proliferator-activated receptor gamma activation ameliorates liver fibrosis-differential action of transcription factor EB and autophagy on hepatocytes and stellate cells. Hepatol. Commun. 2023, 7, e0154. [Google Scholar] [CrossRef]
- Calvier, L.; Chouvarine, P.; Legchenko, E.; Hoffmann, N.; Geldner, J.; Borchert, P.; Jonigk, D.; Mozes, M.M.; Hansmann, G. PPARγ Links BMP2 and TGFβ1 Pathways in Vascular Smooth Muscle Cells, Regulating Cell Proliferation and Glucose Metabolism. Cell Metab. 2017, 25, 1118–1134.e7. [Google Scholar] [CrossRef]
- Sun, K.; Wang, Q.; Huang, X.H. PPAR gamma inhibits growth of rat hepatic stellate cells and TGF beta-induced connective tissue growth factor expression. Acta Pharmacol. Sin. 2006, 27, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Mao, S.; Chen, S.; Zhang, W.; Liu, C. PPARs-Orchestrated Metabolic Homeostasis in the Adipose Tissue. Int. J. Mol. Sci. 2021, 22, 8974. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Zhou, W.; Xu, R.; Xing, L.; Ji, G.; Dang, Y. Natural PPARs agonists for the treatment of nonalcoholic fatty liver disease. Biomed. Pharmacother. 2022, 151, 113127. [Google Scholar] [CrossRef]
- Faghfouri, A.H.; Khajebishak, Y.; Payahoo, L.; Faghfuri, E.; Alivand, M. PPAR-γ agonists: Potential modulators of autophagy in obesity. Eur. J. Pharmacol. 2021, 912, 174562. [Google Scholar] [CrossRef]
- Aibara, D.; Matsuo, K.; Yamano, S.; Matsusue, K. Fat-specific protein 27b is regulated by hepatic peroxisome proliferator-activated receptor γ in hepatic steatosis. Endocr. J. 2020, 67, 37–44. [Google Scholar] [CrossRef]
- Sharma, R.; Luong, Q.; Sharma, V.M.; Harberson, M.; Harper, B.; Colborn, A.; Berryman, D.E.; Jessen, N.; Jørgensen, J.O.L.; Kopchick, J.J.; et al. Growth hormone controls lipolysis by regulation of FSP27 expression. J. Endocrinol. 2018, 239, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.M.; Vestergaard, E.T.; Jessen, N.; Kolind-Thomsen, P.; Nellemann, B.; Nielsen, T.S.; Vendelbo, M.H.; Møller, N.; Sharma, R.; Lee, K.Y.; et al. Growth hormone acts along the PPARγ-FSP27 axis to stimulate lipolysis in human adipocytes. Am. J. Physiol.-Endocrinol. Metab. 2018, 316, E34–E42. [Google Scholar] [CrossRef]
- Hong, F.; Pan, S.; Guo, Y.; Xu, P.; Zhai, Y. PPARs as Nuclear Receptors for Nutrient and Energy Metabolism. Molecules 2019, 24, 2545. [Google Scholar] [CrossRef]
- Zhang, L.; Li, H.-X.; Pan, W.-S.; Ullah Khan, F.; Qian, C.; Qi-Li, F.-R.; Xu, X. Administration of methyl palmitate prevents non-alcoholic steatohepatitis (NASH) by induction of PPAR-α. Biomed. Pharmacother. 2019, 111, 99–108. [Google Scholar] [CrossRef]
- Choudhary, N.S.; Kumar, N.; Duseja, A. Peroxisome Proliferator-Activated Receptors and Their Agonists in Nonalcoholic Fatty Liver Disease. J. Clin. Exp. Hepatol. 2019, 9, 731–739. [Google Scholar] [CrossRef]
- Liss, K.H.H.; Finck, B.N. PPARs and nonalcoholic fatty liver disease. Biochimie 2017, 136, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Boeckmans, J.; Natale, A.; Rombaut, M.; Buyl, K.; Rogiers, V.; De Kock, J.; Vanhaecke, T.; Rodrigues, R.M. Anti-NASH Drug Development Hitches a Lift on PPAR Agonism. Cells 2020, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Honda, Y.; Kessoku, T.; Ogawa, Y.; Tomeno, W.; Imajo, K.; Fujita, K.; Yoneda, M.; Takizawa, T.; Saito, S.; Nagashima, Y.; et al. Pemafibrate, a novel selective peroxisome proliferator-activated receptor alpha modulator, improves the pathogenesis in a rodent model of nonalcoholic steatohepatitis. Sci. Rep. 2017, 7, 42477. [Google Scholar] [CrossRef]
- Ishibashi, S.; Arai, H.; Yokote, K.; Araki, E.; Suganami, H.; Yamashita, S. Efficacy and safety of pemafibrate (K-877), a selective peroxisome proliferator-activated receptor α modulator, in patients with dyslipidemia: Results from a 24-week, randomized, double blind, active-controlled, phase 3 trial. J. Clin. Lipidol. 2018, 12, 173–184. [Google Scholar] [CrossRef]
- Lebovitz, H.E. Thiazolidinediones: The Forgotten Diabetes Medications. Curr. Diabetes Rep. 2019, 19, 151. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.P.; Caffrey, R.; Marioneaux, J.; Santhekadur, P.K.; Bhat, M.; Alonso, C.; Koduru, S.V.; Philip, B.; Jain, M.R.; Giri, S.R.; et al. The PPAR α/γ Agonist Saroglitazar Improves Insulin Resistance and Steatohepatitis in a Diet Induced Animal Model of Nonalcoholic Fatty Liver Disease. Sci. Rep. 2020, 10, 9330. [Google Scholar] [CrossRef]
- Gawrieh, S.; Noureddin, M.; Loo, N.; Mohseni, R.; Awasty, V.; Cusi, K.; Kowdley, K.V.; Lai, M.; Schiff, E.; Parmar, D.; et al. Saroglitazar, a PPAR-α/γ Agonist, for Treatment of NAFLD: A Randomized Controlled Double-Blind Phase 2 Trial. Hepatology 2021, 74, 1809–1824. [Google Scholar] [CrossRef]
- Wettstein, G.; Luccarini, J.M.; Poekes, L.; Faye, P.; Kupkowski, F.; Adarbes, V.; Defrêne, E.; Estivalet, C.; Gawronski, X.; Jantzen, I.; et al. The new-generation pan-peroxisome proliferator-activated receptor agonist IVA337 protects the liver from metabolic disorders and fibrosis. Hepatol. Commun. 2017, 1, 524–537. [Google Scholar] [CrossRef]
- Lange, N.F.; Graf, V.; Caussy, C.; Dufour, J.F. PPAR-Targeted Therapies in the Treatment of Non-Alcoholic Fatty Liver Disease in Diabetic Patients. Int. J. Mol. Sci. 2022, 23, 4305. [Google Scholar] [CrossRef]
- Sztolsztener, K.; Chabowski, A.; Harasim-Symbor, E.; Bielawiec, P.; Konstantynowicz-Nowicka, K. Arachidonic Acid as an Early Indicator of Inflammation during Non-Alcoholic Fatty Liver Disease Development. Biomolecules 2020, 10, 1133. [Google Scholar] [CrossRef] [PubMed]
- Narala, V.; Adapala, R.; Madathilparambil, S.; Brock, T.; Peters-Golden, M.; Reddy, R. Leukotriene B4 Is a Physiologically Relevant Endogenous Peroxisome Proliferator-activated Receptor-Agonist. J. Biol. Chem. 2010, 285, 22067–22074. [Google Scholar] [CrossRef]
- Bhatt, L.; Roinestad, K.; Van, T.; Springman, E.B. Recent advances in clinical development of leukotriene B4 pathway drugs. Semin. Immunol. 2017, 33, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, E.R.; Allin, K.H.; Madsbad, S.; Fenger, M. Phosphatidylcholine and its relation to apolipoproteins A-1 and B changes after Roux-en-Y gastric bypass: A cohort study. Lipids Health Dis. 2019, 18, 169. [Google Scholar] [CrossRef]
- Mao, L.; Wang, M.; Li, Y.; Liu, Y.; Wang, J.; Xue, C. Eicosapentaenoic acid-containing phosphatidylcholine promotes osteogenesis:mechanism of up-regulating Runx2 and ERK-mediated phosphorylation of PPARγ at serine 112. J. Funct. Foods 2019, 52, 73–80. [Google Scholar] [CrossRef]
- Toupchian, O.; Abdollahi, S.; Salehi-Abargouei, A.; Heshmati, J.; Clark, C.C.T.; Sheikhha, M.H.; Fallahzadeh, H.; Mozaffari-Khosravi, H. The effects of resveratrol supplementation on PPARα, p16, p53, p21 gene expressions, and sCD163/sTWEAK ratio in patients with type 2 diabetes mellitus: A double-blind controlled randomized trial. Phytother. Res. 2021, 35, 3205–3213. [Google Scholar] [CrossRef] [PubMed]
- Okudaira, N.; Ishizaka, Y.; Tamamori-Adachi, M. Resveratrol blocks retrotransposition of LINE-1 through PPAR α and sirtuin-6. Sci. Rep. 2022, 12, 7772. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, N.; Cuffe, J.S.M.; Holland, O.J.; Perkins, A.V.; McAinch, A.J.; Hryciw, D.H. Linoleic Acid Increases Prostaglandin E2 Release and Reduces Mitochondrial Respiration and Cell Viability in Human Trophoblast-Like Cells. Cell. Physiol. Biochem. 2019, 52, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.G.; Phipps, R.P. Prostaglandin D2, its metabolite 15-d-PGJ2, and peroxisome proliferator activated receptor-γ agonists induce apoptosis in transformed, but not normal, human T lineage cells. Immunology 2002, 105, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Raman, P.; Kaplan, B.L.F.; Thompson, J.T.; Heuvel, J.P.V.; Kaminski, N.E. 15-Deoxy-Δ12,14-prostaglandin J2-Glycerol Ester, a Putative Metabolite of 2-Arachidonyl Glycerol, Activates Peroxisome Proliferator Activated Receptor γ. Mol. Pharmacol. 2011, 80, 201–209. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, T.M.; Pontsler, A.V.; Silva, A.R.; Hilaire, A.S.; Xu, Y.; Hinshaw, J.C.; Zimmerman, G.A.; Hama, K.; Aoki, J.; Arai, H.; et al. Identification of an intracellular receptor for lysophosphatidic acid (LPA): LPA is a transcellular PPARγ agonist. Proc. Natl. Acad. Sci. USA 2003, 100, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Laurin, J.; Lindor, K.D.; Crippin, J.S.; Gossard, A.; Gores, G.J.; Ludwig, J.; Rakela, J.; McGill, D.B. Ursodeoxycholic acid or clofibrate in the treatment of non-alcohol-induced steatohepatitis: A pilot study. Hepatology 1996, 23, 1464–1467. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Miranda, C.; Pérez-Carreras, M.; Colina, F.; López-Alonso, G.; Vargas, C.; Solís-Herruzo, J.A. A pilot trial of fenofibrate for the treatment of non-alcoholic fatty liver disease. Dig. Liver Dis. 2008, 40, 200–205. [Google Scholar] [CrossRef]
- Fabbrini, E.; Mohammed, B.S.; Korenblat, K.M.; Magkos, F.; McCrea, J.; Patterson, B.W.; Klein, S. Effect of Fenofibrate and Niacin on Intrahepatic Triglyceride Content, Very Low-Density Lipoprotein Kinetics, and Insulin Action in Obese Subjects with Nonalcoholic Fatty Liver Disease. J. Clin. Endocrinol. Metab. 2010, 95, 2727–2735. [Google Scholar] [CrossRef]
- Sasaki, Y.; Shimada, T.; Iizuka, S.; Suzuki, W.; Makihara, H.; Teraoka, R.; Tsuneyama, K.; Hokao, R.; Aburada, M. Effects of bezafibrate in nonalcoholic steatohepatitis model mice with monosodium glutamate-induced metabolic syndrome. Eur. J. Pharmacol. 2011, 662, 1–8. [Google Scholar] [CrossRef]
- Basaranoglu, M.; Acbay, O.; Sonsuz, A. A controlled trial of gemfibrozil in the treatment of patients with nonalcoholic steatohepatitis. J. Hepatol. 1999, 31, 384. [Google Scholar] [CrossRef] [PubMed]
- Ida, S.; Kaneko, R.; Murata, K. Efficacy and safety of pemafibrate administration in patients with dyslipidemia: A systematic review and meta-analysis. Cardiovasc. Diabetol. 2019, 18, 38. [Google Scholar] [CrossRef]
- Blair, H.A. Pemafibrate: First Global Approval. Drugs 2017, 77, 1805–1810. [Google Scholar] [CrossRef]
- Hatanaka, T.; Kakizaki, S.; Saito, N.; Nakano, Y.; Nakano, S.; Hazama, Y.; Yoshida, S.; Hachisu, Y.; Tanaka, Y.; Kashiwabara, K.; et al. Impact of Pemafibrate in Patients with Hypertriglyceridemia and Metabolic Dysfunction-associated Fatty Liver Disease Pathologically Diagnosed with Non-alcoholic Steatohepatitis: A Retrospective, Single-arm Study. Intern. Med. 2021, 60, 2167–2174. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, S.; Tahara, T.; Lefor, A.K.; Ogura, M. Pemafibrate decreases markers of hepatic inflammation in patients with non-alcoholic fatty liver disease. Clin. Exp. Hepatol. 2020, 6, 270–274. [Google Scholar] [CrossRef]
- Shinozaki, S.; Tahara, T.; Kawarai Lefor, A.; Ogura, M. Pemafibrate improves hepatic inflammation, function and fibrosis in patients with non-alcoholic fatty liver disease: A one-year observational study. Clin. Exp. Hepatol. 2021, 7, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, A.; Eguchi, Y.; Yoneda, M.; Imajo, K.; Tamaki, N.; Suganami, H.; Nojima, T.; Tanigawa, R.; Iizuka, M.; Iida, Y.; et al. Randomised clinical trial: Pemafibrate, a novel selective peroxisome proliferator-activated receptor α modulator (SPPARMα), versus placebo in patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2021, 54, 1263–1277. [Google Scholar] [CrossRef] [PubMed]
- Veiga, F.M.S.; Graus-Nunes, F.; Rachid, T.L.; Barreto, A.B.; Mandarim-de-Lacerda, C.A.; Souza-Mello, V. Anti-obesogenic effects of WY14643 (PPAR-α agonist): Hepatic mitochondrial enhancement and suppressed lipogenic pathway in diet-induced obese mice. Biochimie 2017, 140, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.J.; Winegar, D.A.; Plunket, K.D.; Moore, L.B.; Lewis, M.C.; Wilson, J.G.; Sundseth, S.S.; Koble, C.S.; Wu, Z.; Chapman, J.M.; et al. A Ureido-Thioisobutyric Acid (GW9578) Is a Subtype-Selective PPARα Agonist with Potent Lipid-Lowering Activity. J. Med. Chem. 1999, 42, 3785–3788. [Google Scholar] [CrossRef]
- Okishio, S.; Yamaguchi, K.; Ishiba, H.; Tochiki, N.; Yano, K.; Takahashi, A.; Kataoka, S.; Okuda, K.; Seko, Y.; Liu, Y.; et al. PPARα agonist and metformin co-treatment ameliorates NASH in mice induced by a choline-deficient, amino acid-defined diet with 45% fat. Sci. Rep. 2020, 10, 19578. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.J.; Stuart, L.W.; Hurley, K.P.; Lewis, M.C.; Winegar, D.A.; Wilson, J.G.; Wilkison, W.O.; Ittoop, O.R.; Willson, T.M. Identification of a subtype selective human PPARα agonist through parallel-array synthesis. Bioorg. Med. Chem. Lett. 2001, 11, 1225–1227. [Google Scholar] [CrossRef] [PubMed]
- Aleshin, S.; Grabeklis, S.; Hanck, T.; Sergeeva, M.; Reiser, G. Peroxisome Proliferator-Activated Receptor (PPAR)-γ Positively Controls and PPARα Negatively Controls Cyclooxygenase-2 Expression in Rat Brain Astrocytes through a Convergence on PPARβ/δ via Mutual Control of PPAR Expression Levels. Mol. Pharmacol. 2009, 76, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Masubuchi, Y. Metabolic and Non-Metabolic Factors Determining Troglitazone Hepatotoxicity: A Review. Drug Metab. Pharmacokinet. 2006, 21, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, S.H.; Hespenheide, E.E.; Redick, J.A.; Iezzoni, J.C.; Battle, E.H.; Sheppard, B.L. A Pilot Study of A Thiazolidinedione, Troglitazone, in Nonalcoholic Steatohepatitis. Am. J. Gastroenterol. 2001, 96, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Idilman, R.; Mizrak, D.; Corapcioglu, D.; Bektas, M.; Doganay, B.; Sayki, M.; Coban, S.; Erden, E.; Soykan, I.; Emral, R.; et al. Clinical trial: Insulin-sensitizing agents may reduce consequences of insulin resistance in individuals with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2008, 28, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Akyüz, F.; Demir, K.; Özdil, S.; Aksoy, N.; Poturoğlu, Ş.; İbrişim, D.; Kaymakoğlu, S.; Beşışık, F.; Boztaş, G.; Çakaloğlu, Y.; et al. The Effects of Rosiglitazone, Metformin, and Diet with Exercise in Nonalcoholic Fatty Liver Disease. Dig. Dis. Sci. 2007, 52, 2359–2367. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Charlotte, F.; Bernhardt, C.; Giral, P.; Halbron, M.; LeNaour, G.; Hartmann-Heurtier, A.; Bruckert, E.; Poynard, T.; Group, L.S. Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: Results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology 2010, 51, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Sohn, K.-A.K.; Cruciani-Guglielmacci, C.; Kassis, N.; Clément, L.; Ouali, F.; Caüzac, M.; Lebègue, N.; Berthelot, P.; Caignard, D.-H.; Pégorier, J.-P.; et al. S26948, a new specific peroxisome proliferator activated receptor gamma modulator improved in vivo hepatic insulin sensitivity in 48h lipid infused rats. Eur. J. Pharmacol. 2009, 608, 104–111. [Google Scholar] [CrossRef]
- Dunn, F.L.; Higgins, L.S.; Fredrickson, J.; DePaoli, A.M. Selective modulation of PPARγ activity can lower plasma glucose without typical thiazolidinedione side-effects in patients with Type 2 diabetes. J. Diabetes Its Complicat. 2011, 25, 151–158. [Google Scholar] [CrossRef]
- DePaoli, A.M.; Higgins, L.S.; Henry, R.R.; Mantzoros, C.; Dunn, F.L.; for the INT131-007 Study Group. Can a Selective PPARγ Modulator Improve Glycemic Control in Patients with Type 2 Diabetes with Fewer Side Effects Compared with Pioglitazone? Diabetes Care 2014, 37, 1918–1923. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-h.; Kim, J.H.; Kim, S.R.; Jin, H.Y.; Rhee, E.-J.; Cho, Y.M.; Lee, B.-W. Lobeglitazone, a Novel Thiazolidinedione, Improves Non-Alcoholic Fatty Liver Disease in Type 2 Diabetes: Its Efficacy and Predictive Factors Related to Responsiveness. J. Korean Med. Sci. 2017, 32, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.S.; Tan, W.R.; Low, Z.S.; Marvalim, C.; Lee, J.Y.; Tan, N.S. Exploration and Development of PPAR Modulators in Health and Disease: An Update of Clinical Evidence. Int. J. Mol. Sci. 2019, 20, 5055. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, A.; Almotairy, A.R.Z.; Henidi, H.; Alshehri, O.Y.; Aldughaim, M.S. Nanoparticles as Drug Delivery Systems: A Review of the Implication of Nanoparticles’ Physicochemical Properties on Responses in Biological Systems. Polymers 2023, 15, 1596. [Google Scholar] [CrossRef] [PubMed]
- Elumalai, K.; Srinivasan, S.; Shanmugam, A. Review of the efficacy of nanoparticle-based drug delivery systems for cancer treatment. Biomed. Technol. 2024, 5, 109–122. [Google Scholar] [CrossRef]
- Yao, Y.; Zhou, Y.; Liu, L.; Xu, Y.; Chen, Q.; Wang, Y.; Wu, S.; Deng, Y.; Zhang, J.; Shao, A. Nanoparticle-Based Drug Delivery in Cancer Therapy and Its Role in Overcoming Drug Resistance. Front. Mol. Biosci. 2020, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Afzal, O.; Altamimi, A.S.A.; Nadeem, M.S.; Alzarea, S.I.; Almalki, W.H.; Tariq, A.; Mubeen, B.; Murtaza, B.N.; Iftikhar, S.; Riaz, N.; et al. Nanoparticles in Drug Delivery: From History to Therapeutic Applications. Nanomaterials 2022, 12, 4494. [Google Scholar] [CrossRef] [PubMed]
- Ogawara, K.; Yoshida, M.; Higaki, K.; Kimura, T.; Shiraishi, K.; Nishikawa, M.; Takakura, Y.; Hashida, M. Hepatic uptake of polystyrene microspheres in rats: Effect of particle size on intrahepatic distribution. J. Control. Release 1999, 59, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.H.; Li, F.C.; Souris, J.S.; Yang, C.S.; Tseng, F.G.; Lee, H.S.; Chen, C.T.; Dong, C.Y.; Lo, L.W. Visualizing dynamics of sub-hepatic distribution of nanoparticles using intravital multiphoton fluorescence microscopy. ACS Nano 2012, 6, 4122–4131. [Google Scholar] [CrossRef]
- Bottger, R.; Pauli, G.; Chao, P.H.; Al Fayez, N.; Hohenwarter, L.; Li, S.D. Lipid-based nanoparticle technologies for liver targeting. Adv. Drug Deliv. Rev. 2020, 154-155, 79–101. [Google Scholar] [CrossRef]
- Zhang, Y.N.; Poon, W.; Tavares, A.J.; McGilvray, I.D.; Chan, W.C.W. Nanoparticle-liver interactions: Cellular uptake and hepatobiliary elimination. J. Control. Release 2016, 240, 332–348. [Google Scholar] [CrossRef] [PubMed]
- Champion, J.A.; Mitragotri, S. Shape induced inhibition of phagocytosis of polymer particles. Pharm. Res. 2009, 26, 244–249. [Google Scholar] [CrossRef]
- Byrne, J.D.; Betancourt, T.; Brannon-Peppas, L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv. Drug Deliv. Rev. 2008, 60, 1615–1626. [Google Scholar] [CrossRef]
- Danhier, F.; Feron, O.; Preat, V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J. Control. Release 2010, 148, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Love, K.T.; Dorkin, J.R.; Sirirungruang, S.; Zhang, Y.; Chen, D.; Bogorad, R.L.; Yin, H.; Chen, Y.; Vegas, A.J.; et al. Lipopeptide nanoparticles for potent and selective siRNA delivery in rodents and nonhuman primates. Proc. Natl. Acad. Sci. USA 2014, 111, 3955–3960. [Google Scholar] [CrossRef]
- Huang, X.; Leroux, J.C.; Castagner, B. Well-Defined Multivalent Ligands for Hepatocytes Targeting via Asialoglycoprotein Receptor. Bioconjug. Chem. 2017, 28, 283–295. [Google Scholar] [CrossRef]
- D’Souza, A.A.; Devarajan, P.V. Asialoglycoprotein receptor mediated hepatocyte targeting—Strategies and applications. J. Control. Release 2015, 203, 126–139. [Google Scholar] [CrossRef]
- Lin, A.; Liu, Y.; Huang, Y.; Sun, J.; Wu, Z.; Zhang, X.; Ping, Q. Glycyrrhizin surface-modified chitosan nanoparticles for hepatocyte-targeted delivery. Int. J. Pharm. 2008, 359, 247–253. [Google Scholar] [CrossRef]
- Hashiba, K.; Sato, Y.; Harashima, H. pH-labile PEGylation of siRNA-loaded lipid nanoparticle improves active targeting and gene silencing activity in hepatocytes. J. Control. Release 2017, 262, 239–246. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Guo, W.; Deng, F.; Chen, K.; Jiang, Y.; Dong, M.; Peng, L.; Chen, X. Targeted delivery of microRNA 146b mimic to hepatocytes by lactosylated PDMAEMA nanoparticles for the treatment of NAFLD. Artif. Cells Nanomed. Biotechnol. 2018, 46, 217–228. [Google Scholar] [CrossRef]
- Cheng, Q.; Wei, T.; Farbiak, L.; Johnson, L.T.; Dilliard, S.A.; Siegwart, D.J. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing. Nat. Nanotechnol. 2020, 15, 313–320. [Google Scholar] [CrossRef] [PubMed]
- de Bleser, P.J.; Jannes, P.; van Buul-Offers, S.C.; Hoogerbrugge, C.M.; van Schravendijk, C.F.; Niki, T.; Rogiers, V.; van den Brande, J.L.; Wisse, E.; Geerts, A. Insulinlike growth factor-II/mannose 6-phosphate receptor is expressed on CCl4-exposed rat fat-storing cells and facilitates activation of latent transforming growth factor-beta in cocultures with sinusoidal endothelial cells. Hepatology 1995, 21, 1429–1437. [Google Scholar] [CrossRef]
- Bai, X.; Su, G.; Zhai, S. Recent Advances in Nanomedicine for the Diagnosis and Therapy of Liver Fibrosis. Nanomaterials 2020, 10, 1945. [Google Scholar] [CrossRef]
- Yang, D. Research progress of receptor targeted hepatic stellate cell in treatment of liver fibrosis. Zhonghua Gan Zang Bing Za Zhi 2018, 26, 630–632. [Google Scholar] [CrossRef]
- Lee, Y.S.; Jeong, W.I. Retinoic acids and hepatic stellate cells in liver disease. J. Gastroenterol. Hepatol. 2012, 27 (Suppl. S2), 75–79. [Google Scholar] [CrossRef]
- Sato, Y.; Murase, K.; Kato, J.; Kobune, M.; Sato, T.; Kawano, Y.; Takimoto, R.; Takada, K.; Miyanishi, K.; Matsunaga, T.; et al. Resolution of liver cirrhosis using vitamin A-coupled liposomes to deliver siRNA against a collagen-specific chaperone. Nat. Biotechnol. 2008, 26, 431–442. [Google Scholar] [CrossRef]
- Giannitrapani, L.; Soresi, M.; Bondi, M.L.; Montalto, G.; Cervello, M. Nanotechnology applications for the therapy of liver fibrosis. World J. Gastroenterol. 2014, 20, 7242–7251. [Google Scholar] [CrossRef]
- Adrian, J.E.; Poelstra, K.; Scherphof, G.L.; Molema, G.; Meijer, D.K.; Reker-Smit, C.; Morselt, H.W.; Kamps, J.A. Interaction of targeted liposomes with primary cultured hepatic stellate cells: Involvement of multiple receptor systems. J. Hepatol. 2006, 44, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Gong, Y.; Pi, Y.; Liu, X.; Gao, L.; Kang, L.; Wang, J.; Yang, F.; Tang, J.; Lu, W.; et al. pPB Peptide-Mediated siRNA-Loaded Stable Nucleic Acid Lipid Nanoparticles on Targeting Therapy of Hepatic Fibrosis. Mol. Pharm. 2018, 15, 53–62. [Google Scholar] [CrossRef]
- Liu, C.H.; Chan, K.M.; Chiang, T.; Liu, J.Y.; Chern, G.G.; Hsu, F.F.; Wu, Y.H.; Liu, Y.C.; Chen, Y. Dual-Functional Nanoparticles Targeting CXCR4 and Delivering Antiangiogenic siRNA Ameliorate Liver Fibrosis. Mol. Pharm. 2016, 13, 2253–2262. [Google Scholar] [CrossRef] [PubMed]
- Ritz, T.; Krenkel, O.; Tacke, F. Dynamic plasticity of macrophage functions in diseased liver. Cell Immunol. 2018, 330, 175–182. [Google Scholar] [CrossRef]
- He, C.; Yin, L.; Tang, C.; Yin, C. Multifunctional polymeric nanoparticles for oral delivery of TNF-α siRNA to macrophages. Biomaterials 2013, 34, 2843–2854. [Google Scholar] [CrossRef]
- Ravichandran, K.S. Beginnings of a good apoptotic meal: The find-me and eat-me signaling pathways. Immunity 2011, 35, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Pan, W.; Wang, Y.; Lei, W.; Feng, B.; Du, C.; Wang, X.J. Enhanced efficacy of curcumin with phosphatidylserine-decorated nanoparticles in the treatment of hepatic fibrosis. Drug Deliv. 2018, 25, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Guo, J.; Liu, Y.; Wang, M.; Liu, Z.; Gao, Y.; Huang, L. Nano delivery of simvastatin targets liver sinusoidal endothelial cells to remodel tumor microenvironment for hepatocellular carcinoma. J. Nanobiotechnology 2022, 20, 9. [Google Scholar] [CrossRef]
- Gil, M.; Khouri, L.; Raurell, I.; Rafael, D.; Andrade, F.; Abasolo, I.; Schwartz, S., Jr.; Martinez-Gomez, M.; Salcedo, M.T.; Pericas, J.M.; et al. Optimization of Statin-Loaded Delivery Nanoparticles for Treating Chronic Liver Diseases by Targeting Liver Sinusoidal Endothelial Cells. Pharmaceutics 2023, 15, 2463. [Google Scholar] [CrossRef] [PubMed]
- Tee, J.K.; Ng, L.Y.; Koh, H.Y.; Leong, D.T.; Ho, H.K. Titanium Dioxide Nanoparticles Enhance Leakiness and Drug Permeability in Primary Human Hepatic Sinusoidal Endothelial Cells. Int. J. Mol. Sci. 2019, 20, 35. [Google Scholar] [CrossRef]
- Fan, Q.Q.; Zhang, C.L.; Qiao, J.B.; Cui, P.F.; Xing, L.; Oh, Y.K.; Jiang, H.L. Extracellular matrix-penetrating nanodrill micelles for liver fibrosis therapy. Biomaterials 2020, 230, 119616. [Google Scholar] [CrossRef]
- Duong, H.T.; Dong, Z.; Su, L.; Boyer, C.; George, J.; Davis, T.P.; Wang, J. The use of nanoparticles to deliver nitric oxide to hepatic stellate cells for treating liver fibrosis and portal hypertension. Small 2015, 11, 2291–2304. [Google Scholar] [CrossRef]
- Lin Ts, T.; Gao, D.Y.; Liu, Y.C.; Sung, Y.C.; Wan, D.; Liu, J.Y.; Chiang, T.; Wang, L.; Chen, Y. Development and characterization of sorafenib-loaded PLGA nanoparticles for the systemic treatment of liver fibrosis. J. Control. Release 2016, 221, 62–70. [Google Scholar] [CrossRef]
- Hassan, R.; Tammam, S.N.; Safy, S.E.; Abdel-Halim, M.; Asimakopoulou, A.; Weiskirchen, R.; Mansour, S. Prevention of hepatic stellate cell activation using JQ1- and atorvastatin-loaded chitosan nanoparticles as a promising approach in therapy of liver fibrosis. Eur. J. Pharm. Biopharm. 2019, 134, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Zhu, M.; Zhou, S.; Feng, W.; Chen, H. Rapamycin-Loaded mPEG-PLGA Nanoparticles Ameliorate Hepatic Steatosis and Liver Injury in Non-alcoholic Fatty Liver Disease. Front. Chem. 2020, 8, 407. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.N.; Baiyisaiti, A.; Wong, C.W.; Hsu, S.H.; Qi, R. Polyurethane Nanoparticle-Loaded Fenofibrate Exerts Inhibitory Effects on Nonalcoholic Fatty Liver Disease in Mice. Mol. Pharm. 2018, 15, 4550–4557. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Zhang, P.; Zhao, T.; Jia, M.; Yin, P.; Li, W.; Zhang, Z.R.; Fu, Y.; Gong, T. Golgi Apparatus-Targeted Chondroitin-Modified Nanomicelles Suppress Hepatic Stellate Cell Activation for the Management of Liver Fibrosis. ACS Nano 2019, 13, 3910–3923. [Google Scholar] [CrossRef]
- Ji, D.; Wang, Q.; Zhao, Q.; Tong, H.; Yu, M.; Wang, M.; Lu, T.; Jiang, C. Co-delivery of miR-29b and germacrone based on cyclic RGD-modified nanoparticles for liver fibrosis therapy. J. Nanobiotechnol. 2020, 18, 86. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, H.J.; Zamboni, C.G.; Hassan, L.F.; Radant, N.P.; Jacob, D.; Mease, R.C.; Minn, I.; Tzeng, S.Y.; Gabrielson, K.L.; Bhardwaj, P.; et al. Polymeric nanoparticles for dual-targeted theranostic gene delivery to hepatocellular carcinoma. Sci. Adv. 2022, 8, eabo6406. [Google Scholar] [CrossRef]
- Hu, M.; Wang, Y.; Liu, Z.; Yu, Z.; Guan, K.; Liu, M.; Wang, M.; Tan, J.; Huang, L. Hepatic macrophages act as a central hub for relaxin-mediated alleviation of liver fibrosis. Nat. Nanotechnol. 2021, 16, 466–477. [Google Scholar] [CrossRef]
- Li, Y.; Pu, S.; Liu, Q.; Li, R.; Zhang, J.; Wu, T.; Chen, L.; Li, H.; Yang, X.; Zou, M.; et al. An integrin-based nanoparticle that targets activated hepatic stellate cells and alleviates liver fibrosis. J. Control. Release 2019, 303, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Han, M.; Xu, J.; Geng, S.; Zhang, Y.; Ye, X.; Gou, J.; Yin, T.; He, H.; Tang, X. Asialoglycoprotein receptor-targeted liposomes loaded with a norcantharimide derivative for hepatocyte-selective targeting. Int. J. Pharm. 2017, 520, 98–110. [Google Scholar] [CrossRef]
- Ribera, J.; Vilches, C.; Sanz, V.; de Miguel, I.; Portoles, I.; Cordoba-Jover, B.; Prat, E.; Nunes, V.; Jimenez, W.; Quidant, R.; et al. Treatment of Hepatic Fibrosis in Mice Based on Targeted Plasmonic Hyperthermia. ACS Nano 2021, 15, 7547–7562. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, M.; Fu, L.; Lin, J.; Zhou, X.; Zhou, P.; Huang, P.; Hu, H.; Han, Y. Liver-targeted delivery of TSG-6 by calcium phosphate nanoparticles for the management of liver fibrosis. Theranostics 2020, 10, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Afroze, S.H.; Munshi, M.K.; Martinez, A.K.; Uddin, M.; Gergely, M.; Szynkarski, C.; Guerrier, M.; Nizamutdinov, D.; Dostal, D.; Glaser, S. Activation of the renin-angiotensin system stimulates biliary hyperplasia during cholestasis induced by extrahepatic bile duct ligation. Am. J. Physiol. Gastrointest Liver Physiol. 2015, 308, G691–G701. [Google Scholar] [CrossRef] [PubMed]
- Vilar-Gomez, E.; Vuppalanchi, R.; Gawrieh, S.; Ghabril, M.; Saxena, R.; Cummings, O.W.; Chalasani, N. Vitamin E Improves Transplant-Free Survival and Hepatic Decompensation among Patients with Nonalcoholic Steatohepatitis and Advanced Fibrosis. Hepatology 2020, 71, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Pfister, D.; Nunez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef] [PubMed]
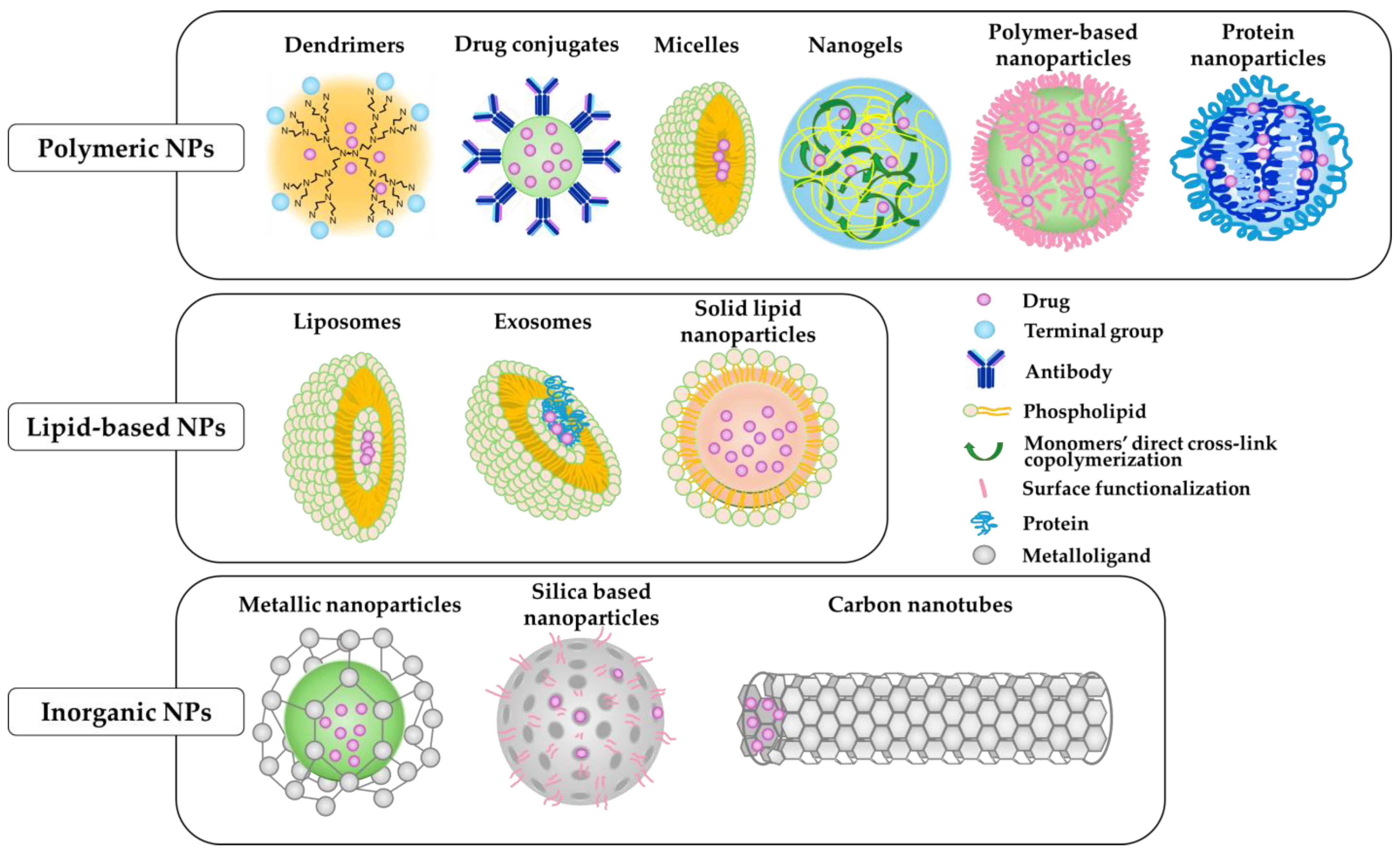
| Types of Nanoparticles | Advantages | Disadvantages |
|---|---|---|
| Polymeric NPs |
|
|
| Lipid-based NPs |
|
|
| Inorganic NPs |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vu, H.T.; Nguyen, V.D.; Ikenaga, H.; Matsubara, T. Application of PPAR Ligands and Nanoparticle Technology in Metabolic Steatohepatitis Treatment. Biomedicines 2024, 12, 1876. https://doi.org/10.3390/biomedicines12081876
Vu HT, Nguyen VD, Ikenaga H, Matsubara T. Application of PPAR Ligands and Nanoparticle Technology in Metabolic Steatohepatitis Treatment. Biomedicines. 2024; 12(8):1876. https://doi.org/10.3390/biomedicines12081876
Chicago/Turabian StyleVu, Hung Thai, Vien Duc Nguyen, Hiroko Ikenaga, and Tsutomu Matsubara. 2024. "Application of PPAR Ligands and Nanoparticle Technology in Metabolic Steatohepatitis Treatment" Biomedicines 12, no. 8: 1876. https://doi.org/10.3390/biomedicines12081876