
Insulin Resistance, a Risk Factor for Alzheimer’s Disease: Pathological Mechanisms and a New Proposal for a Preventive Therapeutic Approach
{kind=link}
{kind=link}
Abstract
:1. Introduction
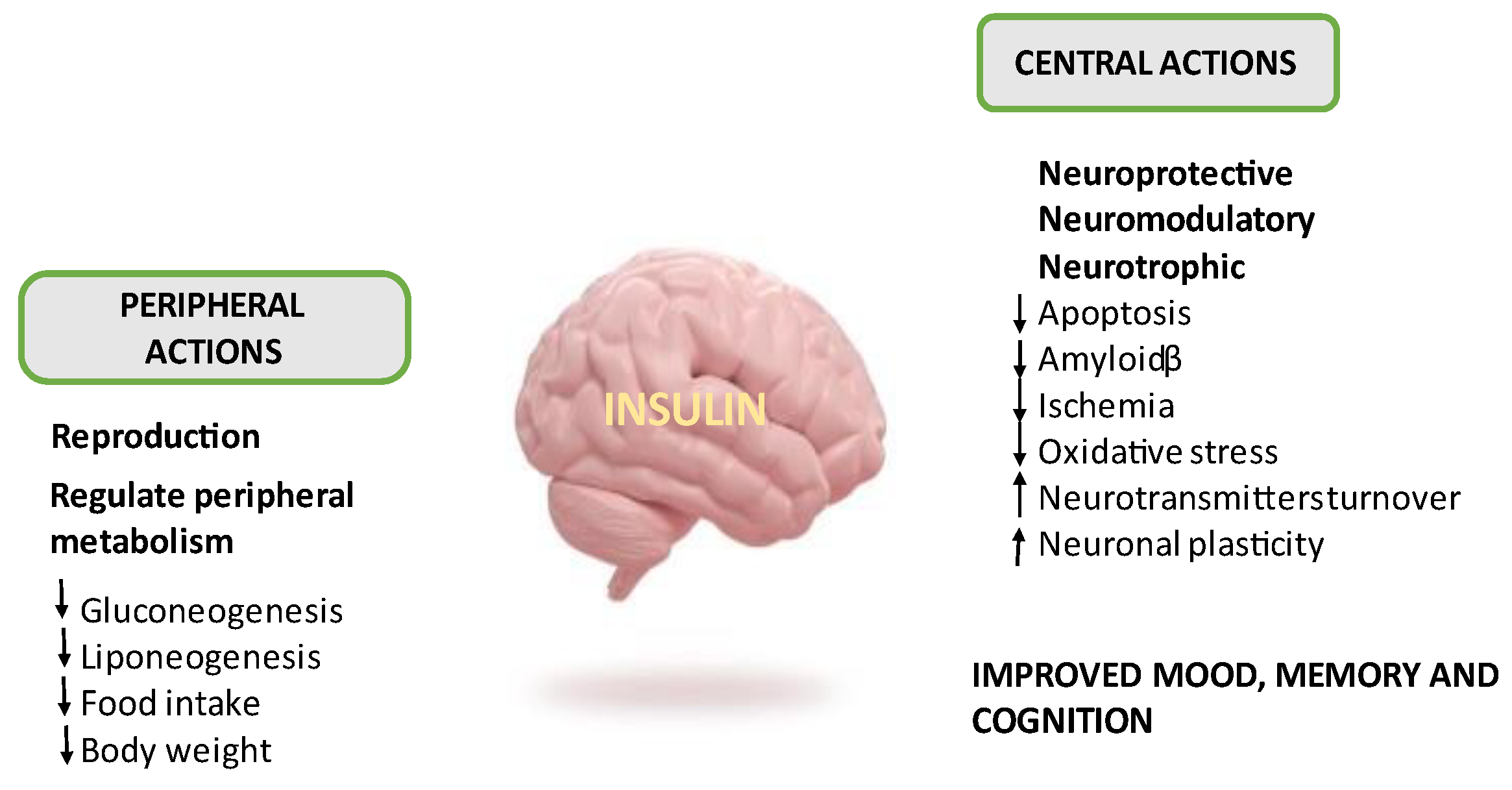
2. Insulin, Brain and Insulin Resistance
3. Glucose Metabolism and Dysregulation of Brain Energetics
4. Aβ, Tau, Apolipoprotein E and IR
5. Mitochondria, Oxidative Stress, IR and Neuroinflammation
6. Vascular Damage
7. Potential Therapeutic Approach
7.1. Metformin
7.2. PPARɣ Agonists
7.3. Glucagon-Like Peptide-1 Receptor Agonists
7.4. Sodium-Glucose Cotransporter 2 Inhibitors
7.5. Phosphodiesterase 5 Inhibitors
7.6. Berberine
7.7. Quercetin
7.8. L-Arginine
7.9. Klotho Protein as Therapeutic Target of Neurodegenerative Diseases
7.10. Inhaled Insulin
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- 2023 Alzheimer’s disease facts and figures. Alzheimers Dement. 2023, 19, 1598–1695. [CrossRef] [PubMed]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. IDF Diabetes Atlas Committee. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res Clin Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed]
- Freeman, A.M.; Acevedo, L.A.; Pennings, N. Insulin Resistance [Updated 2023 Aug 17]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Crane, P.K.; Walker, R.; Hubbard, R.A.; Li, G.; Nathan, D.M.; Zheng, H.; Haneuse, S.; Craft, S.; Montine, T.J.; Kahn, S.E.; et al. Glucose levels and risk of dementia. N. Engl. J. Med. 2013, 369, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wu, C.; Zhang, X.; Wang, L.; Sun, J.; Zhuge, F. Insulin resistance is a risk factor for mild cognitive impairment in elderly adults with T2DM. Open Life Sci. 2019, 14, 255–261. [Google Scholar] [CrossRef]
- Ott, A.; Stolk, R.P.; van Harskamp, F.; Pols, H.A.; Hofman, A.; Breteler, M.M. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 1999, 53, 1937–1942. [Google Scholar] [CrossRef] [PubMed]
- Rhea, E.M.; Banks, W.A.; Raber, J. Insulin Resistance in Peripheral Tissues and the Brain: A Tale of Two Sites. Biomedicines 2022, 10, 1582. [Google Scholar] [CrossRef] [PubMed]
- Janson, J.; Laedtke, T.; Parisi, J.E.; O’Brien, P.; Petersen, R.C.; Butler, P.C. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 2004, 53, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Jaspan, J.B.; Huang, W.; Kastin, A.J. Transport of insulin across the blood-brain barrier: Saturability at euglycemic doses of insulin. Peptides 1997, 18, 1423–1429. [Google Scholar] [CrossRef]
- Baura, G.D.; Foster, D.M.; Porte, D.; Kahn, S.E.; Bergman, R.N.; Cobelli, C.; Schwartz, M.W. Saturable transport of insulin from plasma into the central nervous system of dogs in vivo. A mechanism for regulated insulin delivery to the brain. J. Clin. Investig. 1993, 92, 1824–1830. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.; Thomas, P.; Pemberton, S.; Babin, A.; Noonan, C.; Weaver, R.; Banks, W.A.; Rhea, E.M. Central nervous system insulin signaling can influence the rate of insulin influx into brain. Fluids Barriers CNS 2023, 20, 28. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sánchez-Zúñiga, M.J.; Carrillo-Esper, R.; Sánchez-Pérez, H.; González-Chávez, A.; Elizondo-Argueta, S. Brain insulin circuit. From the basics to impact on the clinic. Cir Cir. 2020, 88, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Rhea, E.M.; Rask-Madsen, C.; Banks, W.A. Insulin transport across the blood–brain barrier can occur independently of the insulin receptor. J. Physiol. 2018, 596, 4753–4765. [Google Scholar] [CrossRef]
- Devaskar, S.U.; Giddings, S.J.; Rajakumar, P.A.; Carnaghi, L.R.; Menon, R.K.; Zahm, D.S. Insulin gene expression and insulin synthesis in mammalian neuronal cells. J. Biol. Chem. 1994, 269, 8445–8454. [Google Scholar] [CrossRef]
- Mazucanti, C.H.; Liu, Q.R.; Lang, D.; Huang, N.; O’Connell, J.F.; Camandola, S.; Egan, J.M. Release of insulin produced by the choroid plexis is regulated by serotonergic signaling. JCI Insight. 2019, 4, e131682. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Heidenreich, K.A.; Zahniser, N.R.; Berhanu, P.; Brandenburg, D.; Olefsky, J.M. Structural differences between insulin receptors in the brain and peripheral target tissues. J. Biol. Chem. 1983, 258, 8527–8530. [Google Scholar] [CrossRef] [PubMed]
- Kleinridders, A.; Ferris, H.A.; Cai, W.; Kahn, C.R. Insulin action in brain regulates systemic metabolism and brain function. Diabetes 2014, 63, 2232–2243. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Pessin, J.E. Insulin-Mediated PI3K and AKT Signaling. In The Liver: Biology And Pathobiology; Wiley: Hoboken, NJ, USA, 2020; pp. 485–495. [Google Scholar]
- Goodner, C.J.; Hom, F.G.; Berrie, M.A. Investigation of the effect of insulin upon regional brain glucose metabolism in the rat in vivo. Endocrinology 1980, 107, 1827–1832. [Google Scholar] [CrossRef]
- Van Der Heide, L.P.; Kamal, A.; Artola, A.; Gispen, W.H.; Ramakers, G.M. Insulin modulates hippocampal activity-dependent synaptic plasticity in a N-methyl-d-aspartate receptor and phosphatidyl-inositol-3-kinase-dependent manner. J. Neurochem. 2005, 94, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Shymko, R.M.; Dumont, E.; Meyts, P.D.; Dumont, J.E. Timing-dependence of insulin-receptor mitogenic versus metabolic signalling: A plausible model based on coincidence of hormone and effector binding. Biochem. J. 1999, 339, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Sakaguchi, M.; Kleinridders, A.; Gonzalez-Del Pino, G.; Dreyfuss, J.M.; O’Neill, B.T.; Kahn, C.R. Domain-dependent effects of insulin and IGF-1 receptors on signalling and gene expression. Nat. Commun. 2017, 8, 14892. [Google Scholar] [CrossRef]
- Suzuki, R.; Lee, K.; Jing, E.; Biddinger, S.B.; McDonald, J.G.; Montine, T.J.; Kahn, C.R. Diabetes and insulin in regulation of brain cholesterol metabolism. Cell Metab. 2010, 12, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, R.; Haeri, A.; Dargahi, L.; Mohamed, Z.; Ahmadiani, A. Insulin in the brain: Sources, localization and functions. Mol. Neurobiol. 2013, 47, 145–171. [Google Scholar] [CrossRef] [PubMed]
- Porte, D., Jr.; Seeley, R.J.; Woods, S.C.; Baskin, D.G.; Figlewicz, D.P.; Schwartz, M.W. Obesity, diabetes and the central nervous system. Diabetologia 1998, 41, 863–881. [Google Scholar] [CrossRef] [PubMed]
- Ryu, B.R.; Ko, H.W.; Jou, I.; Noh, J.S.; Gwag, B.J. Phosphatidylinositol 3-kinase-mediated regulation of neuronal apoptosis and necrosis by insulin and IGF-I. J. Neurobiol. 1999, 39, 536–546. [Google Scholar] [CrossRef]
- Rensink, A.A.; Otte-Höller, I.; de Boer, R.; Bosch, R.R.; ten Donkelaar, H.J.; de Waal, R.M.; Kremer, B. Insulin inhibits amyloid β-induced cell death in cultured human brain pericytes. Neurobiol. Aging 2004, 25, 93–103. [Google Scholar] [CrossRef]
- Nash, A.I. Crosstalk between insulin and dopamine signaling: A basis for the metabolic effects of antipsychotic drugs. J. Chem. Neuroanat. 2017, 83, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Guénette, S. Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Ohtsuki, S.; Murata, S.; Katsukura, Y.; Suzuki, H.; Funaki, M.; Terasaki, T. Involvement of insulin-degrading enzyme in insulin-and atrial natriuretic peptide-sensitive internalization of amyloid-β peptide in mouse brain capillary endothelial cells. J. Alzheimer’s Dis. 2014, 38, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Xiang, Z.; Haroutunian, V.; Buxbaum, J.D.; Stetka, B.; Pasinetti, G.M. Insulin degrading enzyme activity selectively decreases in the hippocampal formation of cases at high risk to develop Alzheimer’s disease. Neurobiol. Aging 2007, 28, 824–830. [Google Scholar] [CrossRef] [PubMed]
- Rhea, E.M.; Leclerc, M.; Yassine, H.N.; Capuano, A.W.; Tong, H.; Petyuk, V.A.; Macauley, S.L.; Fioramonti, X.; Carmichael, O.; Calon, F.; et al. State of the Science on Brain Insulin Resistance and Cognitive Decline Due to Alzheimer’s Disease. Aging Dis. 2023, 15, 1688. [Google Scholar] [CrossRef]
- Leclerc, M.; Bourassa, P.; Tremblay, C.; Caron, V.; Sugère, C.; Edmond, V.; Calon, F. Cerebrovascular insulin receptors are defective in Alzheimer’s disease. Brain 2023, 146, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Arnold, S.E. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [PubMed]
- Konishi, M.; Sakaguchi, M.; Lockhart, S.M.; Cai, W.; Li, M.E.; Homan, E.P.; Kahn, C.R. Endothelial insulin receptors differentially control insulin signaling kinetics in peripheral tissues and brain of mice. Proc. Natl. Acad. Sci. USA 2017, 114, E8478–E8487. [Google Scholar] [CrossRef] [PubMed]
- Rhea, E.M.; Banks, W.A. Role of the blood-brain barrier in central nervous system insulin resistance. Front. Neurosci. 2019, 13, 457034. [Google Scholar] [CrossRef] [PubMed]
- Starr, J.M.; Wardlaw, J.; Ferguson, K.; MacLullich, A.; Deary, I.J.; Marshall, I. Increased blood–brain barrier permeability in type II diabetes demonstrated by gadolinium magnetic resonance imaging. J. Neurol. Neurosurg. Psychiatry 2003, 74, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.E.; Lucki, I.; Brookshire, B.R.; Carlson, G.C.; Browne, C.A.; Kazi, H.; Kim, S.F. High fat diet produces brain insulin resistance, synaptodendritic abnormalities and altered behavior in mice. Neurobiol. Dis. 2014, 67, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Liang, X.; Gu, H.; Hu, Y.; Zhao, Z.; Yang, X.Y.; Qian, C.; Yang, Y.; Teng, G.J. Cerebral perfusion alterations in type 2 diabetes and its relation to insulin resistance and cognitive dysfunction. Brain Imaging Behav. 2017, 11, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.Y.; Ahima, R.S.; Nathan, D.M. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Watson, L.S.; Wilken-Resman, B.; Williams, A.; DiLucia, S.; Sanchez, G.; McLeod, T.L.; Sims-Robinson, C. Hyperinsulinemia alters insulin receptor presentation and internalization in brain microvascular endothelial cells. Diab. Vasc. Dis. Res. 2022, 19, 14791641221118626. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Longato, L.; Tong, M.; Wands, J.R. Insulin resistance and neurodegeneration: Roles of obesity, type 2 diabetes mellitus and non-alcoholic steatohepatitis. Curr. Opin. Investig. Drugs 2009, 10, 1049. [Google Scholar]
- Sripetchwandee, J.; Chattipakorn, N.; Chattipakorn, S.C. Links between obesity-induced brain insulin resistance, brain mitochondrial dysfunction, and dementia. Front. Endocrinol. 2018, 9, 359824. [Google Scholar] [CrossRef]
- Frisardi, V.; Solfrizzi, V.; Seripa, D.; Capurso, C.; Santamato, A.; Sancarlo, D.; Panza, F. Metabolic-cognitive syndrome: A cross-talk between metabolic syndrome and Alzheimer’s disease. Ageing Res. Rev. 2010, 9, 399–417. [Google Scholar] [CrossRef] [PubMed]
- Tong, M.; De la Monte, S.M. Mechanisms of ceramide-mediated neurodegeneration. J. Alzheimer’s Dis. 2009, 16, 705–714. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Di Domenico, F.; Barone, E. Elevated risk of type 2 diabetes for development of Alzheimer disease: A key role for oxidative stress in brain. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 1693–1706. [Google Scholar] [CrossRef] [PubMed]
- Whitmer, R.A. Type 2 diabetes and risk of cognitive impairment and dementia. Curr. Neurol. Neurosci. Rep. 2007, 7, 373–380. [Google Scholar] [CrossRef]
- Ferreira, S.T.; Klein, W.L. The Aβ oligomer hypothesis for synapse failure and memory loss in Alzheimer’s disease. Neurobiol Learn Mem. 2011, 96, 529–543. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhao, W.Q.; De Felice, F.G.; Fernandez, S.; Chen, H.; Lambert, M.P.; Quon, M.J.; Krafft, G.A.; Klein, W.L. Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. 2008, 22, 246–260. [Google Scholar] [CrossRef] [PubMed]
- Hasselbach SGKnudsen, G.M.; Videbaek, C.; Pinborg, L.H.; Schmidt, J.F.; Holm, S.; Paulson, O.B. No effect of insulin on glucose blood-brain barrier transport and cerebral metabolism in humans. Diabetes 1999, 48, 1915–1921. [Google Scholar] [CrossRef]
- Seaquist, E.R.; Damberg, G.S.; Tkac, I.; Gruetter, R. The effect of insulin on in vivo cerebral glucose concentrations and rates of glucose transport/metabolism in humans. Diabetes 2001, 50, 2203–2209. [Google Scholar] [CrossRef] [PubMed]
- Simpson, I.A.; Dwyer, D.; Malide, D.; Moley, K.H.; Travis, A.; Vannucci, S.J. The facilitative glucose transporter GLUT3: 20 years of distinction. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E242–E253. [Google Scholar] [CrossRef]
- Milstein, J.L.; Ferris, H.A. The brain as an insulin-sensitive metabolic organ. Mol. Metab. 2021, 52, 101234. [Google Scholar] [CrossRef] [PubMed]
- Simpson, I.A.; Carruthers, A.; Vannucci, S.J. Supply and demand in cerebral energy metabolism: The role of nutrient transporters. J. Cereb. Blood Flow Metab. 2007, 27, 1766–1791. [Google Scholar] [CrossRef]
- Dienel, G.A. Fueling and imaging brain activation. ASN Neuro 2012, 4, AN20120021. [Google Scholar] [CrossRef]
- Bentsen, M.A.; Mirzadeh, Z.; Schwartz, M.W. Revisiting how the brain senses glucose—And why. Cell Metab. 2019, 29, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Bouzier-Sore, A.K.; Merle, M.; Magistretti, P.J.; Pellerin, L. Feeding active neurons:(re) emergence of a nursing role for astrocytes. J. Physiol. 2002, 96, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Benomar, Y.; Naour, N.; Aubourg, A.; Bailleux, V.; Gertler, A.; Djiane, J.; Taouis, M. Insulin and leptin induce Glut4 plasma membrane translocation and glucose uptake in a human neuronal cell line by a phosphatidylinositol 3-kinase-dependent mechanism. Endocrinology 2006, 147, 2550–2556. [Google Scholar] [CrossRef]
- Reno, C.M.; Puente, E.C.; Sheng, Z.; Daphna-Iken, D.; Bree, A.J.; Routh, V.H.; Kahn, B.B.; Fisher, S.J. Brain GLUT4 Knockout Mice Have Impaired Glucose Tolerance, Decreased Insulin Sensitivity, and Impaired Hypoglycemic Counterregulation. Diabetes 2017, 66, 587–597. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Frere, S.; Slutsky, I. Alzheimer’s disease: From firing instability to homeostasis network collapse. Neuron 2018, 97, 32–58. [Google Scholar] [CrossRef]
- Kapogiannis, D.; Mattson, M.P. Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer’s disease. Lancet Neurol. 2011, 10, 187–198. [Google Scholar] [CrossRef]
- Mosconi, L.; Pupi, A.; De Leon, M.J. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2008, 1147, 180–195. [Google Scholar] [CrossRef]
- Adlimoghaddam, A.; Snow, W.M.; Stortz, G.; Perez, C.; Djordjevic, J.; Goertzen, A.L.; Albensi, B.C. Regional hypometabolism in the 3xTg mouse model of Alzheimer’s disease. Neurobiol. Dis. 2019, 127, 264–277. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, W.; Maudsley, S.; Hull, W.; Havolli, E.; Boshoff, E.; Hill, M.D.; Goetghebeur, P.J.; Harrison, D.C.; Nizami, S.; Bedford, D.C.; et al. The oDGal mouse: A novel, physiologically relevant rodent model of sporadic Alzheimer’s disease. Int. J. Mol. Sci. 2023, 24, 6953. [Google Scholar] [CrossRef] [PubMed]
- Hirono, N.; Mori, E.; Ishii, K.; Ikejiri, Y.; Imamura, T.; Shimomura, T.; Hashimoto, M.; Yamashita, H.; Sasaki, M. Frontal lobe hypometabolism and depression in Alzheimer’s disease. Neurology 1998, 50, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Daulatzai, M.A. Cerebral hypoperfusion and glucose hypometabolism: Key pathophysiological modulators promote neurodegeneration, cognitive impairment, and Alzheimer’s disease. J. Neurosci. Res. 2017, 95, 943–972. [Google Scholar] [CrossRef]
- Förster, S.; Grimmer, T.; Miederer, I.; Henriksen, G.; Yousefi, B.H.; Graner, P.; Wester, H.J.; Förstl, H.; Kurz, A.; Dickerson, B.C.; et al. Regional expansion of hypometabolism in Alzheimer’s disease follows amyloid deposition with temporal delay. Biol. Psychiatry 2012, 71, 792–797. [Google Scholar] [CrossRef] [PubMed]
- Drzezga, A.; Lautenschlager, N.; Siebner, H.; Riemenschneider, M.; Willoch, F.; Minoshima, S.; Schwaiger, M.; Kurz, A. Cerebral metabolic changes accompanying conversion of mild cognitive impairment into Alzheimer’s disease: A PET follow-up study. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 1104–1113. [Google Scholar] [PubMed]
- Small, G.W.; Ercoli, L.M.; Silverman, D.H.; Huang, S.C.; Komo, S.; Bookheimer, S.Y.; Lavretsky, H.; Miller, K.; Siddarth, P.; Rasgon, N.L.; et al. Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 6037–6042. [Google Scholar] [CrossRef]
- Duarte, A.I.; Moreira, P.I.; Oliveira, C.R. Insulin in central nervous system: More than just a peripheral hormone. J. Aging Res. 2012, 2012, 384017. [Google Scholar] [CrossRef] [PubMed]
- McNay, E.C.; Pearson-Leary, J. GluT4: A central player in hippocampal memory and brain insulin resistance. Exp. Neurol. 2020, 323, 113076. [Google Scholar] [CrossRef]
- Raut, S.; Bhalerao, A.; Powers, M.; Gonzalez, M.; Mancuso, S.; Cucullo, L. Hypometabolism, Alzheimer’s Disease, and Possible Therapeutic Targets: An Overview. Cells 2023, 12, 2019. [Google Scholar] [CrossRef]
- Simpson, I.A.; Chundu, K.R.; Davies-Hill, T.; Honer, W.G.; Davies, P. Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer’s disease. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1994, 35, 546–551. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X. Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett. 2008, 582, 359–364. [Google Scholar] [CrossRef]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Harr, S.D.; Simonian, N.A.; Hyman, B.T. Functional alterations in Alzheimer’s disease: Decreased glucose transporter 3 immunoreactivity in the perforant pathway terminal zone. J. Neuropathol. Exp. Neurol. 1995, 54, 38–41. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Varma, V.R.; Varma, S.; Casanova, R.; Dammer, E.; Pletnikova, O.; Chia, C.W.; Egan, J.M.; Ferrucci, L.; Troncoso, J.; et al. Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 318–329. [Google Scholar] [CrossRef]
- Zhang, H.; Wei, W.; Zhao, M.; Ma, L.; Jiang, X.; Pei, H.; Cao, Y.; Li, H. Interaction between Aβ and Tau in the Pathogenesis of Alzheimer’s Disease. Int. J. Biol. Sci. 2021, 17, 2181–2192. [Google Scholar] [CrossRef]
- Ramos-Rodriguez, J.J.; Spires-Jones, T.; Pooler, A.M.; Lechuga-Sancho, A.M.; Bacskai, B.J.; Garcia-Alloza, M. Progressive neuronal pathology and synaptic loss induced by prediabetes and type 2 diabetes in a mouse model of Alzheimer’s disease. Mol. Neurobiol. 2017, 54, 3428–3438. [Google Scholar] [CrossRef]
- Lee, C.C.; Kuo, Y.M.; Huang, C.C.; Hsu, K.S. Insulin rescues amyloid β-induced impairment of hippocampal long-term potentiation. Neurobiol. Aging 2009, 30, 377–387. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Vieira, M.N.; Bomfim, T.R.; Decker, H.; Velasco, P.T.; Lambert, M.P.; Viola, K.L.; Zhao, W.Q.; Ferreira, S.T.; Klein, W.L. Protection of synapses against Alzheimer’s-linked toxins: Insulin signaling prevents the pathogenic binding of Aβ oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 1971–1976. [Google Scholar] [CrossRef]
- Steen, E.; Terry, B.M.; JRivera, E.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de La Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease–is this type 3 diabetes? J. Alzheimer’s Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef]
- Medeiros, R.; Baglietto-Vargas, D.; LaFerla, F.M. The role of tau in Alzheimer’s disease and related disorders. CNS Neurosci. Ther. 2011, 17, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Sperber, B.R.; Leight, S.; Goedert, M.; Lee, V.M. Glycogen synthase kinase-3 beta phosphorylates tau protein at multiple sites in intact cells. Neurosci. Lett. 1995, 197, 149–153. [Google Scholar] [CrossRef]
- Kim, J.; Basak, J.M.; Holtzman, D.M. The role of apolipoprotein E in Alzheimer’s disease. Neuron 2009, 63, 287–303. [Google Scholar] [CrossRef]
- Xu, P.T.; Gilbert, J.R.; Qiu, H.L.; Ervin, J.; Rothrock-Christian, T.R.; Hulette, C.; Schmechel, D.E. Specific regional transcription of apolipoprotein E in human brain neurons. Am. J. Pathol. 1999, 154, 601–611. [Google Scholar] [CrossRef]
- Mahley, R.W.; Huang, Y. Apolipoprotein e sets the stage: Response to injury triggers neuropathology. Neuron 2012, 76, 871–885. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Ebbert, M.T.; Baker, K.E.; Cook, C.; Wang, X.; Sens, J.P.; Kocher, J.P.; Petrucelli, L.; Fryer, J.D. Microglial translational profiling reveals a convergent APOE pathway from aging, amyloid, and tau. J. Exp. Med. 2018, 215, 2235–2245. [Google Scholar] [CrossRef]
- Lanfranco, M.F.; Sepulveda, J.; Kopetsky, G.; Rebeck, G.W. Expression and secretion of apoE isoforms in astrocytes and microglia during inflammation. Glia 2021, 69, 1478–1493. [Google Scholar] [CrossRef]
- van der Lee, S.J.; Wolters, F.J.; Ikram, M.K.; Hofman, A.; Ikram, M.A.; Amin, N.; van Duijn, C.M. The effect of APOE and other common genetic variants on the onset of Alzheimer’s disease and dementia: A community-based cohort study. Lancet Neurol. 2018, 17, 434–444. [Google Scholar] [CrossRef]
- Bales, K.R.; Dodart, J.C.; DeMattos, R.B.; Holtzman, D.M.; Paul, S.M. Apolipoprotein E, amyloid, and Alzheimer disease. Mol. Interv. 2002, 2, 363. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Fagan, A.M.; Mackey, B.; Tenkova, T.; Sartorius, L.; Paul, S.M.; Bales, K.; Hsiao Ashe, K.; Irizarry, M.C.; Hyman, B.T. Apolipoprotein E facilitates neuritic and cerebrovascular plaque formation in an Alzheimer’s disease model. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2000, 47, 739–747. [Google Scholar] [CrossRef]
- Kok, E.; Haikonen, S.; Luoto, T.; Huhtala, H.; Goebeler, S.; Haapasalo, H.; Karhunen, P.J. Apolipoprotein E–dependent accumulation of Alzheimer disease–related lesions begins in middle age. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2009, 65, 650–657. [Google Scholar] [CrossRef]
- Garai, K.; Verghese, P.B.; Baban, B.; Holtzman, D.M.; Frieden, C. The binding of apolipoprotein E to oligomers and fibrils of amyloid-β alters the kinetics of amyloid aggregation. Biochemistry 2014, 53, 6323–6331. [Google Scholar] [CrossRef] [PubMed]
- Tai, L.M.; Bilousova, T.; Jungbauer, L.; Roeske, S.K.; Youmans, K.L.; Yu, C.; Poon, W.W.; Cornwell, L.B.; Miller, C.A.; Vinters, H.V.; et al. Levels of soluble apolipoprotein E/amyloid-β (Aβ) complex are reduced and oligomeric Aβ increased with APOE4 and Alzheimer disease in a transgenic mouse model and human samples. J. Biol. Chem. 2013, 288, 5914–5926. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain—Implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Wadhwani, A.R.; Affaneh, A.; Van Gulden, S.; Kessler, J.A. Neuronal apolipoprotein E4 increases cell death and phosphorylated tau release in alzheimer disease. Ann. Neurol. 2019, 85, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Harris, F.M.; Brecht, W.J.; Xu, Q.; Mahley, R.W.; Huang, Y. Increased tau phosphorylation in apolipoprotein E4 transgenic mice is associated with activation of extracellular signal-regulated kinase: Modulation by zinc. J. Biol. Chem. 2004, 279, 44795–44801. [Google Scholar] [CrossRef] [PubMed]
- Filippini, N.; Ebmeier, K.P.; MacIntosh, B.J.; Trachtenberg, A.J.; Frisoni, G.B.; Wilcock, G.K.; Beckmann, C.F.; Smith, S.M.; Matthews, P.M.; Mackay, C.E. Differential effects of the APOE genotype on brain function across the lifespan. Neuroimage 2011, 54, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Thambisetty, M.; Beason-Held, L.; An, Y.; Kraut, M.A.; Resnick, S.M. APOE ε4 genotype and longitudinal changes in cerebral blood flow in normal aging. Arch. Neurol. 2010, 67, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.A.; Torres, E.R.; Weber Boutros, S.; Patel, E.; Akinyeke, T.; Alkayed, N.J.; Raber, J. Apolipoprotein E4 mediates insulin resistance-associated cerebrovascular dysfunction and the post-prandial response. J. Cereb. Blood Flow Metab. 2019, 39, 770–781. [Google Scholar] [CrossRef]
- Zhao, N.; Liu, C.C.; Van Ingelgom, A.J.; Martens, Y.A.; Linares, C.; Knight, J.A.; Painter, M.M.; Sullivan, P.M.; Bu, G. Apolipoprotein E4 impairs neuronal insulin signaling by trapping insulin receptor in the endosomes. Neuron 2017, 96, 115–129. [Google Scholar] [CrossRef]
- Simonovitch, S.; Schmukler, E.; Masliah, E.; Pinkas-Kramarski, R.; Michaelson, D.M. The effects of APOE4 on mitochondrial dynamics and proteins in vivo. J. Alzheimer’s Dis. 2019, 70, 861–875. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.J.; Stein, J.L.; Hua, X.; Lee, S.; Hibar, D.P.; Leow, A.D.; Dinov, I.D.; Toga, A.W.; Saykin, A.J.; Shen, L.; et al. Alzheimer’s Disease Neuroimaging Initiative. A commonly carried allele of the obesity-related FTO gene is associated with reduced brain volume in the healthy elderly. Proc. Natl. Acad. Sci. USA 2010, 107, 8404–8409. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Du, X.; Jiang, H.; Xie, J. Ghrelin and Neurodegenerative Disorders-a Review. Mol. Neurobiol. 2017, 54, 1144–1155. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.M.; Mostafa, H.; Khonah, H.M.J. Insulin resistance in Alzheimer’s disease. The genetica and metabolomics links. Clin. Chim. Acta 2023, 539, 215–236. [Google Scholar] [CrossRef] [PubMed]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Dubinin, M.V. Diabetes mellitus, mitochondrial dysfunction and Ca2+-dependent permeability transition pore. Int. J. Mol. Sci. 2020, 21, 6559. [Google Scholar] [CrossRef] [PubMed]
- Skelly, D.T.; Hennessy, E.; Dansereau, M.A.; Cunningham, C. A systematic analysis of the peripheral and CNS effects of systemic LPS, IL-1β, TNF-α and IL-6 challenges in C57BL/6 mice. PLoS ONE 2013, 8, e69123. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Patti, M.E.; Butte, A.J.; Crunkhorn, S.; Cusi, K.; Berria, R.; Kashyap, S.; Miyazaki, Y.; Kohane, I.; Costello, M.; Saccone, R.; et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA 2003, 100, 8466–8471. [Google Scholar] [CrossRef]
- Qin, W.; Haroutunian, V.; Katsel, P.; Cardozo, C.P.; Ho, L.; Buxbaum, J.D.; Pasinetti, G.M. PGC-1α expression decreases in the Alzheimer disease brain as a function of dementia. Arch. Neurol. 2009, 66, 352–361. [Google Scholar] [CrossRef]
- Srivastava, S.; Diaz, F.; Iommarini, L.; Aure, K.; Lombes, A.; Moraes, C.T. PGC-1alpha/beta induced expression partially compesates for respiratory chain defects in cells from patients with mitochondrial disorders. Hum. Mol. Genet. 2009, 18, 180512. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, J.; Markesbery, W.R.; Lovell, M.A. Increased oxidative damage in nuclear and mitochondrial DNA in mild cognitive impairment. J. Neurochem. 2006, 96, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Fournet, M.; Bonté, F.; Desmoulière, A. Glycation damage: A possible hub for major pathophysiological disorders and aging. Aging Dis. 2018, 9, 880. [Google Scholar] [CrossRef] [PubMed]
- Sirangelo, I.; Iannuzzi, C. Understanding the role of protein glycation in the amyloid aggregation process. Int. J. Mol. Sci. 2021, 22, 6609. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Rudnicka-Nawrot, M.; Richey, P.L.; Praprotnik, D.; Mulvihill, P.; Miller, C.A.; Sayre, L.M.; Perry, G. Carbonyl-related posttranslational modification of neurofilament protein in the neurofibrillary pathology of Alzheimer’s disease. J. Neurochem. 1995, 64, 2660–2666. [Google Scholar] [CrossRef]
- Rolland, S.G.; Motori, E.; Memar, N.; Hench, J.; Frank, S.; Winklhofer, K.F.; Conradt, B. Impaired complex IV activity in response to loss of LRPPRC function can be compensated by mitochondrial hyperfusion. Proc. Natl. Acad. Sci. USA 2013, 110, E2967–E2976. [Google Scholar] [CrossRef]
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Et Biophys. Acta (BBA) Bioenerg. 2012, 1817, 1833–1838. [Google Scholar] [CrossRef]
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef]
- Maciejczyk, M.; Żebrowska, E.; Chabowski, A. Insulin resistance and oxidative stress in the brain: What’s new? Int. J. Mol. Sci. 2019, 20, 874. [Google Scholar] [CrossRef] [PubMed]
- Verdile, G.; Keane, K.N.; Cruzat, V.F.; Medic, S.; Sabale, M.; Rowles, J.; Wijesekara, N.; Martins, R.N.; Fraser, P.E.; Newsholme, P. Inflammation and oxidative stress: The molecular connectivity between insulin resistance, obesity, and Alzheimer’s disease. Mediat. Inflamm. 2015, 2015, 105828. [Google Scholar] [CrossRef]
- Finkel, T. Oxygen radicals and signaling. Curr. Opin. Cell Biol. 1998, 10, 248–253. [Google Scholar] [CrossRef]
- Nemoto, S.; Takeda, K.; Yu, Z.X.; Ferrans, V.J.; Finkel, T. Role for mitochondrial oxidants as regulators of cellular metabolism. Mol. Cell. Biol. 2000, 20, 7311–7318. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Trzyna, W.C.; McClintock, D.S.; Schumacker, P.T. Role of oxidants in NF-κB activation and TNF-α gene transcription induced by hypoxia and endotoxin. J. Immunol. 2000, 165, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Collins, Y.; Chouchani, E.T.; James, A.M.; Menger, K.E.; Cochemé, H.M.; Murphy, M.P. Mitochondrial redox signalling at a glance. J. Cell Sci. 2012, 125, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Rehani, K.; Jope, R.S.; Michalek, S.M. Toll-like receptor–mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat. Immunol. 2005, 6, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Federico, A.; Cardaioli, E.; Da Pozzo, P.; Formichi, P.; Gallus, G.N.; Radi, E. Mitochondria, oxidative stress and neurodegeneration. J. Neurol. Sci. 2012, 322, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Ruegsegger, G.N.; Vanderboom, P.M.; Dasari, S.; Klaus, K.A.; Kabiraj, P.; McCarthy, C.B.; Lucchinetti, C.F.; Nair, K.S. Exercise and metformin counteract altered mitochondrial function in the insulin-resistant brain. JCI insight 2019, 4, e130681. [Google Scholar] [CrossRef]
- Maciejczyk, M.; Żebrowska, E.; Zalewska, A.; Chabowski, A. Redox balance, antioxidant defense, and oxidative damage in the hypothalamus and cerebral cortex of rats with high fat diet-induced insulin resistance. Oxidative Med. Cell. Longev. 2018, 2018, 6940515. [Google Scholar] [CrossRef]
- Di Filippo, M.; Chiasserini, D.; Tozzi, A.; Picconi, B.; Calabresi, P. Mitochondria and the link between neuroinflammation and neurodegeneration. J. Alzheimer’s Dis. 2010, 20, S369–S379. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, H.M.; Carl, S.M.; Greenlief, A.C.; Festoff, B.W.; Swerdlow, R.H. Bioenergetic dysfunction and inflammation in Alzheimer’s disease: A possible connection. Front. Aging Neurosci. 2014, 6, 311. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Sprenger, H.G.; MacVicar, T.; Bahat, A.; Fiedler, K.U.; Hermans, S.; Ehrentraut, D.; Ried, K.; Milenkovic, D.; Bonekamp, N.; Larsson, N.G.; et al. Cellular pyrimidine imbalance triggers mitochondrial DNA–dependent innate immunity. Nat. Metab. 2021, 3, 636–650. [Google Scholar] [CrossRef]
- Zhao, W.; Beers, D.R.; Bell, S.; Wang, J.; Wen, S.; Baloh, R.H.; Appel, S.H. TDP-43 activates microglia through NF-κB and NLRP3 inflammasome. Exp. Neurol. 2015, 273, 24–35. [Google Scholar] [CrossRef]
- Hamelin, L.; Lagarde, J.; Dorothée, G.; Leroy, C.; Labit, M.; Comley, R.A.; Clinical IMABio3 Team. Early and protective microglial activation in Alzheimer’s disease: A prospective study using 18 F-DPA-714 PET imaging. Brain 2016, 139, 1252–1264. [Google Scholar] [CrossRef]
- Jiang, C.; Li, G.; Huang, P.; Liu, Z.; Zhao, B. The Gut Microbiota and Alzheimer’s Disease. J. Alzheimers. Dis. 2017, 58, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lazar, E.; Sherzai, A.; Adeghate, J.; Sherzai, D. Gut dysbiosis, insulin resistance and Alzheimer’s disease: Review of a novel approach to neurodegeneration. Front. Biosci. (Schol. Ed.) 2021, 13, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Iturria-Medina, Y.; Sotero, R.C.; Toussaint, P.J.; Mateos-Pérez, J.M.; Evans, A.C. Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat. Commun. 2016, 7, 11934. [Google Scholar] [CrossRef]
- Cuadrado-Godia, E.; Dwivedi, P.; Sharma, S.; Santiago, A.O.; Gonzalez, J.R.; Balcells, M.; Laird, J.; Turk, M.; Suri, H.S.; Nicolaides, A.; et al. Cerebral small vessel disease: A review focusing on pathophysiology, biomarkers, and machine learning strategies. J. Stroke 2018, 20, 302. [Google Scholar] [CrossRef]
- Cortes-Canteli, M.; Iadecola, C. Alzheimer’s disease and vascular aging: JACC focus seminar. J. Am. Coll. Cardiol. 2020, 75, 942–951. [Google Scholar] [CrossRef]
- Kawahito, S.; Kitahata, H.; Oshita, S. Problems associated with glucose toxicity: Role of hyperglycemia-induced oxidative stress. World J. Gastroenterol. 2009, 15, 4137. [Google Scholar] [CrossRef]
- Lau, L.H.; Lew, J.; Borschmann, K.; Thijs, V.; Ekinci, E.I. Prevalence of diabetes and its effects on stroke outcomes: A meta-analysis and literature review. J. Diabetes Investig. 2019, 10, 780–792. [Google Scholar] [CrossRef]
- Claassen, J.A.H.R.; Thiessens, D.H.J.; Panerai, R.B.; Faraci, F.M. Regulation of cerebral blood flow in humans: Physiology and clinical implications of autoregulation. Physiol. Rev. 2021, 101, 1487–1559. [Google Scholar] [CrossRef] [PubMed]
- Biffi, A.; Greenberg, S.M. Cerebral amyloid angiopathy: A systematic review. J. Clin. Neurol. 2011, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Abraham, H.M.; Wolfson, L.; Moscufo, N.; Guttmann, C.R.; Kaplan, R.F.; White, W.B. Cardiovascular risk factors and small vessel disease of the brain: Blood pressure, white matter lesions, and functional decline in older persons. J. Cereb. Blood Flow Metab. 2016, 36, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Herman, R.; Kravos, N.A.; Jensterle, M.; Janež, A.; Dolžan, V. Metformin and Insulin Resistance: A Review of the Underlying Mechanisms behind Changes in GLUT4-Mediated Glucose Transport. Int. J. Mol. Sci. 2022, 23, 1264. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hsu, C.C.; Wahlqvist, M.L.; Lee, M.S.; Tsai, H.N. Incidence of dementia is increased in type 2diabetes and reduced by the use of sulfonylureas and metformin. J Alzheimer’s Dis. 2011, 24, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, S.C.; Ferguson, E.L.; Choudhary, V.; Ranatunga, D.K.; Oni-Orisan, A.; Hayes-Larson, E.; Folle, A.D.; Mayeda, E.R.; Whitmer, R.A.; Gilsanz, P.; et al. Metformin Cessation and Dementia Incidence. JAMA Netw Open. 2023, 6, e2339723. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zheng, J.; Xu, M.; Walker, V.; Yuan, J.; Korologou-Linden, R.; Robinson, J.; Huang, P.; Burgess, S.; Au Yeung, S.L.; Luo, S.; et al. Evaluating the efficacy and mechanism of metformin targets on reducing Alzheimers disease risk in the general population: A Mendelian randomisation study. Diabetologia 2022, 65, 1664–1675. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Combs, C.K.; Johnson, D.E.; Karlo, J.C.; Cannady, S.B.; Landreth, G.E. Inflammatory mechanisms in Alzheimer’s disease: Inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists. J. Neurosci. 2000, 20, 558–567. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Delerive, P.; Fruchart, J.C.; Staels, B. Peroxisome proliferator-activated receptors ininflammation control. J. Endocrinol. 2001, 169, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Seferović, P.M.; Petrie, M.C.; Filippatos, G.S.; Anker, S.D.; Rosano, G.; Bauersachs, J.; Paulus, W.J.; Komajda, M.; Cosentino, F.; De Boer, R.A.; et al. Type 2 diabetes mellitus and heart failure: A position statement from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2018, 20, 853–872. [Google Scholar] [CrossRef] [PubMed]
- McClean, P.L.; Parthsarathy, V.; Faivre, E.; Hölscher, C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease. J Neurosci. 2011, 31, 6587–6594. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, Y.; Duffy, K.B.; Ottinger, M.A.; Ray, B.; Bailey, J.A.; Holloway, H.W.; Tweedie, D.; Perry, T.; Mattson, M.P.; Kapogiannis, D.; et al. GLP-1 receptor stimulation reduces amyloid-beta peptide accumulation and cytotoxicity in cellular and animal models of Alzheimer’s disease. J. Alzheimers. Dis. 2010, 19, 1205–1219. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nørgaard, C.H.; Friedrich, S.; Hansen, C.T.; Gerds, T.; Ballard, C.; Møller, D.V.; Knudsen, L.B.; Kvist, K.; Zinman, B.; Holm, E.; et al. Treatment with glucagon-like peptide-1 receptor agonists and incidence of dementia: Data from pooled double-blind randomized controlled trials and nationwide disease and prescription registers. Alzheimers Dement 2022, 8, e12268. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kelsey, M.D.; Nelson, A.J.; Green, J.B.; Granger, C.B.; Peterson, E.D.; McGuire, D.K.; Pagidipati, N.J. Guidelines for Cardiovascular Risk Reduction in Patients With Type 2 Diabetes: JACCGuideline Comparison. J. Am. Coll. Cardiol. 2022, 79, 1849–1857. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2022, 145, e895–e1032, Erratum in Circulation 2022, 145, e1033; Erratum in Circulation 2022, 146, e185; Erratum in Circulation 2023, 147, e674. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, Y.; Ogawa, W. SGLT2 inhibitors for genetic and acquired insulin resistance: Considerations for clinical use. J Diabetes Investig. 2020, 11, 1431–1433. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sim, A.Y.; Choi, D.H.; Kim, J.Y.; Kim, E.R.; Goh, A.R.; Lee, Y.H.; Lee, J.E. SGLT2 and DPP4inhibitors improve Alzheimer’s disease-like pathology and cognitive function through distinct mechanisms in a T2D-AD mouse model. Biomed. Pharmacother. 2023, 168, 115755. [Google Scholar] [CrossRef] [PubMed]
- Siao, W.Z.; Lin, T.K.; Huang, J.Y.; Tsai, C.F.; Jong, G.P. The association between sodium-glucosecotransporter 2 inhibitors and incident dementia: A nationwide population-based longitudinal cohort study. Diab. Vasc. Dis. Res. 2022, 19, 14791641221098168. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kuate Defo, A.; Bakula, V.; Pisaturo, A.; Labos, C.; Wing, S.S.; Daskalopoulou, S.S. Diabetes, antidiabetic medications and risk of dementia: A systematic umbrella review and meta-analysis. Diabetes Obes Metab. 2024, 26, 441–462. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Y.; Iskander, C.; Wang, C.; Xiong, L.Y.; Shah, B.R.; Edwards, J.D.; Kapral, M.K.; Herrmann, N.; Lanctôt, K.L.; Masellis, M.; et al. Association of Sodium-Glucose Cotransporter 2 Inhibitors with Time to Dementia: A Population-Based Cohort Study. Diabetes Care 2023, 46, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Shao, H.; Shaaban, C.E.; Yang, K.; Brown, J.; Anton, S.; Wu, Y.; Bress, A.; Donahoo, W.T.; DeKosky, S.T.; et al. Newer glucose-lowering drugs and risk of dementia: A systematic review and meta-analysis of observational studies. J. Am. Geriatr. Soc. 2023, 71, 2096–2106. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- O’Keefe, J.H.R.; O’Keefe, E.L.; Franco, W.G. SGLT inhibitors for improving Healthspan and lifespan. Prog Cardiovasc Dis. 2023, 81, 2–9. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Antinozzi, C.; Sgrò, P.; Di Luigi, L. Advantages of Phosphodiesterase Type 5 Inhibitors in the Management of Glucose Metabolism Disorders: A Clinical and Translational Issue. Int. J. Endocrinol. 2020, 2020, 7078108. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ayala, J.E.; Bracy, D.P.; Julien, B.M.; Rottman, J.N.; Fueger, P.T.; Wasserman, D.H. Chronic treatment with sildenafil improves energy balance and insulin action in high fat-fed conscious mice. Diabetes 2007, 56, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Puzzo, D.; Staniszewski, A.; Deng, S.X.; Privitera, L.; Leznik, E.; Liu, S.; Zhang, H.; Feng, Y.; Palmeri, A.; Landry, D.W.; et al. Phosphodiesterase 5 inhibition improves synaptic function, memory, and amyloid-beta load in an Alzheimer’s disease mouse model. J. Neurosci. 2009, 29, 8075–8086. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- García-Barroso, C.; Ricobaraza, A.; Pascual-Lucas, M.; Unceta, N.; Rico, A.J.; Goicolea, M.A.; Sallés, J.; Lanciego, J.L.; Oyarzabal, J.; Franco, R.; et al. Tadalafil crosses the blood-brain barrier and reverses cognitive dysfunction in a mouse model of AD. Neuropharmacology 2013, 64, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Adesuyan, M.; Jani, Y.H.; Alsugeir, D.; Howard, R.; Ju, C.; Wei, L.; Brauer, R. Phosphodiesterase Type 5 Inhibitors in Men with Erectile Dysfunction and the Risk of Alzheimer Disease: A Cohort Study. Neurology 2024, 102, e209131. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Singh, A.; Dhaneshwar, S.; Mazumder, A. Investigating Neuroprotective Potential of Berberine, Levetiracetam and their Combination in the Management of Alzheimer’s Disease Utilizing Drug Repurposing Strategy. Curr. Rev. Clin. Exp. Pharmacol. 2023, 18, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Zhang, N. Berberine: Pathways to protect neurons. Phytother Res. 2018, 32, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Meng, X.J.; Xu, T.B.; Zhang, X.C.; Zhou, Y.; Tong, Z.F.; Jiang, J.H. Berberine attenuates cognitive dysfunction and hippocampal apoptosis in rats with prediabetes. Chem. Biol. Drug Des. 2024, 103, e14420. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Zhang, J.F.; Song, J.; Bai, Y.; Deng, L.; Feng, C.P.; Xu, X.Y.; Guo, H.X.; Wang, Y.; Gao, X.; et al. Effects of Berberine on Diabetes and Cognitive Impairment in an Animal Model: The Mechanisms of Action. Am. J. Chin. Med. 2021, 49, 1399–1415. [Google Scholar] [CrossRef] [PubMed]
- Baska, A.; Leis, K.; Gałązka, P. Berberine in the Treatment of Diabetes Mellitus: A Review. Endocr. Metab. Immune Disord. Drug Targets 2021, 21, 1379–1386. [Google Scholar] [CrossRef] [PubMed]
- Bellavite, P.; Fazio, S.; Affuso, F. A Descriptive Review of the Action Mechanisms of Berberine, Quercetin and Silymarin on Insulin Resistance/Hyperinsulinemia and Cardiovascular Prevention. Molecules 2023, 28, 4491. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Huang, M.; Chen, S.; Liang, Y.; Guo, Y. The Role of Berberine in the Multi-Target Treatment of Senile Dementia. Curr. Top Med. Chem. 2016, 16, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Gao, Y.; Yu, S.; Sun, X.; Shen, K. Berberine attenuates Aβ42-induced neuronal damage through regulating circHDAC9/miR-142-5p axis in human neuronal cells. Life Sci. 2020, 252, 117637. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, H.K.; Baluchnejadmojarad, T.; Roghani, M.; Khaksari, M.; Norouzi, P.; Ahooie, M.; Mahboobi, F. Berberine ameliorate oxidative stress and astrogliosis in the hippocampus of STZ-induced diabetic rats. Mol. Neurobiol. 2014, 49, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Vermunt, L.; Gordon, B.; Pedrini, S.; Boonkamp, L.; Armstrong, N.; Teunissen, C. Plasma glial fibrillary acidic protein in autosomal dominant Alzheimer’s disease: Associations with β-amyloid-PET, neurodegeneration and cognition. Alzheimer’s Dement. 2022, 19, 2790–2804. [Google Scholar] [CrossRef]
- Durairajan, S.S.K.; Liu, L.F.; Lu, J.H.; Chen, L.L.; Yuan, Q.; Chung, S.K.; Huang, L.; Li, X.S.; Huang, J.D.; Li, M. Berberine ameliorates β-amyloid pathology, gliosis, and cognitive impairment in an Alzheimer’s disease transgenic mouse model. Neurobiol. Aging 2012, 33, 2903–2919. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Dong, S.; Chen, W.; Li, J.; Luo, E.; Ji, J. Berberine alleviates Alzheimer’s disease by regulating the gut microenvironment, restoring the gut barrier and brain-gut axis balance. Phytomedicine 2024, 129, 155624. [Google Scholar] [CrossRef] [PubMed]
- Duc Nguyen, H. Neurotherapeutic Effects of Quercetin and Its Metabolite Compounds on Cognitive Impairment and Parkinson’s Disease: An In Silico Study. Eur. J. Drug Metabm. Pharmacokinet. 2023, 48, 151–169. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.; Ullah, H.; Aschner, M.; Cheang, W.S.; Akkol, E.K. Neuroprotective Effects of Quercetin in Alzheimer’s Disease. Biomolecules 2019, 30, 59. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, D.; Zhao, J.; Li, S.; Shen, G.; Hu, S. Quercetin attenuates domoic acid-induced cognitive deficits in mice. Nutr. Neurosci. 2018, 21, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Watanabe, H.; Tanaka, A.; Nishihira, J.; Murayama, N. Effect of quercetin glycosides on cognitive functions and cerebral blood flow: A randomized, double-blind, and placebo-controlled study. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 8700–8712. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Kishimoto, Y.; Grammatikakis, I.; Gottimukkala, K.; Cutler, R.G.; Zhang, S.; Abdelmohsen, K.; Bohr, V.A.; Misra Sen, J.; Gorospe, M.; et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 2019, 22, 719–728. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bellavite, P. Neuroprotective Potentials of Flavonoids: Experimental Studies and Mechanisms of Action. Antioxidants 2023, 12, 280. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- de Luis, D.; Izaola, O.; de la Fuente, B.; Aller, R. Effect of L-arginine supplementation on insulin resistance and adipocitokines levels in head and neck cancer non diabetic patients after surgery. Nutr Hosp. 2014, 30, 870–875. [Google Scholar] [CrossRef] [PubMed]
- Piatti, P.; Monti, L.D.; Valsecchi, G.; Magni, F.; Setola, E.; Marchesi, F.; Galli-Kienle, M.; Pozza, G.; Alberti, K.G.M. Long-term oral L-arginine administration improves peripheral and hepatic insulin sensitivity in type 2 diabetic patients. Diabetes Care 2001, 24, 875–880. [Google Scholar] [CrossRef]
- Fleszar, M.G.; Wiśniewski, J.; Zboch, M.; Diakowska, D.; Gamian, A.; Krzystek-Korpacka, M. Targeted metabolomic analysis of nitric oxide/L-arginine pathway metabolites in dementia: Association with pathology, severity, and structural brain changes. Sci. Rep. 2019, 9, 13764. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Virarkar, M.; Alappat, L.; Bradford, P.G.; Awad, A.B. L-arginine and nitric oxide in CNS function and neurodegenerative diseases. Crit. Rev. Food Sci. Nutr. 2013, 53, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Geravand, S.; Karami, M.; Sahraei, H.; Rahimi, F. Protective effects of L-arginine on Alzheimer’s disease: Modulating hippocampal nitric oxide levels and memory deficits in aluminum chloride-induced rat model. Eur. J. Pharmacol. 2023, 958, 176030. [Google Scholar] [CrossRef] [PubMed]
- İsmail, S.; Serpil, E.; Esma, Ö.; Durmuş, A.; Erdal, E.; Avni, B.; Savaş, K. Changes in arginine metabolism in advanced Alzheimer’s patients:mExperimental and theoretical analyses. J. Mol. Struct. 2023, 1282, 135254. [Google Scholar]
- Mone, P.; Pansini, A.; Jankauskas, S.S.; Varzideh, F.; Kansakar, U.; Lombardi, A.; Trimarco, V.; Frullone, S.; Santulli, G. L-Arginine Improves Cognitive Impairment in Hypertensive Frail Older Adults. Front. Cardiovasc. Med. 2022, 9, 868521. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kuro-O, M. The Klotho proteins in health and disease. Nat. Rev. Nephrol. 2019, 15, 27–44. [Google Scholar] [CrossRef] [PubMed]
- Prud’homme, G.J.; Kurt, M.; Wang, Q. Pathobiology of the Klotho Antiaging Protein and Therapeutic Considerations. Front. Aging 2022, 3, 931331. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hwang, H.J.; Kim, N.; Herman, A.B.; Gorospe, M.; Lee, J.S. Factors and Pathways Modulating Endothelial Cell Senescence in Vascular Aging. Int. J. Mol. Sci. 2022, 23, 10135. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yan, L.; Hu, X.; Wu, S.; Zhao, S. Serum Klotho and insulin resistance: Insights from a cross-sectional analysis. Medicine 2024, 103, e37971. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Silva, A.P.; Mendes, F.; Pereira, L.; Fragoso, A.; Gonçalves, R.B.; Santos, N.; Rato, F.; Neves, P.L. Klotho levels: Association with insulin resistance and albumin-to-creatinine ratio in type 2 diabetic patients. Int. Urol. Nephrol. 2017, 49, 1809–1814. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.W.; Hung, C.C.; Fang, W.H.; Chen, W.L. Association between Soluble α-Klotho Protein and Metabolic Syndrome in the Adult Population. Biomolecules 2022, 12, 70. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dërmaku-Sopjani, M.; Kolgec, S.; Abazi, S.; Sopjani, M. Significance of the anti-aging protein Klotho. Mol. Membr. Biol. 2013, 30, 369–385. [Google Scholar] [CrossRef] [PubMed]
- Semba, R.D.; Moghekar, A.R.; Hu, J.; Sun, K.; Turner, R.; Ferrucci, L.; O’Brien, R. Klotho in the cerebrospinal fluid of adults with and without Alzheimer’s disease. Neurosci. Lett. 2014, 558, 37–40. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhao, Y.; Zeng, C.Y.; Li, X.H.; Yang, T.T.; Kuang, X.; Du, J.R. Klotho overexpression improves amyloid-β clearance and cognition in the APP/PS1 mouse model of Alzheimer’s disease. Aging Cell. 2020, 19, e13239. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pedersen, A.K.N.; Gormsen, L.C.; Nielsen, S.; Jessen, N.; Bjerre, M. Metformin Improves the Prerequisites for FGF21 Signaling in Patients with Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2024, 109, e552–e561. [Google Scholar] [CrossRef] [PubMed]
- Wolf, L.; Föller, M.; Feger, M. The impact of SGLT2 inhibitors on αKlotho in renal MDCK and HK-2 cells. Front. Endocrinol. 2023, 14, 1069715. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, C.; Jiang, S.; Wang, H.; Wang, Y.; Han, Y.; Jiang, J. Berberine exerts protective effects on cardiac senescence by regulating the Klotho/.SIRT1 signaling pathway. Biomed Pharmacother. 2022, 151, 113097. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Prata, L.G.L.; Gerdes, E.O.W.; Netto, J.M.E.; Pirtskhalava, T.; Giorgadze, N.; Tripathi, U.; Inman, C.L.; Johnson, K.O.; Xue, A.; et al. Orally-active, clinically-translatable senolytics restore α-Klotho in mice and humans. EBioMedicine 2022, 77, 103912. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Alharbi, K.S.; Afzal, O.; Altamimi, A.S.A.; Almalki, W.H.; Kazmi, I.; Al-Abbasi, F.A.; Alzarea, S.I.; Makeen, H.A.; Albratty, M. A study of the molecular mechanism of quercetin and dasatinib combination as senolytic in alleviating age-related and kidney diseases. J. Food Biochem. 2022, 46, e14471. [Google Scholar] [CrossRef] [PubMed]
- Schmid, V.; Kullmann, S.; Gfrörer, W.; Hund, V.; Hallschmid, M.; Lipp, H.P.; Häring, H.U.; Preissl, H.; Fritsche, A.; Heni, M. Safety of intranasal human insulin: A review. Diabetes Obes. Metab. 2018, 20, 1563–1577. [Google Scholar] [CrossRef]
- Salameh, T.S.; Bullock, K.M.; Hujoel, I.A.; Niehoff, M.L.; Wolden-Hanson, T.; Kim, J.; Morley, J.E.; Farr, S.A.; Banks, W.A. Central nervous system delivery of intranasal insulin: Mechanisms of uptake and effects on cognition. J. Alzheimer’s Dis. 2015, 47, 715–728. [Google Scholar] [CrossRef] [PubMed]
- McNay, E.C.; Ong, C.T.; McCrimmon, R.J.; Cresswell, J.; Bogan, J.S.; Sherwin, R.S. Hippocampal memory processes are modulated by insulin and high-fat-induced insulin resistance. Neurobiol Learn Mem. 2010, 93, 546–553. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R.; et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: A pilot clinical trial. Arch. Neurol. 2012, 69, 29–38. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Formiga, F.; Pérez-Maraver, M. Type 3 diabetes mellitus. The revival of inhaled insulin? Endocrinol Nutr. 2014, 61, 173–175. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M. Early intranasal insulin therapy halts progression of neurodegeneration: Progress in Alzheimer’s disease therapeutics. Aging Health 2012, 8, 61–64. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Reger, M.A.; Watson, G.; Green, P.S.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Cherrier, M.M.; Schellenberg, G.D.; Frey Ii, W.H.; et al. Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J. Alzheimer’s Dis. 2008, 13, 323–331. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Claxton, A.; Baker, L.D.; Hanson, A.; Trittschuh, E.H.; Cholerton, B.; Morgan, A.; Callaghan, M.; Arbuckle, M.; Behl, C.; Craft, S. Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s disease dementia. J. Alzheimers Dis. 2015, 44, 897–906, Erratum in J. Alzheimers Dis. 2015, 45, 1269–1270. [Google Scholar] [CrossRef] [PubMed]
- National Library of Medicine. ClinicalTrials.gov. SNIFF-Combo INI+EMPA Trial. ClinicalTrials.gov ID NCT05081219. Wake Forest University Healthy Sciences (Responsible Party). Available online: https://clinicaltrials.gov/search?lat=36.13700800000001&lng=-80.275307&locStr=Wake%20Forest%20University%20Health%20and%20Exercise%20Science%20Department,%20Winston-Salem,%20NC&distance=50&cond=Alzheimer%27s%20Disease&intr=SNIFF-Combo%20INI%2BEMPA%20 (accessed on 16 May 2024).

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Affuso, F.; Micillo, F.; Fazio, S. Insulin Resistance, a Risk Factor for Alzheimer’s Disease: Pathological Mechanisms and a New Proposal for a Preventive Therapeutic Approach. Biomedicines 2024, 12, 1888. https://doi.org/10.3390/biomedicines12081888
Affuso F, Micillo F, Fazio S. Insulin Resistance, a Risk Factor for Alzheimer’s Disease: Pathological Mechanisms and a New Proposal for a Preventive Therapeutic Approach. Biomedicines. 2024; 12(8):1888. https://doi.org/10.3390/biomedicines12081888
Chicago/Turabian StyleAffuso, Flora, Filomena Micillo, and Serafino Fazio. 2024. "Insulin Resistance, a Risk Factor for Alzheimer’s Disease: Pathological Mechanisms and a New Proposal for a Preventive Therapeutic Approach" Biomedicines 12, no. 8: 1888. https://doi.org/10.3390/biomedicines12081888