
The Role of Epithelial-to-Mesenchymal Transition Transcription Factors (EMT-TFs) in Acute Myeloid Leukemia Progression
 , , , ,
, , , ,
Abstract
:1. Definition of Acute Myeloid Leukemia (AML), Genetic Variability, and Classification
2. MLL-AF9 Fusion Protein Oncogenic Mechanisms and Incidence in AML
3. First-Line Treatments for AML May Cause t(9;11)—A Mechanistic Perspective

4. Emergence of Epithelial-to-Mesenchymal Transition (EMT) Factors in the Risk and Progression of AML: The Role of ZEB Transcription Factors
5. Role of ZEB Transcription Factors
6. Role of SNAI Transcription Factors
7. LSD1 and Other Potential Therapeutic Targets
8. Role of SNAI2 in AML
9. Role of TWIST1 in AML
10. Spread of AML Cells
11. Intravasation and Extravasation Mechanisms of AML
12. Overall Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AML | acute myeloid leukemia |
| WHO | World Health Organization |
| MLL-AF9 | Mixed-Lineage Leukemia-AF9 fusion protein |
| EMT | epithelial-to-mesenchymal transition |
| EMT-TFs | epithelial-to-mesenchymal transition transcription factors |
| FAB | French–American–British |
| APL | Acute Promyelocytic Leukemia |
| PML-RARα | Promyelocytic Leukemia-Retinoic Acid Receptor Alpha |
| ATRA | all-trans retinoic acid |
| ATO | arsenic trioxide |
| MDS | myelodysplastic syndrome |
| TP53 | Tumor Protein 53 |
| ELN | Elastin |
| HSCs | hematopoietic stem cells |
| BM | bone marrow |
| PB | peripheral blood |
| RUNX1::RUNX1T1 | Runt-Related Transcription Factor 1–RUNX1 Translocation Partner 1 |
| CBFB::MYH11 | Core-Binding Factor Beta Subunit–Myosin Heavy Chain 11 |
| BCR::ABL1 | Breakpoint Cluster Region–Abelson Tyrosine Kinase 1 |
| CEBPA | CCAAT/Enhancer Binding Protein Alpha |
| VAF | variant allele fraction |
| MLLT3 | MLLT3 super elongation complex subunit |
| NMR | nuclear magnetic resonance |
| H3K4 | histone H3 lysine 4 |
| HSPCs | hematopoietic stem and progenitor cells |
| DOT1L | Disruptor of Telomeric Silencing 1-Like Protein |
| ETO | RUNX1 partner transcriptional co-repressor 1 |
| TOP2 | Topoisomerase II |
| DSBs | double-strand breaks |
| NSCLC | Non-Small Cell Lung Cancer |
| WBCs | White Blood Cells |
| AKT/mTOR | AKT Serine/Threonine Kinase/Mechanistic Target of Rapamycin |
| ZEB | Zinc Finger E-Box Binding Homeobox |
| CTBP | C-Terminal Binding Protein |
| SLc13A3 | Solute Carrier Family 13 Member 3 |
| CD36 | Cluster of Differentiation 36 |
| THBS1 | thrombospondin 1 |
| IL-17 | Interleukin 17 |
| SOCS2 | Suppressor of Cytokine Signaling 2 |
| TGF-β | Transforming Growth Factor Beta |
| CXCR4 | C-X-C Motif Chemokine Receptor 4 |
| CDH2 | cadherin 2 |
| LOX | Lysyl Oxidase |
| COL3A1 | Collagen Type III Alpha 1 Chain |
| MRTF-SRF | Myocardin-Related Transcription Factors and Serum-Response Factor |
| SDF-1 | Stromal Cell-Derived Factor 1 |
| CXCL12 | C-X-C Motif Chemokine Ligand 12 |
| NPM1 | Nucleophosmin 1 |
| FLT3-ITD | Fms-Like Tyrosine Kinase 3-Internal Tandem Duplication |
| LDH | Lactate Dehydrogenase |
| LDHA | Lactate Dehydrogenase A |
| SF3B1 | splicing factor 3b subunit 1 |
| circRNAs | Circular RNAs |
| MALAT1 | Metastasis-Associated Lung Adenocarcinoma Transcript 1 |
| Hox | Homeobox |
| LSD1 | lysine-specific demethylase 1 |
| GFI1 | Growth Factor Independent 1 |
| miRNA | microRNA |
| CRISPR/Cas9 | Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR Associated Protein 9 |
| lncRNA | long non-coding RNA |
| TGF | Transforming Growth Factor |
| CD8 | Cluster of Differentiation 8 |
| TGF-β | Transforming Growth Factor Beta |
| VEGF | Vascular Endothelial Growth Factor |
| SETDB1 | SET Domain Bifurcated Histone Lysine Methyltransferase 1 |
| N-WASP | Neural Wiskott–Aldrich Syndrome Protein |
| Tks4 | Tyrosine Kinase Substrate With Four SH3 Domains |
| Tks5 | SH3 and PX Domains 2A Protein |
| G-CSF | Granulocyte Colony-Stimulating Factor |
| IL-6 | Interleukin 6 |
| LDS1 | lysine-specific histone demethylase 1A |
| GATA2 | GATA Binding Protein 2 |
| MECOM | MDS1 and EVI1 Complex Locus |
| DEK | DEK oncogene |
| NUP214 | Nucleoporin 214 |
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Chennamadhavuni, A.; Lyengar, V.; Mukkamalla, S.K.R.; Shimanovsky, A. Leukemia. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Clarkson, B.; Strife, A.; Wisniewski, D.; Lambek, C.L.; Liu, C. Chronic myelogenous leukemia as a paradigm of early cancer and possible curative strategies. Leukemia 2003, 17, 1211–1262. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, G.; Hochhaus, A.; Schultheis, B.; Hehlmann, R. Chronic myelogenous leukemia: Molecular and cellular aspects. J. Cancer Res. Clin. Oncol. 1998, 124, 643–660. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.S.; Viera, A.J.; Mead, M.D. Leukemia: An overview for primary care. Am. Fam. Physician 2014, 89, 731–738. [Google Scholar] [PubMed]
- Vakiti, A.; Reynolds, S.B.; Mewawalla, P. Acute Myeloid Leukemia. In StatPearls [Internet]; StatPearls: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK507875/ (accessed on 6 August 2024).
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates ofincidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed]
- Reikvam, H.; Hatfield, K.J.; Kittang, A.O.; Hovland, R.; Bruserud, Ø. Acute Myeloid Leukemia with the t(8;21) Translocation: Clinical Consequences and Biological Implications. BioMed Res. Int. 2011, 2011, 104631. [Google Scholar] [CrossRef] [PubMed]
- Tallman, M.S.; Kim, H.T.; Paietta, E.; Bennett, J.M.; Dewald, G.; Cassileth, P.A.; Wiernik, P.H.; Rowe, J.M. Acute Monocytic Leukemia (French-American-British classification M5) Does Not Have a Worse Prognosis Than Other Subtypes of Acute Myeloid Leukemia: A Report From the Eastern Cooperative Oncology Group. J. Clin. Oncol. 2004, 22, 1276–1286. [Google Scholar] [CrossRef]
- De Rossi, G.; Avvisati, G.; Coluzzi, S.; Fenu, S.; LoCoco, F.; Lopez, M.; Nanni, M.; Pasqualetti, D.; Mandelli, F. Immunological definition of acute promyelocyte leukemia (FAB M3): A study of 39 cases. Eur. J. Haematol. 1990, 45, 168–171. [Google Scholar] [CrossRef]
- Randolph, T.R. Acute promyelocytic leukemia (AML-M3)—Part 1: Pathophysiology, clinical diagnosis, and differentiation therapy. Clin. Lab. Sci. 2000, 13, 98–105. [Google Scholar] [PubMed]
- Randolph, T.R. Acute promyelocytic leukemia (AML-M3)—Part 2: Molecular defect, DNA diagnosis, and proposed models of leukemogenesis and differentiation therapy. Clin. Lab. Sci. 2000, 13, 106–116. [Google Scholar] [PubMed]
- Saeed, S.; Logie, C.; Stunnenberg, H.G.; Martens, J.H. Genome-wide functions of PML-RARalpha in acute promyelocytic leukaemia. Br. J. Cancer 2011, 104, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Delaunay, J.; Vey, N.; Leblanc, T.; Fenaux, P.; Rigal-Huguet, F.; Witz, F.; Lamy, T.; Auvrignon, A.; Blaise, D.; Pigneux, A.; et al. Prognosis of inv(16)/t(16;16) acute myeloid leukemia (AML): A survey of 110 cases from the French AML Intergroup. Blood 2003, 102, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Plantier, I.; Lai, J.L.; Wattel, E.; Bauters, F.; Fenaux, P. Inv(16) may be one of the only ‘favorable’ factors in acute myeloid leukemia: A report on 19 cases with prolonged follow-up. Leuk. Res. 1994, 18, 885–888. [Google Scholar] [CrossRef] [PubMed]
- Varotto, E.; Munaretto, E.; Stefanachi, F.; Della Torre, F.; Buldini, B. Acute myeloid leukemia with the t(8;21) translocation: Clinical consequences and biological implications. J. Biomed. Biotechnol. Front. Pediatr. 2022, 10, 911093. [Google Scholar]
- De Boer, J.; Walf-Vorderwülbecke, V.; Williams, O. In focus: MLL-rearranged leukemia. Leukemia 2013, 27, 1224–1228. [Google Scholar] [CrossRef] [PubMed]
- Cowell, I.G.; Austin, C.A. DNA fragility at the KMT2A/MLL locus: Insights from old and new technologies. Open Biol. 2023, 13, 220232. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Hofmann, J.; Burmeister, T.; Gröger, D.; Park, T.S.; Emerenciano, M.P.; De Oliveira, M.P.; Renneville, A.; Villarese, P.; Macintyre, E.; et al. The MLL recombinome of acute leukemias in 2013. Leukemia 2013, 27, 2165–2176. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Z.; Polski, J.M.; Kasyan, A.; Medeiros, L.J. Acute erythroid leukemia. Arch. Pathol. Lab. Med. 2010, 134, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Cervera, N.; Lhoumeau, A.-C.; Adélaïde, J.; Guille, A.; Murati, A.; Mozziconacci, M.-J.; Vey, N.; Birnbaum, D.; Gelsi-Boyer, V. Acute erythroid leukemias have a distinct molecular hierarchy from non-erythroid acute myeloid leukemias. Haematologica 2020, 105, e340–e342. [Google Scholar] [CrossRef] [PubMed]
- Santos, F.P.; Faderl, S.; Garcia-Manero, G.; Koller, C.; Beran, M.; O’Brien, S.; Pierce, S.; Freireich, E.J.; Huang, X.; Borthakur, G.; et al. Adult acute erythroleukemia: An analysis of 91 patients treated at a single institution. Leukemia 2009, 23, 2275–2280. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gassmann, W.; Löffler, H. Acute megakaryoblastic leukemia. Leuk. Lymphoma 1995, 18, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Dima, D.; Oprita, L.; Rosu, A.-M.; Trifa, A.; Selicean, C.; Moisoiu, V.; Frinc, I.; Zdrenghea, M.; Tomuleasa, C. Adult acute megakaryoblastic leukemia: Rare association with cytopenias of undetermined significance and p210 and p190 BCR–ABL transcripts. OncoTargets Ther. 2017, 10, 5047–5051. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.M. Classification of acute myeloid leukemia. Blood Res. 2020, 55, S1–S4. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A. The 2016 WHO classification of acute myeloid leukemia: What the practicing clinician needs to know. Semin. Hematol. 2019, 56, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellström-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef] [PubMed]
- Marks, J.A.; Wang, X.; Fenu, E.M.; Bagg, A.; Lai, C. TP53 in AML and MDS: The new (old) kid on the block. Blood Rev. 2023, 60, 101055. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.G.; Iqbal, S.; Renard, C.; Chan, R.J.; Hasegawa, K.; Hu, H.; Tse, P.; Yan, J.; Zoratti, M.J.; Xie, F.; et al. Treatment outcomes for newly diagnosed, treatment-naive TP53-mutated acute myeloid leukemia: A systematic review and meta-analysis. J. Hematol. Oncol. 2023, 16, 19. [Google Scholar] [CrossRef] [PubMed]
- Shimony, S.; Stahl, M.; Stone, R.M. Acute myeloid leukemia: 2023 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2023, 98, 502–526. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.; Baer, C.; Hutter, S.; Dicker, F.; Meggendorfer, M.; Pohlkamp, C.; Kern, W.; Haferlach, T.; Haferlach, C.; Hoermann, G. AML classification in the year 2023: How to avoid a Babylonian confusion of languages. Leukemia 2023, 37, 1413–1420. [Google Scholar] [CrossRef]
- Cicconi, L.; Platzbecker, U.; Avvisati, G.; Paoloni, F.; Thiede, C.; Vignetti, M.; Fazi, P.; Ferrara, F.; Divona, M.; Albano, F.; et al. Long-term results of all-trans retinoic acid and arsenic trioxide in non-high-risk acute promyelocytic leukemia: Update of the APL0406 Italian-German randomized trial. Leukemia 2020, 34, 914–918. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.A.; Montesinos, P. Risk-Adapted Treatment for Low- and Intermediate-Risk Acute Promyelocytic Leukemia. Clin. Lymphoma Myeloma Leuk. 2010, 10 (Suppl. S3), S130–S134. [Google Scholar] [CrossRef] [PubMed]
- Cicconi, L.; Testi, A.M.; Montesinos, P.; Rego, E.; Zhu, H.H.; Takahashi, H.; Dworzak, M.; Estey, E.; Schwarer, A.; Esteve, J.; et al. Characteristics and outcome of acute myeloid leukemia with uncommon retinoic acid receptor-alpha (RARA) fusion variants. Blood Cancer J. 2021, 11, 167. [Google Scholar] [CrossRef]
- Fan, G.L.; Jiang, P.J.; Yuan, M. Clinical Prognostic Factors Analysis of Initially Treated AML Children with t (8;21)/RUNX1-RUNX1T1. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2020, 28, 1510–1515. [Google Scholar]
- Schnittger, S.; Bacher, U.; Haferlach, C.; Kern, W.; Haferlach, T. Rare CBFB-MYH11 fusion transcripts in AML with inv (16)/t (16;16) are associated with therapy-related AML M4eo, atypical cytomorphology, atypical immunophenotype, atypical additional chromosomal rearrangements and low white blood cell count: A study on 162 patients. Leukemia 2007, 21, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Labopin, M.; Gérard, S.; Yakoub-Agha, I.; Blau, I.W.; Aljurf, M.; Forcade, E.; Gedde-Dahl, T.; Burns, D.; Vydra, J.; et al. Lower relapse incidence with haploidentical versus matched sibling or unrelated donor hematopoietic cell transplantation for core-binding factor AML patients in CR2: A study from the Global Committee and the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation. Am. J. Hematol. 2024, 99, 1290–1299. [Google Scholar] [CrossRef] [PubMed]
- Van Weelderen, R.E.; Harrison, C.J.; Klein, K.; Jiang, Y.; Abrahamsson, J.; Alonzo, T.; Aplenc, R.; Arad-Cohen, N.; Bart-Delabesse, E.; Buldini, B.; et al. Optimized cytogenetic risk-group stratification of KMT2A-rearranged pediatric acute myeloid leukemia. Blood Adv. 2024, 8, 3200–3213. [Google Scholar] [CrossRef] [PubMed]
- Chiriches, C.; Nicolaisen, N.; Wieske, M.; Elhaddad, H.; Mehmetbeyoglu, E.; Alvares, C.; Becher, D.; Hole, P.; Ottmann, O.G.; Ruthardt, M. Understanding a high-risk acute myeloid leukemia by analyzing the interactome of its major driver mutation. PLOS Genet. 2022, 18, e1010463. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.; Tsai, X.C.; Lin, C.; Tien, F.; Kuo, Y.; Lee, W.; Peng, Y.; Liu, M.; Tseng, M.; Hsu, C.; et al. Validation of the prognostic significance of the 2022 European LeukemiaNet risk stratification system in intensive chemotherapy treated aged 18 to 65 years patients with de novo acute myeloid leukemia. Am. J. Hematol. 2023, 98, 760–769. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, D.; Chi, S.; Uchiyama, S.; Nakamura, H.; Guo, Y.-M.; Yamauchi, N.; Yuda, J.; Minami, Y. Molecular Classification and Overcoming Therapy Resistance for Acute Myeloid Leukemia with Adverse Genetic Factors. Int. J. Mol. Sci. 2022, 23, 5950. [Google Scholar] [CrossRef] [PubMed]
- Rørvik, S.D.; Torkildsen, S.; Bruserud, Ø.; Tvedt, T.H.A. Acute myeloid leukemia with rare recurring translocations—An overview of the entities included in the international consensus classification. Ann. Hematol. 2024, 103, 1103–1119. [Google Scholar] [CrossRef] [PubMed]
- Branford, S.; Yeung, D.T.; Parker, W.T.; Roberts, N.D.; Purins, L.; Braley, J.A.; Altamura, H.K.; Yeoman, A.L.; Georgievski, J.; Jamison, B.A.; et al. Prognosis for patients with CML and >10% BCR-ABL1 after 3 months of imatinib depends on the rate of BCR-ABL1 decline. Blood 2014, 124, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Othman, J.; Potter, N.; Ivey, A.; Tazi, Y.; Papaemmanuil, E.; Jovanovic, J.; Freeman, S.D.; Gilkes, A.F.; E Gale, R.; Rapoz-D’Silva, T.; et al. Molecular, clinical and therapeutic determinants of outcome in NPM1 mutated AML. Blood J. 2024. [Google Scholar] [CrossRef] [PubMed]
- Tien, F.-M.; Hou, H.-A. CEBPA mutations in acute myeloid leukemia: Implications in risk stratification and treatment. Int. J. Hematol. 2024, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.L.; Wu, Y.B.; Song, X.L.; Chen, Y.; Lu, Y.; Lai, X.Y.; Shi, J.M.; Liu, L.Z.; Zhao, Y.M.; Yu, J.; et al. Efficacy and prognostic factors of allogeneic hematopoietic stem cell transplantation in the treatment of secondary acute myeloid leukemia. Chin. J. Hematol. 2024, 45, 41–47. [Google Scholar] [CrossRef]
- Tsai, X.C.-H.; Sun, K.-J.; Lo, M.-Y.; Tien, F.-M.; Kuo, Y.-Y.; Tseng, M.-H.; Peng, Y.-L.; Chuang, Y.-K.; Ko, B.-S.; Tang, J.-L.; et al. Poor prognostic implications of myelodysplasia-related mutations in both older and younger patients with de novo AML. Blood Cancer J. 2023, 13, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kawankar, N.; Vundinti, B.R. Cytogenetic abnormalities in myelodysplastic syndrome: An overview. Hematology 2011, 16, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, I.; Lenk, M.; Haferlach, T.; Stengel, A.; Hutter, S.; Baer, C.; Meggendorfer, M.; Kern, W.; Haferlach, C. AML, NOS and AML-MRC as defined by multilineage dysplasia share a common mutation pattern which is distinct from AML-MRC as defined by MDS-related cytogenetics. Leukemia 2022, 36, 1939–1942. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Dong, Z.; Wan, D.; Cao, W.; Xing, H.; Liu, Z.; Fan, J.; Wang, H.; Lu, R.; Zhang, Y.; et al. Clinical characteristics, treatment, and prognosis of 118 cases of myeloid sarcoma. Sci. Rep. 2022, 12, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Yokoyama, A. The molecular functions of common and atypical MLL fusion protein complexes. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194548. [Google Scholar] [CrossRef] [PubMed]
- Iacobucci, I.; Mullighan, C.G. KMT2A-rearranged leukemia: The shapeshifter. Blood 2022, 140, 1833–1835. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H. The complex activities of the SET1/MLL complex core subunits in development and disease. Biochim Biophys Acta Gene Regul Mech. 2020, 1863, 194560. [Google Scholar] [CrossRef] [PubMed]
- Thiel, A.T.; Blessington, P.; Zou, T.; Feather, D.; Wu, X.; Yan, J.; Zhang, H.; Liu, Z.; Ernst, P.; Koretzky, G.A.; et al. MLL-AF9-induced leukemogenesis requires coexpression of the wild-type Mll allele. Cancer Cell 2010, 17, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Slany, R.K. The molecular biology of mixed lineage leukemia. Haematologica 2009, 94, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Vedadi, M.; Blazer, L.; Eram, M.S.; Barsyte-Lovejoy, D.; Arrowsmith, C.H.; Hajian, T. Targeting human SET1/MLL family of proteins. Protein Sci. 2017, 26, 662–676. [Google Scholar] [CrossRef]
- Yang, W.; Ernst, P. SET/MLL family proteins in hematopoiesis and leukemia. Int. J. Hematol. 2016, 105, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, M.S.; Patel, A. Mixed lineage leukemia: A structure–function perspective of the MLL1 protein. FEBS J. 2010, 277, 1832–1842. [Google Scholar] [CrossRef] [PubMed]
- Winters, A.C.; Bernt, K.M. MLL-Rearranged Leukemias—An Update on Science and Clinical Approaches. Front. Pediatr. 2017, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Song, Y. Structure, function and inhibition of critical protein–protein interactions involving mixed lineage leukemia 1 and its fusion oncoproteins. J. Hematol. Oncol. 2021, 14, 1–33. [Google Scholar] [CrossRef]
- Heuts, B.M.H.; Arza-Apalategi, S.; Alkema, S.G.; Tijchon, E.; Jussen, L.; Bergevoet, S.M.; van der Reijden, B.A.; Martens, J.H.A. Inducible MLL-AF9 Expression Drives an AML Program during Human Pluripotent Stem Cell-Derived Hematopoietic Differentiation. Cells 2023, 12, 1195. [Google Scholar] [CrossRef]
- Schoch, C.; Schnittger, S.; Klaus, M.; Kern, W.; Hiddemann, W.; Haferlach, T. AML with 11q23/MLL abnormalities as defined by the WHO classification: Incidence, partner chromosomes, FAB subtype, age distribution, and prognostic impact in an unselected series of 1897 cytogenetically analyzed AML cases. Blood 2003, 102, 2395–2402. [Google Scholar] [CrossRef]
- Hagag, A.A.; Shebl, S.S.; El-Fadaly, N.H. Frequency of 11q23/MLL gene rearrangement in Egyptian childhood acute myeloblastic leukemia: Biologic and clinical significance. South Asian J. Cancer 2014, 3, 206–208. [Google Scholar] [CrossRef]
- Marschalek, R. MLL, Life Sciences; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Kabra, A.; Bushweller, J. The Intrinsically Disordered Proteins MLLT3 (AF9) and MLLT1 (ENL)—Multimodal Transcriptional Switches With Roles in Normal Hematopoiesis, MLL Fusion Leukemia, and Kidney Cancer. J. Mol. Biol. 2022, 434, 167117. [Google Scholar] [CrossRef]
- GeneCards. MLLT3 Gene. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=MLLT3 (accessed on 6 August 2024).
- Yi, Y.; Ge, S. Targeting the histone H3 lysine 79 methyltransferase DOT1L in MLL-rearranged leukemias. J. Hematol. Oncol. 2022, 15, 1–21. [Google Scholar] [CrossRef]
- Olsen, S.N.; Godfrey, L.; Healy, J.P.; Choi, Y.A.; Kai, Y.; Hatton, C.; Perner, F.; Haarer, E.L.; Nabet, B.; Yuan, G.C.; et al. MLL::AF9 degradation induces rapid changes in transcriptional elongation and subsequent loss of an active chromatin landscape. Mol. Cell 2022, 82, 1140–1155. [Google Scholar] [CrossRef]
- Stavropoulou, V.; Peters, A.H.F.M.; Schwaller, J. Aggressive leukemia driven by MLL-AF9. Mol. Cell. Oncol. 2017, 5, e1241854. [Google Scholar] [CrossRef]
- Francis, J.C.; Gardiner, J.R.; Renaud, Y.; Chauhan, R.; Weinstein, Y.; Gomez-Sanchez, C.; Lefrançois-Martinez, A.-M.; Bertherat, J.; Val, P.; Swain, A. HOX genes promote cell proliferation and are potential therapeutic targets in adrenocortical tumours. Br. J. Cancer 2020, 124, 805–816. [Google Scholar] [CrossRef]
- Myers, P. Hox Genes in Development: The Hox Code. Nat. Educ. 2008, 1, 2. [Google Scholar]
- Hubert, K.A.; Wellik, D.M. Hox genes in development and beyond. Development 2023, 150, dev192476. [Google Scholar] [CrossRef]
- Lappin, T.R.; Grier, G.D.; Thompson, A.; Halliday, H.L. HOX genes: Seductive science, mysterious mechanisms., HOX genes: Seductive science, mysterious mechanisms. Ulster Med. J. 2006, 75, 23–31. [Google Scholar] [PubMed Central]
- Luo, Z.; Rhie, S.K.; Farnham, P.J. The Enigmatic HOX Genes: Can We Crack Their Code? Cancers 2019, 11, 323. [Google Scholar] [CrossRef] [PubMed]
- A Alharbi, R.; Pettengell, R.; Pandha, H.S.; Morgan, R. The role of HOX genes in normal hematopoiesis and acute leukemia. Leukemia 2012, 27, 1000–1008. [Google Scholar] [CrossRef] [PubMed]
- Nagy, Á.; Ősz, Á.; Budczies, J.; Krizsán, S.; Szombath, G.; Demeter, J.; Bödör, C.; Győrffy, B. Elevated HOX gene expression in acute myeloid leukemia is associated with NPM1 mutations and poor survival. J. Adv. Res. 2019, 20, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Aryal, S.; Zhang, Y.; Wren, S.; Li, C.; Lu, R. Molecular regulators of HOXA9 in acute myeloid leukemia. FEBS J. 2021, 290, 321–339. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-L.; Qin, Z.-Y.; Hu, F.; Wang, Y.; Dai, Y.-J.; Liang, Y. The Role of the HOXA Gene Family in Acute Myeloid Leukemia. Genes 2019, 10, 621. [Google Scholar] [CrossRef] [PubMed]
- Lindblad, O.; Chougule, R.A.; Moharram, S.A.; Kabir, N.N.; Sun, J.; Kazi, J.U.; Rönnstrand, L. The role of HOXB2 and HOXB3 in acute myeloid leukemia. Biochem. Biophys. Res. Commun. 2015, 467, 742–747. [Google Scholar] [CrossRef]
- Umeda, S.; Yamamoto, K.; Murayama, T.; Hidaka, M.; Kurata, M.; Ohshima, T.; Suzuki, S.; Sugawara, E.; Kawano, F.; Kitagawa, M. Prognostic significance of HOXB4 in de novo acute myeloid leukemia. Hematology 2012, 17, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Afonja, O.; Smith, J.E., Jr.; Cheng, D.M.; Goldenberg, A.S.; Amorosi, E.; Shimamoto, T.; Nakamura, S.; Ohyashiki, K.; Ohyashiki, J.; Toyama, K.; et al. MEIS1 and HOXA7 genes in human acute myeloid leukemia. Leuk. Res. 2000, 24, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Lachowiez, C.A.; Loghavi, S.; Kadia, T.M.; Daver, N.; Borthakur, G.; Pemmaraju, N.; Naqvi, K.; Alvarado, Y.; Yilmaz, M.; Short, N.; et al. Outcomes of older patients with NPM1-mutated AML: Current treatments and the promise of venetoclax-based regimens. Blood Adv. 2020, 4, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Prange, K.H.M.; Mandoli, A.; Kuznetsova, T.; Wang, S.Y.; Sotoca, A.M.; Marneth, A.E.; van der Reijden, B.A.; Stunnenberg, H.G.; Martens, J.H. MLL-AF9 and MLL-AF4 oncofusion proteins bind a distinct enhancer repertoire and target the RUNX1 program in 11q23 acute myeloid leukemia. Oncogene 2017, 36, 3346–3356. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, R.; Pianigiani, G.; Sciabolacci, S.; Perriello, V.M.; Marra, A.; Cardinali, V.; Pierangeli, S.; Milano, F.; Gionfriddo, I.; Brunetti, L.; et al. Current status and future perspectives in targeted therapy of NPM1-mutated AML. Leukemia 2022, 36, 2351–2367. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Liesveld, J.L. NPM 1 Mutations in AML—The Landscape in 2023. Cancers 2023, 15, 1177. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Song, C.-M.; Liu, S.; Chen, L.-M.; Xue, S.-F.; Huang, S.-H.; Lin, H.; Liu, G.-H. ZFX-mediated upregulation of CEBPA-AS1 contributes to acute myeloid leukemia progression through miR-24-3p/CTBP2 axis. Cell Biol. Toxicol. 2023, 39, 2631–2645. [Google Scholar] [CrossRef]
- Chen, X.; Burkhardt, D.B.; Hartman, A.A.; Hu, X.; Eastman, A.E.; Sun, C.; Wang, X.; Zhong, M.; Krishnaswamy, S.; Guo, S. MLL-AF9 initiates transformation from fast-proliferating myeloid progenitors. Nat. Commun. 2019, 10, 5767. [Google Scholar] [CrossRef] [PubMed]
- Fortune, J.M.; Osheroff, N. Topoisomerase II as a target for anticancer drugs: When enzymes stop being nice. Prog. Nucleic Acid. Res. Mol. Biol. 2000, 64, 221–253. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA Topoisomerases and Their Poisoning by Anticancer and Antibacterial Drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Anand, U.; Dey, A.; Chandel, A.K.S.; Sanyal, R.; Mishra, A.; Pandey, D.K.; De Falco, V.; Upadhyay, A.; Kandimalla, R.; Chaudhary, A.; et al. Cancer chemotherapy and beyond: Current status, drug candidates, associated risks and progress in targeted therapeutics. Genes Dis. 2023, 10, 1367–1401. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef] [PubMed]
- Kerr, A.J.; Dodwell, D.; McGale, P.; Holt, F.; Duane, F.; Mannu, G.; Darby, S.C.; Taylor, C.W. Adjuvant and neoadjuvant breast cancer treatments: A systematic review of their effects on mortality. Cancer Treat. Rev. 2022, 105, 102375. [Google Scholar] [CrossRef] [PubMed]
- Cowell, I.G.; Austin, C.A. Do transcription factories and TOP2B provide a recipe for chromosome translocations in therapy-related leukemia? Cell Cycle 2012, 11, 3143–3144. [Google Scholar] [CrossRef]
- Pedersen-Bjergaard, J.; Andersen, M.K.; Christiansen, D.H.; Nerlov, C. Genetic pathways in therapy-related myelodysplasia and acute myeloid leukemia. Blood 2002, 99, 1909–1912. [Google Scholar] [CrossRef] [PubMed]
- Kollmannsberger, C.; Beyer, J.; Droz, J.P.; Harstrick, A.; Hartmann, J.T.; Biron, P.; Flechon, A.; Schoffski, P.; Kuczyk, M.; Schmoll, H.J.; et al. Secondary leukemia following high cumulative doses of etoposide in patients treated for advanced germ cell tumors. J. Clin. Oncol. 1998, 16, 3386–3391. [Google Scholar] [CrossRef] [PubMed]
- Pedersen-Bjergaard, J.; Daugaard, G.; Hansen, S.W.; Philip, P.; Larsen, S.O.; Rorth, M. Increased risk of myelodysplasia and leukaemia after etoposide, cisplatin, and bleomycin for germ-cell tumours. Lancet 1991, 338, 359–363. [Google Scholar] [CrossRef]
- Howard, R.; Gilbert, E.; Lynch, C.F.; Hall, P.; Storm, H.; Holowaty, E.; Pukkala, E.; Langmark, F.; Kaijser, M.; Andersson, M.; et al. Risk of leukemia among survivors of testicular cancer: A population-based study of 42,722 patients. Ann. Epidemiol. 2008, 18, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Nakamura, T.; Nakanishi, H.; Oishi, M.; Hongo, F.; Okihara, K.; Mizutani, S.; Kuroda, J.; Ukimura, O. Therapy-related acute myeloid leukemia and myelodysplastic syndrome among refractory germ cell tumor patients. Int. J. Urol. 2018, 25, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Richiardi, L.; Scelo, G.; Boffetta, P.; Hemminki, K.; Pukkala, E.; Olsen, J.H.; Weiderpass, E.; Tracey, E.; Brewster, D.H.; McBride, M.L.; et al. Second malignancies among survivors of germ-cell testicular cancer: A pooled analysis between 13 cancer registries. Int. J. Cancer 2007, 120, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Travis, L.B.; Andersson, M.; Gospodarowicz, M.; van Leeuwen, F.E.; Bergfeldt, K.; Lynch, C.F.; Curtis, R.E.; Kohler, B.A.; Wiklund, T.; Storm, H.; et al. Treatment-associated leukemia following testicular cancer. J. Natl. Cancer Inst. 2000, 92, 1165–1171. [Google Scholar] [CrossRef] [PubMed]
- Winick, N.J.; McKenna, R.W.; Shuster, J.J.; Schneider, N.R.; Borowitz, M.J.; Bowman, W.P.; Jacaruso, D.; Kamen, B.A.; Buchanan, G.R. Secondary acute myeloid leukemia in children with acute lymphoblastic leukemia treated with etoposide. J. Clin. Oncol. 1993, 11, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Ratain, M.J.; Kaminer, L.S.; Bitran, J.D.; Larson, R.A.; Le Beau, M.M.; Skosey, C.; Purl, S.; Hoffman, P.C.; Wade, J.; Vardiman, J.W.; et al. Acute nonlymphocytic leukemia following etoposide and cisplatin combination chemotherapy for advanced non-small-cell carcinoma of the lung. Blood 1987, 70, 1412–1417. [Google Scholar] [CrossRef] [PubMed]
- Kushner, B.H.; Kramer, K.; Modak, S.; Qin, L.X.; Yataghena, K.; Jhanwar, S.C.; Cheung, N.K. Reduced risk of secondary leukemia with fewer cycles of dose-intensive induction chemotherapy in patients with neuroblastoma. Pediatr. Blood Cancer 2009, 53, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Le Deley, M.C.; Vassal, G.; Taibi, A.; Shamsaldin, A.; Leblanc, T.; Hartmann, O. High cumulative rate of secondary leukemia after continuous etoposide treatment for solid tumors in children and young adults. Pediatr. Blood Cancer 2005, 45, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Burmeister, T.; Groger, D.; Tsaur, G.; Fechina, L.; Renneville, A.; Sutton, R.; Venn, N.C.; Emerenciano, M.; Pombo-de-Oliveira, M.S.; et al. The MLL recombinome of acute leukemias in 2017. Leukemia 2018, 32, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Rowley, J.D.; Olney, H.J. International workshop on the relationship of prior therapy to balanced chromosome aberrations in therapy-related myelodysplastic syndromes and acute leukemia: Overview report. Genes Chromosomes Cancer 2002, 33, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.L.; Warren, A.J.; Pannell, R.; Forster, A.; Lavenir, I.; Corral, J.; Smith, A.J.; Rabbitts, T.H. The mll-AF9 gene fusion in mice controls myeloproliferation and specifies acute myeloid leukaemogenesis. EMBO J. 1999, 18, 3564–3574. [Google Scholar] [CrossRef] [PubMed]
- Ghisi, M.; Kats, L.; Masson, F.; Li, J.; Kratina, T.; Vidacs, E.; Gilan, O.; Doyle, M.A.; Newbold, A.; Bolden, J.E.; et al. Id2 and E Proteins Orchestrate the Initiation and Maintenance of MLL-Rearranged Acute Myeloid Leukemia. Cancer Cell 2016, 30, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Krivtsov, A.V.; Twomey, D.; Feng, Z.; Stubbs, M.C.; Wang, Y.; Faber, J.; Levine, J.E.; Wang, J.; Hahn, W.C.; Gilliland, D.G.; et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 2006, 442, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef]
- Bian, X.; Liu, R.; Meng, Y.; Xing, D.; Xu, D.; Lu, Z. Lipid metabolism and cancer. J. Exp. Med. 2021, 218, e20201606. [Google Scholar] [CrossRef] [PubMed]
- Nistico, C.; Chiarella, E. An Overview on Lipid Droplets Accumulation as Novel Target for Acute Myeloid Leukemia Therapy. Biomedicines 2023, 11, 3186. [Google Scholar] [CrossRef]
- Verbrugge, S.E.; Al, M.; Assaraf, Y.G.; Kammerer, S.; Chandrupatla, D.M.; Honeywell, R.; Musters, R.P.; Giovannetti, E.; O’Toole, T.; Scheffer, G.L.; et al. Multifactorial resistance to aminopeptidase inhibitor prodrug CHR2863 in myeloid leukemia cells: Down-regulation of carboxylesterase 1, drug sequestration in lipid droplets and pro-survival activation ERK/Akt/mTOR. Oncotarget 2016, 7, 5240–5257. [Google Scholar] [CrossRef] [PubMed]
- Bosc, C.; Broin, N.; Fanjul, M.; Saland, E.; Farge, T.; Courdy, C.; Batut, A.; Masoud, R.; Larrue, C.; Skuli, S.; et al. Autophagy regulates fatty acid availability for oxidative phosphorylation through mitochondria-endoplasmic reticulum contact sites. Nat. Commun. 2020, 11, 4056. [Google Scholar] [CrossRef] [PubMed]
- Esmaeili, S.; Salari, S.; Kaveh, V.; Ghaffari, S.H.; Bashash, D. Alteration of PPAR-GAMMA (PPARG.; PPARgamma) and PTEN gene expression in acute myeloid leukemia patients and the promising anticancer effects of PPARgamma stimulation using pioglitazone on AML cells. Mol. Genet. Genom. Med. 2021, 9, e1818. [Google Scholar] [CrossRef] [PubMed]
- Stavropoulou, V.; Kaspar, S.; Brault, L.; Sanders, M.A.; Juge, S.; Morettini, S.; Tzankov, A.; Iacovino, M.; Lau, I.J.; Milne, T.A.; et al. MLL-AF9 Expression in Hematopoietic Stem Cells Drives a Highly Invasive AML Expressing EMT-Related Genes Linked to Poor Outcome. Cancer Cell 2016, 30, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef]
- Vandamme, N.; Denecker, G.; Bruneel, K.; Blancke, G.; Akay, Ö.; Taminau, J.; De Coninck, J.; De Smedt, E.; Skrypek, N.; Van Loocke, W.; et al. The EMT Transcription Factor ZEB2 Promotes Proliferation of Primary and Metastatic Melanoma While Suppressing an Invasive, Mesenchymal-Like Phenotype. Cancer Res. 2020, 80, 2983–2995. [Google Scholar] [CrossRef] [PubMed]
- Goossens, S.; Radaelli, E.; Blanchet, O.; Durinck, K.; Van der Meulen, J.; Peirs, S.; Taghon, T.; Tremblay, C.S.; Costa, M.; Farhang Ghahremani, M.; et al. ZEB2 drives immature T-cell lymphoblastic leukaemia development via enhanced tumour-initiating potential and IL-7 receptor signalling. Nat. Commun. 2015, 6, 5794. [Google Scholar] [CrossRef] [PubMed]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef]
- Arumugam, T.; Ramachandran, V.; Fournier, K.F.; Wang, H.; Marquis, L.; Abbruzzese, J.L.; Gallick, G.E.; Logsdon, C.D.; McConkey, D.J.; Choi, W. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009, 69, 5820–5828. [Google Scholar] [CrossRef] [PubMed]
- Sayan, A.E.; Griffiths, T.R.; Pal, R.; Browne, G.J.; Ruddick, A.; Yagci, T.; Edwards, R.; Mayer, N.J.; Qazi, H.; Goyal, S.; et al. SIP1 protein protects cells from DNA damage-induced apoptosis and has independent prognostic value in bladder cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 14884–14889. [Google Scholar] [CrossRef]
- Verschueren, K.; Remacle, J.E.; Collart, C.; Kraft, H.; Baker, B.S.; Tylzanowski, P.; Nelles, L.; Wuytens, G.; Su, M.T.; Bodmer, R.; et al. SIP1, a novel zinc finger/homeodomain repressor, interacts with Smad proteins and binds to 5′-CACCT sequences in candidate target genes. J. Biol. Chem. 1999, 274, 20489–20498. [Google Scholar] [CrossRef] [PubMed]
- Postigo, A.A.; Dean, D.C. ZEB represses transcription through interaction with the corepressor CtBP. Proc. Natl. Acad. Sci. USA 1999, 96, 6683–6688. [Google Scholar] [CrossRef] [PubMed]
- Drápela, S.; Bouchal, J.; Jolly, M.K.; Culig, Z.; Souček, K. ZEB1: A Critical Regulator of Cell Plasticity, DNA Damage Response, and Therapy Resistance. Front. Mol. Biosci. 2020, 7, 36. [Google Scholar] [CrossRef] [PubMed]
- Goossens, S.; Janzen, V.; Bartunkova, S.; Yokomizo, T.; Drogat, B.; Crisan, M.; Haigh, K.; Seuntjens, E.; Umans, L.; Riedt, T.; et al. The EMT regulator Zeb2/Sip1 is essential for murine embryonic hematopoietic stem/progenitor cell differentiation and mobilization. Blood 2011, 117, 5620–5630. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Riedt, T.; Goossens, S.; Carrillo Garcia, C.; Szczepanski, S.; Brandes, M.; Pieters, T.; Dobrosch, L.; Gutgemann, I.; Farla, N.; et al. The EMT transcription factor Zeb2 controls adult murine hematopoietic differentiation by regulating cytokine signaling. Blood 2017, 129, 460–472. [Google Scholar] [CrossRef] [PubMed]
- Almotiri, A.; Alzahrani, H.; Menendez-Gonzalez, J.B.; Abdelfattah, A.; Alotaibi, B.; Saleh, L.; Greene, A.; Georgiou, M.; Gibbs, A.; Alsayari, A.; et al. Zeb1 modulates hematopoietic stem cell fates required for suppressing acute myeloid leukemia. J. Clin. Investig. 2021, 131, e129115. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Farkas, C.; Benyoucef, A.; Carmichael, C.; Haigh, K.; Wong, N.; Huylebroeck, D.; Stemmler, M.P.; Brabletz, S.; Brabletz, T.; et al. Interplay between the EMT transcription factors ZEB1 and ZEB2 regulates hematopoietic stem and progenitor cell differentiation and hematopoietic lineage fidelity. PLoS Biol. 2021, 19, e3001394. [Google Scholar] [CrossRef]
- Omilusik, K.D.; Best, J.A.; Yu, B.; Goossens, S.; Weidemann, A.; Nguyen, J.V.; Seuntjens, E.; Stryjewska, A.; Zweier, C.; Roychoudhuri, R.; et al. Transcriptional repressor ZEB2 promotes terminal differentiation of CD8+ effector and memory T cell populations during infection. J. Exp. Med. 2015, 212, 2027–2039. [Google Scholar] [CrossRef]
- Scott, C.L.; Soen, B.; Martens, L.; Skrypek, N.; Saelens, W.; Taminau, J.; Blancke, G.; Van Isterdael, G.; Huylebroeck, D.; Haigh, J.; et al. The transcription factor Zeb2 regulates development of conventional and plasmacytoid DCs by repressing Id2. J. Exp. Med. 2016, 213, 897–911. [Google Scholar] [CrossRef]
- Van Helden, M.J.; Goossens, S.; Daussy, C.; Mathieu, A.L.; Faure, F.; Marcais, A.; Vandamme, N.; Farla, N.; Mayol, K.; Viel, S.; et al. Terminal NK cell maturation is controlled by concerted actions of T-bet and Zeb2 and is essential for melanoma rejection. J. Exp. Med. 2015, 212, 2015–2025. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.L.; T’Jonck, W.; Martens, L.; Todorov, H.; Sichien, D.; Soen, B.; Bonnardel, J.; De Prijck, S.; Vandamme, N.; Cannoodt, R.; et al. The Transcription Factor ZEB2 Is Required to Maintain the Tissue-Specific Identities of Macrophages. The Transcription Factor ZEB2 Is Required to Maintain the Tissue-Specific Identities of Macrophages. Immunity 2018, 49, 312–325.e5. [Google Scholar] [CrossRef] [PubMed]
- Soen, B.; Vandamme, N.; Berx, G.; Schwaller, J.; Van Vlierberghe, P.; Goossens, S. ZEB Proteins in Leukemia: Friends, Foes, or Friendly Foes? Hemasphere 2018, 2, e43. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Wei, H.; Wang, H.; Wang, Z.; Li, J.; Ou, Y.; Xiao, X.; Wang, W.; Chang, A.; Sun, W.; et al. Zeb1-induced metabolic reprogramming of glycolysis is essential for macrophage polarization in breast cancer. Cell Death Dis. 2022, 13, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Q.; He, T.; Wang, Y.; Lu, T.; Wang, Z.; Xiao, N. The transcription factor Zeb1 controls homeostasis and function of type 1 conventional dendritic cells. Nat. Commun. 2023, 14, 6639. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, S.; Brabletz, T. The ZEB/miR-200 feedback loop—A motor of cellular plasticity in development and cancer? EMBO Rep. 2010, 11, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Mar, B.G.; Zhang, H.; Puram, R.V.; Vazquez, F.; Weir, B.A.; Hahn, W.C.; Ebert, B.; Pellman, D. The EMT regulator ZEB2 is a novel dependency of human and murine acute myeloid leukemia. Blood 2017, 129, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.D.; Zhang, L.C.; Zhang, T.J.; Gu, Y.; Wu, D.H.; Zhang, W.; Ma, J.C.; Wen, X.M.; Guo, H.; Lin, J.; et al. Dysregulation of miR-200s clusters as potential prognostic biomarkers in acute myeloid leukemia. J. Transl. Med. 2018, 16, 135. [Google Scholar] [CrossRef] [PubMed]
- De Conti, G.; Gruszka, A.M.; Valli, D.; Cammarata, A.U.; Righi, M.; Mazza, M.; Pelicci, P.G. A Novel Platform to Test In Vivo Single Gene Dependencies in t(8,21) and t(15,17) AML Confirms Zeb2 as Leukemia Target. Cancers 2020, 12, 3768. [Google Scholar] [CrossRef]
- Di Giacomo, D.; La Starza, R.; Gorello, P.; Pellanera, F.; Kalender Atak, Z.; De Keersmaecker, K.; Pierini, V.; Harrison, C.J.; Arniani, S.; Moretti, M.; et al. 14q32 rearrangements deregulating BCL11B mark a distinct subgroup of T-lymphoid and myeloid immature acute leukemia. Blood 2021, 138, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhong, L.; Dan, W.; Chu, X.; Luo, X.; Liu, C.; Wan, P.; Lu, Y.; Liu, Z.; Zhang, Z.; et al. MiR-454-3p promotes apoptosis and autophagy of AML cells by targeting ZEB2 and regulating AKT/mTOR pathway. Hematology 2023, 28, 2223874. [Google Scholar] [CrossRef]
- Shi, X.; Li, J.; Ma, L.; Wen, L.; Wang, Q.; Yao, H.; Ruan, C.; Wu, D.; Zhang, X.; Chen, S. Overexpression of ZEB2-AS1 lncRNA is associated with poor clinical outcomes in acute myeloid leukemia. Oncol. Lett. 2019, 17, 4935–4947. [Google Scholar] [CrossRef] [PubMed]
- Chiarella, E.; Aloisio, A.; Scicchitano, S.; Todoerti, K.; Cosentino, E.G.; Lico, D.; Neri, A.; Amodio, N.; Bond, H.M.; Mesuraca, M. ZNF521 Enhances MLL-AF9-Dependent Hematopoietic Stem Cell Transformation in Acute Myeloid Leukemias by Altering the Gene Expression Landscape. Int. J. Mol. Sci. 2021, 22, 10814. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Feng, Y.; Hu, S.; Du, Y.; Xu, X.; Zhang, M.; Peng, X.; Chen, F. ZEB1 serves as an oncogene in acute myeloid leukaemia via regulating the PTEN/PI3K/AKT signalling pathway by combining with P53. J. Cell. Mol. Med. 2021, 25, 5295–5304. [Google Scholar] [CrossRef] [PubMed]
- Bassani, B.; Simonetti, G.; Cancila, V.; Fiorino, A.; Ciciarello, M.; Piva, A.; Khorasani, A.M.; Chiodoni, C.; Lecis, D.; Gulino, A.; et al. ZEB1 shapes AML immunological niches, suppressing CD8 T cell activity while fostering Th17 cell expansion. Cell Rep. 2024, 43, 113794. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Ye, A.; Bi, L.; Wu, J.; Yu, K.; Zhang, S. Th17 cells and interleukin-17 increase with poor prognosis in patients with acute myeloid leukemia. Cancer Sci. 2014, 105, 933–942. [Google Scholar] [CrossRef]
- Trsova, I.; Hrustincova, A.; Krejcik, Z.; Kundrat, D.; Holoubek, A.; Staflova, K.; Janstova, L.; Vanikova, S.; Szikszai, K.; Klema, J.; et al. Expression of circular RNAs in myelodysplastic neoplasms and their association with mutations in the splicing factor gene SF3B1. Mol. Oncol. 2023, 17, 2565–2583. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Fu, L.; Hong, P.; Feng, W. MALAT-1 regulates the AML progression by promoting the m6A modification of ZEB1. Acta Biochim. Pol. 2023, 70, 37–43. [Google Scholar] [CrossRef]
- Li, W.; Wang, Q.; Su, Q.; Ma, D.; An, C.; Ma, L.; Liang, H. Honokiol suppresses renal cancer cells’ metastasis via dual-blocking epithelial-mesenchymal transition and cancer stem cell properties through modulating miR-141/ZEB2 signaling. Mol. Cells 2014, 37, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Metge, B.J.; Alsheikh, H.A.M.; Kammerud, S.C.; Chen, D.; Das, D.; Nebane, N.M.; Bostwick, J.R.; Shevde, L.A.; Samant, R.S. Targeting EMT using low-dose Teniposide by downregulating ZEB2-driven activation of RNA polymerase I in breast cancer. Cell Death Dis. 2024, 15, 322. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Rubinstein, L.; Ungerleider, R.S. Therapy-related acute myeloid leukemia following treatment with epipodophyllotoxins: Estimating the risks. Med. Pediatr. Oncol. 1994, 23, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Lu, Y.; Lin, L.; Liu, C.; Ma, X.; Chen, X.; Zhou, Z.; Hu, Z.; Pu, J.; Chen, G.; et al. Harnessing endogenous transcription factors directly by small molecules for chemically induced pluripotency inception. Proc. Natl. Acad. Sci. USA 2023, 120, e2215155120. [Google Scholar] [CrossRef] [PubMed]
- Kahlert, U.D.; Joseph, J.V.; Kruyt, F.A.E. EMT- and MET-related processes in nonepithelial tumors: Importance for disease progression, prognosis, and therapeutic opportunities. Mol. Oncol. 2017, 11, 860–877. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.M.; Weinberg, R.A.; Lees, J.A.; Guen, V.J. Emerging Mechanisms by which EMT Programs Control Stemness. Trends Cancer 2020, 6, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, P.G.; Moreno-Bueno, G.; Cano, A. Contribution of Epithelial Plasticity to Therapy Resistance. J. Clin. Med. 2019, 8, 676. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.G.; Langer, E.M.; Lindsley, R.C.; Cai, M.; Murphy, T.L.; Kyba, M.; Murphy, K.M. Snail and the microRNA-200 family act in opposition to regulate epithelial-to-mesenchymal transition and germ layer fate restriction in differentiating ESCs. Stem Cells 2011, 29, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.; Ayyanathan, K. Snail/Gfi-1 (SNAG) family zinc finger proteins in transcription regulation, chromatin dynamics, cell signaling, development, and disease. Cytokine Growth Factor Rev. 2013, 24, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Beauchemin, H.; Moroy, T. Multifaceted Actions of GFI1 and GFI1B in Hematopoietic Stem Cell Self-Renewal and Lineage Commitment. Front. Genet. 2020, 11, 591099. [Google Scholar] [CrossRef] [PubMed]
- Pioli, P.D.; Weis, J.H. Snail transcription factors in hematopoietic cell development: A model of functional redundancy. Exp. Hematol. 2014, 42, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Pioli, P.D.; Whiteside, S.K.; Weis, J.J.; Weis, J.H. Snai2 and Snai3 transcriptionally regulate cellular fitness and functionality of T cell lineages through distinct gene programs. Immunobiology 2016, 221, 618–633. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, C.L.; Wang, J.; Nguyen, T.; Kolawole, O.; Benyoucef, A.; De Maziere, C.; Milne, A.R.; Samuel, S.; Gillinder, K.; Hediyeh-Zadeh, S.; et al. The EMT modulator SNAI1 contributes to AML pathogenesis via its interaction with LSD1. Blood 2020, 136, 957–973. [Google Scholar] [CrossRef] [PubMed]
- Hartung, E.E.; Singh, K.; Berg, T. LSD1 inhibition modulates transcription factor networks in myeloid malignancies. Front. Oncol. 2023, 13, 1149754. [Google Scholar] [CrossRef] [PubMed]
- Malinge, S. SNAIL trail in myeloid malignancies. Blood 2020, 136, 920–921. [Google Scholar] [CrossRef] [PubMed]
- Goossens, S.; Peirs, S.; Van Loocke, W.; Wang, J.; Takawy, M.; Matthijssens, F.; Sonderegger, S.E.; Haigh, K.; Nguyen, T.; Vandamme, N.; et al. Oncogenic ZEB2 activation drives sensitivity toward KDM1A inhibition in T-cell acute lymphoblastic leukemia. Blood 2017, 129, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Noce, B.; Di Bello, E.; Fioravanti, R.; Mai, A. LSD1 inhibitors for cancer treatment: Focus on multi-target agents and compounds in clinical trials. Front. Pharmacol. 2023, 14, 1120911. [Google Scholar] [CrossRef] [PubMed]
- Goossens, S.; Wang, J.; Tremblay, C.S.; De Medts, J.; T’Sas, S.; Nguyen, T.; Saw, J.; Haigh, K.; Curtis, D.J.; Van Vlierberghe, P.; et al. ZEB2 and LMO2 drive immature T-cell lymphoblastic leukemia via distinct oncogenic mechanisms. Haematologica 2019, 104, 1608–1616. [Google Scholar] [CrossRef] [PubMed]
- Benyoucef, A.; Haigh, K.; Cuddihy, A.; Haigh, J.J. JAK/BCL2 inhibition acts synergistically with LSD1 inhibitors to selectively target ETP-ALL. Leukemia 2022, 36, 2802–2816. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, L.; Wu, C.; Yin, G.; Zhu, P.; Zhou, Y.; Hong, Y.; Ni, H.; Qian, Z.; Wu, W.S. Inhibition of Slug effectively targets leukemia stem cells via the Slc13a3/ROS signaling pathway. Leukemia 2020, 34, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Dorn, D.C.; Kou, C.A.; Png, K.J.; Moore, M.A. The effect of cantharidins on leukemic stem cells. Int. J. Cancer 2009, 124, 2186–2199. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Guo, D.; Zhao, Y.Y.; Dong, C.Y.; Liu, X.Y.; Yang, B.X.; Wang, S.W.; Wang, L.; Liu, Q.G.; Ren, Q.; et al. TWIST-1 promotes cell growth, drug resistance and progenitor clonogenic capacities in myeloid leukemia and is a novel poor prognostic factor in acute myeloid leukemia. Oncotarget 2015, 6, 20977–20992. [Google Scholar] [CrossRef] [PubMed]
- Gong, A.; Wang, X.; Wang, X.; Zhao, Y.; Cui, Y. Twist1 Promoter Methylation Regulates the Proliferation and Apoptosis of Acute Myeloid Leukemia Cells via PI3K/AKT Pathway. Indian J. Hematol. Blood Transfus. 2023, 39, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Ottone, T.; Silvestrini, G.; Piazza, R.; Travaglini, S.; Gurnari, C.; Marchesi, F.; Nardozza, A.M.; Fabiani, E.; Attardi, E.; Guarnera, L.; et al. Expression profiling of extramedullary acute myeloid leukemia suggests involvement of epithelial-mesenchymal transition pathways. Leukemia 2023, 37, 2383–2394. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Yin, J.; You, N.; Zhu, W.; Guo, N.; Liu, X.; Zhang, P.; Huang, W.; Xie, Y.; Ren, Q.; et al. Twist family BHLH transcription factor 1 is required for the maintenance of leukemia stem cell in MLL-AF9(+) acute myeloid leukemia. Haematologica 2024, 109, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, Y.; Yang, F.; Feng, S.; Chang, K.; Yu, X.; Guan, F.; Li, X. Clonal MDS/AML cells with enhanced TWIST1 expression reprogram the differentiation of bone marrow MSCs. Redox Biol. 2023, 67, 102900. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Yang, S.; Lu, W.; Liu, J.; Wei, Y.; Guo, H.; Zhang, Y.; Shi, J. Increased NFATC4 Correlates With Poor Prognosis of AML Through Recruiting Regulatory T Cells. Front. Genet. 2020, 11, 573124. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, Y.; Feng, S.; Chang, K.; Yu, X.; Yang, F.; Huang, H.; Wang, Y.; Li, X.; Guan, F. Reciprocal regulation of TWIST1 and OGT determines the decitabine efficacy in MDS/AML. Cell Commun. Signal. 2023, 21, 255. [Google Scholar] [CrossRef] [PubMed]
- Eckardt, J.N.; Stolzel, F.; Kunadt, D.; Rollig, C.; Stasik, S.; Wagenfuhr, L.; Johrens, K.; Kuithan, F.; Kramer, A.; Scholl, S.; et al. Molecular profiling and clinical implications of patients with acute myeloid leukemia and extramedullary manifestations. J. Hematol. Oncol. 2022, 15, 60. [Google Scholar] [CrossRef] [PubMed]
- Tettamanti, S.; Pievani, A.; Biondi, A.; Dotti, G.; Serafini, M. Catch me if you can: How AML and its niche escape immunotherapy. Leukemia 2022, 36, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Cancilla, D.; Rettig, M.P.; DiPersio, J.F. Targeting CXCR4 in AML and ALL. Front. Oncol. 2020, 10, 1672. [Google Scholar] [CrossRef] [PubMed]
- Hanoun, M.; Zhang, D.; Mizoguchi, T.; Pinho, S.; Pierce, H.; Kunisaki, Y.; Lacombe, J.; Armstrong, S.A.; Duhrsen, U.; Frenette, P.S. Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell 2014, 15, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.; Hockney, S.; Blaschuk, O.W.; Pal, D. Targeting N-cadherin (CDH2) and the malignant bone marrow microenvironment in acute leukaemia. Expert Rev. Mol. Med. 2023, 25, e16. [Google Scholar] [CrossRef] [PubMed]
- Kunadt, D.; Kramer, M.; Dill, C.; Altmann, H.; Wagenfuhr, L.; Mohr, B.; Thiede, C.; Rollig, C.; Schetelig, J.; Bornhauser, M.; et al. Lysyl oxidase expression is associated with inferior outcome and Extramedullary disease of acute myeloid leukemia. Biomark. Res. 2020, 8, 20. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, R.J.; Azadniv, M.; Guo, N.; Acklin, J.; Lacagnina, K.; Coppage, M.; Liesveld, J.L. Phenotypic, genotypic, and functional characterization of normal and acute myeloid leukemia-derived marrow endothelial cells. Exp. Hematol. 2016, 44, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.; Huang, X.; Guo, X.; Zheng, Z.; Wei, T.; Chen, B. Metastasis Related Epithelial-Mesenchymal Transition Signature Predicts Prognosis and Response to Chemotherapy in Acute Myeloid Leukemia. Drug Des. Dev. Ther. 2023, 17, 1651–1663. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Gao, Y.; Li, K.; Lin, A.; Jiang, Z. Genomic analysis of biomarkers related to the prognosis of acute myeloid leukemia. Oncol. Lett. 2020, 20, 1824–1834. [Google Scholar] [CrossRef] [PubMed]
- Stefanidakis, M.; Karjalainen, K.; Jaalouk, D.E.; Gahmberg, C.G.; O’Brien, S.; Pasqualini, R.; Arap, W.; Koivunen, E. Role of leukemia cell invadosome in extramedullary infiltration. Blood 2009, 114, 3008–3017. [Google Scholar] [CrossRef] [PubMed]
- Paz, H.; Pathak, N.; Yang, J. Invading one step at a time: The role of invadopodia in tumor metastasis. Oncogene 2014, 33, 4193–4202. [Google Scholar] [CrossRef] [PubMed]
- Whiteley, A.E.; Price, T.T.; Cantelli, G.; Sipkins, D.A. Leukaemia: A model metastatic disease. Nat. Rev. Cancer 2021, 21, 461–475. [Google Scholar] [CrossRef] [PubMed]
- Augoff, K.; Hryniewicz-Jankowska, A.; Tabola, R. Invadopodia: Clearing the way for cancer cell invasion. Ann. Transl. Med. 2020, 8, 902. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.L.; Qiao, Y.D.; Li, J.Y.; Li, X.M.; Zhang, D.; Zhang, X.J.; Zhu, X.H.; Zhou, W.J.; Shi, J.; Wang, W.; et al. Cortactin recruits FMNL2 to promote actin polymerization and endosome motility in invadopodia formation. Cancer Lett. 2018, 419, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Gligorijevic, B.; Wyckoff, J.; Yamaguchi, H.; Wang, Y.; Roussos, E.T.; Condeelis, J. N-WASP-mediated invadopodium formation is involved in intravasation and lung metastasis of mammary tumors. J. Cell Sci. 2012, 125 Pt 3, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, S.; Abdullah, C.; Buschman, M.D.; Diaz, B.; Courtneidge, S.A. The role of Tks adaptor proteins in invadopodia formation, growth and metastasis of melanoma. Oncotarget 2016, 7, 78473–78486. [Google Scholar] [CrossRef]
- Chen, Y.C.; Baik, M.; Byers, J.T.; Chen, K.T.; French, S.W.; Diaz, B. TKS5-positive invadopodia-like structures in human tumor surgical specimens. Exp. Mol. Pathol. 2019, 106, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Kudlik, G.; Takacs, T.; Radnai, L.; Kurilla, A.; Szeder, B.; Koprivanacz, K.; Mero, B.L.; Buday, L.; Vas, V. Advances in Understanding TKS4 and TKS5: Molecular Scaffolds Regulating Cellular Processes from Podosome and Invadopodium Formation to Differentiation and Tissue Homeostasis. Int. J. Mol. Sci. 2020, 21, 8117. [Google Scholar] [CrossRef] [PubMed]
- Daubon, T.; Rochelle, T.; Bourmeyster, N.; Genot, E. Invadopodia and rolling-type motility are specific features of highly invasive p190(bcr-abl) leukemic cells. Eur. J. Cell Biol. 2012, 91, 978–987. [Google Scholar] [CrossRef] [PubMed]
- Poincloux, R.; Vincent, C.; Labrousse, A.; Castandet, J.; Rigo, M.; Cougoule, C.; Bordier, C.; Le Cabec, V.; Maridonneau-Parini, I. Re-arrangements of podosome structures are observed when Hck is activated in myeloid cells. Eur. J. Cell Biol. 2006, 85, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Mukwaya, A.; Jensen, L.; Lagali, N. Relapse of pathological angiogenesis: Functional role of the basement membrane and potential treatment strategies. Exp. Mol. Med. 2021, 53, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Seano, G.; Primo, L. Podosomes and invadopodia: Tools to breach vascular basement membrane. Cell Cycle 2015, 14, 1370–1374. [Google Scholar] [CrossRef] [PubMed]
- Morimatsu, M.; Yamashita, E.; Seno, S.; Sudo, T.; Kikuta, J.; Mizuno, H.; Okuzaki, D.; Motooka, D.; Ishii, M. Migration arrest of chemoresistant leukemia cells mediated by MRTF-SRF pathway. Inflamm. Regen. 2020, 40, 15. [Google Scholar] [CrossRef] [PubMed]
- Auvinen, K.; Jalkanen, S.; Salmi, M. Expression and function of endothelial selectins during human development. Immunology 2014, 143, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Cuellar, T.L.; Herzner, A.M.; Zhang, X.; Goyal, Y.; Watanabe, C.; Friedman, B.A.; Janakiraman, V.; Durinck, S.; Stinson, J.; Arnott, D.; et al. Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemia. J. Cell Biol. 2017, 216, 3535–3549. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.; Salari, K.; Yang, S. SETDB1: A perspective into immune cell function and cancer immunotherapy. Immunology 2023, 169, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Farge, T.; Nakhle, J.; Lagarde, D.; Cognet, G.; Polley, N.; Castellano, R.; Nicolau, M.L.; Bosc, C.; Sabatier, M.; Sahal, A.; et al. CD36 Drives Metastasis and Relapse in Acute Myeloid Leukemia. Cancer Res. 2023, 83, 2824–2838. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, W.; Yan, X.J.; Lin, X.Q.; Li, W.; Mi, J.Q.; Li, J.M.; Zhu, J.; Chen, Z.; Chen, S.J. DNMT3A mutation leads to leukemic extramedullary infiltration mediated by TWIST1. J. Hematol. Oncol. 2016, 9, 106. [Google Scholar] [CrossRef] [PubMed]
- Ponomaryov, T.; Peled, A.; Petit, I.; Taichman, R.S.; Habler, L.; Sandbank, J.; Arenzana-Seisdedos, F.; Magerus, A.; Caruz, A.; Fujii, N.; et al. Induction of the chemokine stromal-derived factor-1 following DNA damage improves human stem cell function. J. Clin. Investig. 2000, 106, 1331–1339. [Google Scholar] [CrossRef] [PubMed]
- Voermans, C.; van Heese, W.P.; de Jong, I.; Gerritsen, W.R.; van Der Schoot, C.E. Migratory behavior of leukemic cells from acute myeloid leukemia patients. Leukemia 2002, 16, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Saavedra, E.; Tang, R.; Gu, Y.; Lappin, P.; Trajkovic, D.; Liu, S.H.; Smeal, T.; Fantin, V.; De Botton, S.; et al. Targeting primary acute myeloid leukemia with a new CXCR4 antagonist IgG1 antibody (PF-06747143). Sci. Rep. 2017, 7, 7305. [Google Scholar] [CrossRef] [PubMed]
- Tavor, S.; Petit, I.; Porozov, S.; Avigdor, A.; Dar, A.; Leider-Trejo, L.; Shemtov, N.; Deutsch, V.; Naparstek, E.; Nagler, A.; et al. CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res. 2004, 64, 2817–2824. [Google Scholar] [CrossRef] [PubMed]
- Konoplev, S.; Rassidakis, G.Z.; Estey, E.; Kantarjian, H.; Liakou, C.I.; Huang, X.; Xiao, L.; Andreeff, M.; Konopleva, M.; Medeiros, L.J. Overexpression of CXCR4 predicts adverse overall and event-free survival in patients with unmutated FLT3 acute myeloid leukemia with normal karyotype. Cancer 2007, 109, 1152–1156. [Google Scholar] [CrossRef]
- Muz, B.; Abdelghafer, A.; Markovic, M.; Yavner, J.; Melam, A.; Salama, N.N.; Azab, A.K. Targeting E-selectin to Tackle Cancer Using Uproleselan. Cancers 2021, 13, 335. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Ye, J.; Xia, Y.; Li, M.; Li, G.; Hu, X.; Su, X.; Wang, D.; Zhao, X.; Lu, F.; et al. METTL3 mediates chemoresistance by enhancing AML homing and engraftment via ITGA4. Leukemia 2022, 36, 2586–2595. [Google Scholar] [CrossRef]
- Bakst, R.L.; Tallman, M.S.; Douer, D.; Yahalom, J. How I treat extramedullary acute myeloid leukemia. Blood 2011, 118, 3785–3793. [Google Scholar] [CrossRef]
- Paydas, S.; Zorludemir, S.; Ergin, M. Granulocytic sarcoma: 32 cases and review of the literature. Leuk. Lymphoma 2006, 47, 2527–2541. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Brandwein, J.; Yi, Q.L.; Chun, K.; Patterson, B.; Brien, B. Extramedullary infiltrates of AML are associated with CD56 expression, 11q23 abnormalities and inferior clinical outcome. Leuk. Res. 2004, 28, 1007–1011. [Google Scholar] [CrossRef] [PubMed]
- Novotny, J.R.; Nuckel, H.; Duhrsen, U. Correlation between expression of CD56/NCAM and severe leukostasis in hyperleukocytic acute myelomonocytic leukaemia. Eur. J. Haematol. 2006, 76, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Alegretti, A.P.; Bittar, C.M.; Bittencourt, R.; Piccoli, A.K.; Schneider, L.; Silla, L.M.; Bo, S.D.; Xavier, R.M. The expression of CD56 antigen is associated with poor prognosis in patients with acute myeloid leukemia. Rev. Bras. Hematol. Hemoter. 2011, 33, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Assaad, M.; Kumar, V.; Carmack, A.; Karki, A.; Golden, D. Acute Myeloid Leukemia with Central Nervous System Involvement Following Routine Surgical Procedures: A Bridge between Surgical, Medical, and Neurological Critical Care. Cureus 2022, 14, e21245. [Google Scholar] [CrossRef] [PubMed]
- Abbott, B.L.; Rubnitz, J.E.; Tong, X.; Srivastava, D.K.; Pui, C.H.; Ribeiro, R.C.; Razzouk, B.I. Clinical significance of central nervous system involvement at diagnosis of pediatric acute myeloid leukemia: A single institution’s experience. Leukemia 2003, 17, 2090–2096. [Google Scholar] [CrossRef] [PubMed]
- Alakel, N.; Stolzel, F.; Mohr, B.; Kramer, M.; Oelschlagel, U.; Rollig, C.; Bornhauser, M.; Ehninger, G.; Schaich, M. Symptomatic central nervous system involvement in adult patients with acute myeloid leukemia. Cancer Manag. Res. 2017, 9, 97–102. [Google Scholar] [CrossRef] [PubMed]
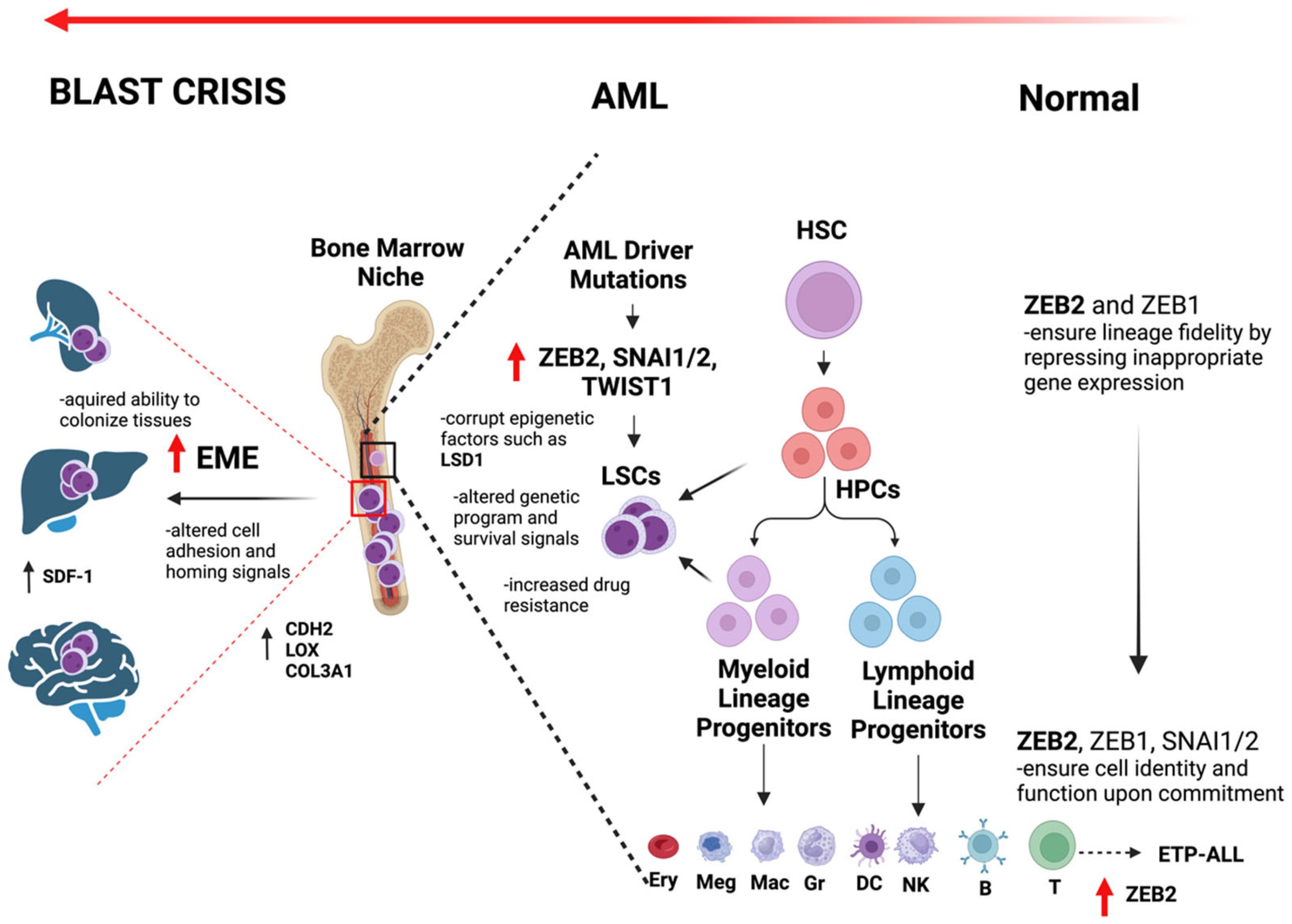

{kind=link}
{kind=link}
| Subtype | Characteristics | Prognosis and Features |
|---|---|---|
| M0 | High percentage of minimally differentiated blasts, negative for peroxidase, confirmed with myeloid markers by flow cytometry. | Poor prognosis, associated with complex chromosomal abnormalities. |
| M1 | Less than 10% promyelocytes, blasts lack granules with distinct nucleoli, >3% positive for myeloperoxidase. | |
| M2 | Presence of mature cells, including cells with Auer rods, associated with t(8;21) translocations and AML1-ETO/ETO-AML1 fusion proteins. | More favorable prognosis when linked with specific translocations. |
| M3 (APL) | Hypergranulated promyelocytes, bilobed nuclei, characterized by the PML-RARα fusion from t(15;17) translocation. Treatable with ATRA and arsenic trioxide (ATO). | Favorable prognosis due to effective treatment options. |
| M4 | Presence of monocytes and promonocytes in bone marrow. The M4Eo variant shows abnormal eosinophils and inv(16) cytogenetic abnormality. | Favorable prognosis for variants like M4Eo depending on WBC levels. |
| M5 | High number of monocytic lineage cells, over 30% blast cells in BM or PB, often associated with 11q abnormalities and MLL gene rearrangements (e.g., MLL-AF9). | Poor prognosis, common extramedullary disease, and linkage to severe clinical features. |
| M6 | Over 50% nucleated erythroid cells in bone marrow with substantial abnormalities, positive for PAS staining and glycophorin A. | Rare, less than 5% of AML cases, associated with severe developmental abnormalities in erythroid cells. |
| M7 | Poor megakaryocytic differentiation, megakaryoblasts with scant cytoplasm and dense chromatin, negative for common stains, confirmed by CD41 and electron microscopy. | Extremely rare (~1% of cases), poor prognosis due to aggressive nature and difficulty in treatment due to morphological variability in the cells. |
| AML Type | Blast % | Genetics | ELN Risk Class (2022) | Literature (2024) | Refs. |
|---|---|---|---|---|---|
| APL with t(15;17)/PML::RARA | >10% | PML:RARA | - | Favorable | [33,34] |
| APL with other RARA rearrangements | >10% | Various RARA rearrangements | - | Variable, depends on the rearrangement | [35] |
| AML with t(8;21)/RUNX1::RUNX1T1 | >10% | RUNX1:RUNX1T1 | Favorable | Favorable | [36] |
| AML with inv(16) or t(16;16)/CBFB::MYH11 | >10% | CBFB:MYH11 | Favorable | Favorable | [37,38] |
| AML with t(9;11)/MLLT3::KMT2A | >10% | MLLT3:KMT2A | Intermediate | Intermediate | [39] |
| AML with other KMT2A rearrangements | >10% | Various KMT2A rearrangements | - | Variable, depends on the rearrangement | [39] |
| AML with t(6;9)/DEK::NUP214 | >10% | DEK:NUP214 | Adverse | Adverse | [40] |
| AML with inv(3) or t(3;3)/GATA2; MECOM | >10% | GATA2; MECOM | Adverse | Adverse | [41] |
| AML with other MECOM rearrangements | >10% | Various MECOM rearrangements | - | Adverse | [42] |
| AML with other rare recurring translocations | >10% | Rare recurring translocations | - | Adverse | [43] |
| AML with t(9;22)/BCR::ABL1 | >10% | BCR:ABL1 | Adverse | Adverse | [44] |
| AML with mutated NPM1 | >10% | Mutated NPM1 | Favorable | Favorable | [45] |
| AML with bZIP CEBPA mutations | >10% | bZIP CEBPA mutations | Favorable | Favorable | [46] |
| AML/MDS with mutated TP53 | 10–19%/>20% | Mutated TP53 | Adverse | Adverse | [47] |
| AML/MDS with myelodysplasia-related gene mutations | 10–19%/>20% | Myelodysplasia-related gene mutations | - | Adverse | [48] |
| AML with myelodysplasia-related cytogenetic abnormalities | 10–19%/>20% | Myelodysplasia-related cytogenetic abnormalities | - | Intermediate | [49] |
| AML not otherwise specified (NOS) | 10–19%/>20% | - | - | - | [50] |
| Myeloid sarcoma | Not specified | - | Adverse | Adverse | [51] |
| MDS with mutated TP53 | 0–9% | Multi-hit TP53 mutation or TP53 mutation (VAF > 10%) and complex karyotype often with loss of 17p | Adverse | Adverse | [29,30] |
| MDS/AML with mutated TP53 | 10–19% | Any somatic TP53 mutation (VAF > 10%) | Adverse | Adverse | [29,30] |
| AML with mutated TP53 | >20% | Any somatic TP53 mutation (VAF > 10%) | Adverse | Adverse | [29,30] |
| Feature | ZEB1 | ZEB2 | SNAI1 | SNAI2 | TWIST1 |
|---|---|---|---|---|---|
| Roles in EMT Processes | Involved in malignant dissemination and metastasis [116,117] | Plays a role in cancer or tumor stem cell properties, development, and treatment resistance [118,119] | Essential for EMT, cancer stemness, and drug resistance [155,156,157] | Promotes leukemogenesis and influences chemotherapy resistance [170,171] | Central to AML pathophysiology; affects growth and drug resistance [172] |
| Roles in Hematopoiesis | Lesser degree of influence compared with ZEB2 [129,130] | Limits inappropriate expression of immune cell programs [131,132,133,134] | Influences stem and progenitor cell functions [158] | Impairs LSC self-renewal, restricts LSC self-renewal via Slc13a3 [170] | Impacts progenitor clonogenic capacities [175] |
| Regulation by the MiR200 Family of miRNAs | Negatively regulated, lower levels in certain AML subtypes [138] | Negatively regulated; absence leads to oncogenic levels [138,139,140] | Relationship in hematopoiesis is unclear [158] | Not specified | Not specified |
| Influence on AML Patient Outcomes | Associated with poor outcomes, essential for leukemic blast invasion [117,130] | Upregulation associated with leukemic blasts [130] | Overexpression contributes to impaired differentiation and enhanced self-renewal [163] | Associated with poor clinical outcomes [171] | Linked to poor prognostic factors; promotes tissue invasion [172,174] |
| Oncofusion Protein Interactions | Upregulated by MLL-AF9 and MLL-AF4 [117] | Upregulated by AML-ETO, MLL-AF9, MLL-AF4, and PML-RARα [116,117] | Not clear | Not specified | Notably involved in extramedullary manifestations [174] |
| Genetic Screening Findings | Deletion may accelerate AML progression [129,130] | Involved in myeloid and lymphoid leukemic transformation [120,139] | Knock-down enhances morphological differentiation and improves survival [163] | Not specified | Essential for viability and self-renewal of LSCs [175] |
| Functional Roles in Immune Cell Differentiation | Plays a role in macrophage differentiation [136] and dendritic cell homeostasis [137] | Ensures immune cell lineage fidelity [131,132,133,134] | Implicated in myeloid development and self-renewal of progenitors [161,162] | Not specified | Influences bone marrow microenvironment interactions [176] |
| Contribution to Leukemic Transformation | Potentially oncogenic, may act as a tumor suppressor [129,130] | Involved in myeloid leukemia transformation [135] | Leads to myeloproliferative disorders and AML transformation [162,163] | Promotes leukemogenesis [170] | Promotes disease initiation and maintenance [175] |
| Potential Therapeutic Targets | Could offer novel approaches for AML treatment if targeted [146,147,148] | Inhibition may improve outcomes [139,140,141,169] | Knockout or inhibition improves survival [163] | Targeting could impair LSC self-renewal and chemoresistance [171] | Targeting TWIST1 could overcome chemoresistance and influence treatment [178] |
| Factor | Survival | Motility | Adherence |
|---|---|---|---|
| SDF-1 | [207] | [208] | |
| METTL-3 | [213] | ||
| Integrin β | [188] | [188] | |
| N-WASP | [193] | ||
| Tks4, Tks5 | [194,195] | ||
| E-selectin | [202] | [202] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuevas, D.; Amigo, R.; Agurto, A.; Heredia, A.A.; Guzmán, C.; Recabal-Beyer, A.; González-Pecchi, V.; Caprile, T.; Haigh, J.J.; Farkas, C. The Role of Epithelial-to-Mesenchymal Transition Transcription Factors (EMT-TFs) in Acute Myeloid Leukemia Progression. Biomedicines 2024, 12, 1915. https://doi.org/10.3390/biomedicines12081915
Cuevas D, Amigo R, Agurto A, Heredia AA, Guzmán C, Recabal-Beyer A, González-Pecchi V, Caprile T, Haigh JJ, Farkas C. The Role of Epithelial-to-Mesenchymal Transition Transcription Factors (EMT-TFs) in Acute Myeloid Leukemia Progression. Biomedicines. 2024; 12(8):1915. https://doi.org/10.3390/biomedicines12081915
Chicago/Turabian StyleCuevas, Diego, Roberto Amigo, Adolfo Agurto, Adan Andreu Heredia, Catherine Guzmán, Antonia Recabal-Beyer, Valentina González-Pecchi, Teresa Caprile, Jody J. Haigh, and Carlos Farkas. 2024. "The Role of Epithelial-to-Mesenchymal Transition Transcription Factors (EMT-TFs) in Acute Myeloid Leukemia Progression" Biomedicines 12, no. 8: 1915. https://doi.org/10.3390/biomedicines12081915