
Animal Models of Retinopathy of Prematurity: Advances and Metabolic Regulators
 , , ,
, , ,
Abstract
:1. Introduction
2. Animal Models of Experimental ROP
2.1. The Mouse Model of Oxygen-Induced Retinopathy
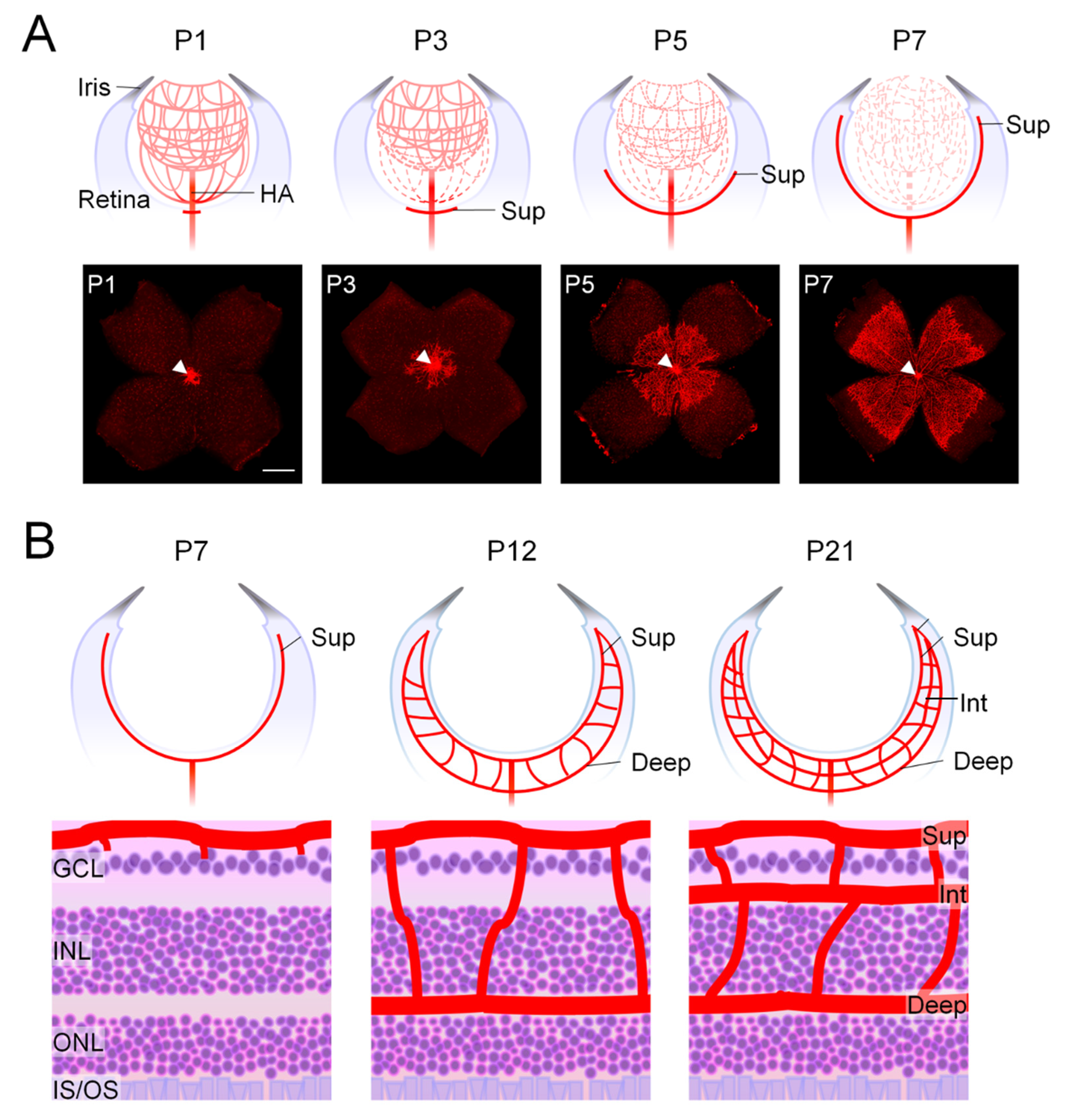
2.1.1. Development of Mouse Retinal Vasculature Occurs Postnatally
2.1.2. The Neonatal Mouse Model of OIR with Consistent High Oxygen Exposure
2.2. The Rat Model of OIR with Cycling Oxygen Treatment
2.3. Hyperglycemia-Associated Murine Models of ROP
2.4. Early Feline and Canine Models of OIR
2.5. The Zebrafish Model of Hypoxia-Induced Retinopathy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Standardized Experiment Protocol | ROP-like Features | Advantages | Limitation | Reference |
|---|---|---|---|---|---|
| Mouse OIR model | Two-phase model. Phase 1: From P7–P12, 75% ± 2% oxygen for five days. Phase 2: Normal room air exposure from P12–P17. | VO phase: Regression of blood vessels in central retinas from P8–P12 under hyperoxic conditions. NV Phase: Ischemia and hypoxia-induced uncontrolled compensatory pathological NV in mid-peripheral retina from P14–P17. |
|
| [28,54,100] |
| Rat OIR model | Two-phase model. Phase 1: From P1–P14, oxygen levels alternate be-tween 50% and 10% every 24 h. Phase 2: Normal room air exposure from P14–P20. | VO phase: Vessel loss and delayed vascular development in peripheral retinas under hyperoxic conditions from P1–P14. NV Phase: Peripheral retinal ischemia and hypoxia stimulates pathological NV starting at P18. |
|
| [27,74] |
| Neonatal hyperglycemia associated murine models of ROP | One-phase rat model. By administering a single injection of streptozotocin (STZ) at P1 in rat model. | Hyperglycemia induction from P2/3–P6 Vascular features: Delayed development of superficial retinal vasculature at P6 under hyperglycemic conditions. |
|
| [32] |
| One-phase mice model. Administration of STZ from P1–P9 in mice. | Hyperglycemia induction from P8–P10 Vascular features: Delayed development of deep retinal vascular layer at P10 under hyperglycemic conditions. |
|
| [33,101] | |
| Zebrafish model of hypoxia-induced retinopathy | By keeping zebrafish in 10% oxygen tank from 3–15 (max) days. | Higher density of vascular sprouts in optic capillary plexus in the hypoxia model. |
|
| [38] |
| Feline OIR model | Two-phase model. Phase 1: From P3–P7, 70–80% oxygen for a minimum of 1–4 days. Phase 2: Returned to normal air with 21% oxygen from P3–P80. | VO phase: complete or partial obliteration of retinal vessels in the VO phase. NV phase: revascularization with intravitreal NV. |
|
| [39,102] |
| Canine OIR model | Two-phase model. Phase 1: 95% to 100% oxygen for 4 days. Phase 2: Returned to room air until P22–P45. | Peripheral VO and NV. Presence of retinal detachment. |
|
| [40,103] |
2.6. Automated Analysis and Quantification of OIR Vascular Features
3. Metabolic Regulators Influencing OIR
3.1. Hypoxia, Hypoxia-Induced Growth Factors and VEGF
3.2. Lipid Metabolism and Modulation: Fatty Acid Oxidation, Polyunsaturated Fatty Acids, and Sphingolipids
3.3. Emerging Role of Amino Acid Metabolism in Angiogenesis and Retinopathy
4. Summary and Future Directions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Coats, D.K. Retinopathy of Prematurity: Involution, Factors Predisposing to Retinal Detachment, and Expected Utility of Preemptive Surgical Reintervention. Trans. Am. Ophthalmol. Soc. 2005, 103, 281–312. [Google Scholar] [CrossRef] [PubMed]
- Hess, J.H. Oxygen Unit for Premature and Very Young Infants. Am. J. Dis. Child. 1934, 47, 916–917. [Google Scholar]
- Silverman, W.A. Retrolental Fibroplasia: A Modern Parable; Grune & Stratton: New York, NY, USA, 1980. [Google Scholar]
- Campbell, K. Intensive Oxygen Therapy as a Possible Cause of Retrolental Fibroplasia; a Clinical Approach. Med. J. Aust. 1951, 2, 48–50. [Google Scholar] [CrossRef]
- Patz, A.; Hoeck, L.E.; de La Cruz, E. Studies on the Effect of High Oxygen Administration in Retrolental Fibroplasia. I. Nursery Observations. Am. J. Ophthalmol. 1952, 35, 1248–1253. [Google Scholar] [CrossRef] [PubMed]
- Kinsey, V.E. Retrolental Fibroplasia; Cooperative Study of Retrolental Fibroplasia and the Use of Oxygen. AMA Arch. Ophthalmol. 1956, 56, 481–543. [Google Scholar] [CrossRef]
- Chen, J.; Smith, L.E. Retinopathy of Prematurity. Angiogenesis 2007, 10, 133–140. [Google Scholar] [CrossRef]
- Hartnett, M.E.; Penn, J.S. Mechanisms and Management of Retinopathy of Prematurity. N. Engl. J. Med. 2012, 367, 2515–2526. [Google Scholar] [CrossRef] [PubMed]
- Hellstrom, A.; Smith, L.E.; Dammann, O. Retinopathy of Prematurity. Lancet 2013, 382, 1445–1457. [Google Scholar] [CrossRef]
- Hardy, P.; Dumont, I.; Bhattacharya, M.; Hou, X.; Lachapelle, P.; Varma, D.R.; Chemtob, S. Oxidants, Nitric Oxide and Prostanoids in the Developing Ocular Vasculature: A Basis for Ischemic Retinopathy. Cardiovasc. Res. 2000, 47, 489–509. [Google Scholar] [CrossRef]
- Schaffer, D.B.; Palmer, E.A.; Plotsky, D.F.; Metz, H.S.; Flynn, J.T.; Tung, B.; Hardy, R.J. Prognostic Factors in the Natural Course of Retinopathy of Prematurity. The Cryotherapy for Retinopathy of Prematurity Cooperative Group. Ophthalmology 1993, 100, 230–237. [Google Scholar] [CrossRef]
- Foos, R.Y. Chronic Retinopathy of Prematurity. Ophthalmology 1985, 92, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Patz, A. Oxygen Studies in Retrolental Fibroplasia. IV. Clinical and Experimental Observations. Am. J. Ophthalmol. 1954, 38, 291–308. [Google Scholar] [CrossRef] [PubMed]
- White, J.E.; Repka, M.X. Randomized Comparison of Diode Laser Photocoagulation Versus Cryotherapy for Threshold Retinopathy of Prematurity: 3-Year Outcome. J. Pediatr. Ophthalmol. Strabismus 1997, 34, 83–87. [Google Scholar] [CrossRef]
- Hwang, C.K.; Hubbard, G.B.; Hutchinson, A.K.; Lambert, S.R. Outcomes after Intravitreal Bevacizumab Versus Laser Photocoagulation for Retinopathy of Prematurity: A 5-Year Retrospective Analysis. Ophthalmology 2015, 122, 1008–1015. [Google Scholar] [CrossRef]
- Stahl, A.; Sukgen, E.A.; Wu, W.C.; Lepore, D.; Nakanishi, H.; Mazela, J.; Moshfeghi, D.M.; Vitti, R.; Athanikar, A.; Chu, K.; et al. Effect of Intravitreal Aflibercept Vs. Laser Photocoagulation on Treatment Success of Retinopathy of Prematurity: The Firefleye Randomized Clinical Trial. J. Am. Med. Assoc. 2022, 328, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Stahl, A.; Nakanishi, H.; Lepore, D.; Wu, W.-C.; Azuma, N.; Jacas, C.; Vitti, R.; Athanikar, A.; Chu, K.; Iveli, P.; et al. Intravitreal Aflibercept Vs. Laser Therapy for Retinopathy of Prematurity: Two-Year Efficacy and Safety Outcomes in the Nonrandomized Controlled Trial Firefleye Next. JAMA Netw. Open 2024, 7, e248383. [Google Scholar] [CrossRef] [PubMed]
- Hellstrom, A.; Smith, L.E.H.; Hard, A.L. ROP: 80 Years after Its Detection—Where Do We Stand and How Long Will We Continue to Laser. Neonatology 2024. ahead of print. [Google Scholar] [CrossRef]
- Raghuveer, T.S.; Zackula, R.E.; Hartnett, M.E. Aflibercept to Treat Retinopathy of Prematurity: Need for More Research. J. Perinatol. 2024. epub ahead of printing. [Google Scholar] [CrossRef]
- Fu, Z.; Nilsson, A.K.; Hellstrom, A.; Smith, L.E.H. Retinopathy of Prematurity: Metabolic Risk Factors. Elife 2022, 11, e80550. [Google Scholar] [CrossRef]
- O’Connor, A.R.; Wilson, C.M.; Fielder, A.R. Ophthalmological Problems Associated with Preterm Birth. Eye 2007, 21, 1254–1260. [Google Scholar] [CrossRef]
- Hong, E.H.; Shin, Y.U.; Cho, H. Retinopathy of Prematurity: A Review of Epidemiology and Current Treatment Strategies. Clin. Exp. Pediatr. 2022, 65, 115–126. [Google Scholar] [CrossRef]
- Kim, S.J.; Port, A.D.; Swan, R.; Campbell, J.P.; Chan, R.V.P.; Chiang, M.F. Retinopathy of Prematurity: A Review of Risk Factors and Their Clinical Significance. Surv. Ophthalmol. 2018, 63, 618–637. [Google Scholar] [CrossRef]
- De las Rivas Ramírez, N.; Luque Aranda, G.; Rius Díaz, F.; Pérez Frías, F.J.; Sánchez Tamayo, T. Risk Factors Associated with Retinopathy of Prematurity Development and Progression. Sci. Rep. 2022, 12, 21977. [Google Scholar] [CrossRef]
- Hartnett, M.E. Pathophysiology of Retinopathy of Prematurity. Annu. Rev. Vis. Sci. 2023, 9, 39–70. [Google Scholar] [CrossRef]
- Dammann, O.; Hartnett, M.E.; Stahl, A. Retinopathy of Prematurity. Dev. Med. Child. Neurol. 2023, 65, 625–631. [Google Scholar] [CrossRef]
- Penn, J.S.; Tolman, B.L.; Lowery, L.A. Variable Oxygen Exposure Causes Preretinal Neovascularization in the Newborn Rat. Investig. Ophthalmol. Vis. Sci. 1993, 34, 576–585. [Google Scholar]
- Smith, L.E.; Wesolowski, E.; McLellan, A.; Kostyk, S.K.; D’Amato, R.; Sullivan, R.; D’Amore, P.A. Oxygen-Induced Retinopathy in the Mouse. Investig. Ophthalmol. Vis. Sci. 1994, 35, 101–111. [Google Scholar]
- Stahl, A.; Connor, K.M.; Sapieha, P.; Chen, J.; Dennison, R.J.; Krah, N.M.; Seaward, M.R.; Willett, K.L.; Aderman, C.M.; Guerin, K.I.; et al. The Mouse Retina as an Angiogenesis Model. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2813–2826. [Google Scholar] [CrossRef] [PubMed]
- Connor, K.M.; Sangiovanni, J.P.; Lofqvist, C.; Aderman, C.M.; Chen, J.; Higuchi, A.; Hong, S.; Pravda, E.A.; Majchrzak, S.; Carper, D.; et al. Increased Dietary Intake of Omega-3-Polyunsaturated Fatty Acids Reduces Pathological Retinal Angiogenesis. Nat. Med. 2007, 13, 868–873. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.E.; Shen, W.; Perruzzi, C.; Soker, S.; Kinose, F.; Xu, X.; Robinson, G.; Driver, S.; Bischoff, J.; Zhang, B.; et al. Regulation of Vascular Endothelial Growth Factor-Dependent Retinal Neovascularization by Insulin-Like Growth Factor-1 Receptor. Nat. Med. 1999, 5, 1390–1395. [Google Scholar] [CrossRef] [PubMed]
- Kermorvant-Duchemin, E.; Pinel, A.C.; Lavalette, S.; Lenne, D.; Raoul, W.; Calippe, B.; Behar-Cohen, F.; Sahel, J.A.; Guillonneau, X.; Sennlaub, F. Neonatal Hyperglycemia Inhibits Angiogenesis and Induces Inflammation and Neuronal Degeneration in the Retina. PLoS ONE 2013, 8, e79545. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Lofqvist, C.A.; Liegl, R.; Wang, Z.; Sun, Y.; Gong, Y.; Liu, C.H.; Meng, S.S.; Burnim, S.B.; Arellano, I.; et al. Photoreceptor Glucose Metabolism Determines Normal Retinal Vascular Growth. EMBO Mol. Med. 2018, 10, 76–90. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Agthe, A.G.; Donohue, P.K.; Lehmann, C.U. Hyperglycemia and Retinopathy of Prematurity in Very Low Birth Weight Infants. J. Perinatol. 2003, 23, 186–194. [Google Scholar] [CrossRef]
- Blanco, C.L.; Baillargeon, J.G.; Morrison, R.L.; Gong, A.K. Hyperglycemia in Extremely Low Birth Weight Infants in a Predominantly Hispanic Population and Related Morbidities. J. Perinatol. 2006, 26, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Kaempf, J.W.; Kaempf, A.J.; Wu, Y.; Stawarz, M.; Niemeyer, J.; Grunkemeier, G. Hyperglycemia, Insulin and Slower Growth Velocity May Increase the Risk of Retinopathy of Prematurity. J. Perinatol. 2011, 31, 251–257. [Google Scholar] [CrossRef]
- Mohamed, S.; Murray, J.C.; Dagle, J.M.; Colaizy, T. Hyperglycemia as a Risk Factor for the Development of Retinopathy of Prematurity. BMC Pediatr. 2013, 13, 78. [Google Scholar] [CrossRef]
- Cao, R.; Jensen, L.D.; Söll, I.; Hauptmann, G.; Cao, Y. Hypoxia-Induced Retinal Angiogenesis in Zebrafish as a Model to Study Retinopathy. PLoS ONE 2008, 3, e2748. [Google Scholar] [CrossRef]
- Ashton, N.; Ward, B.; Serpell, G. Effect of Oxygen on Developing Retinal Vessels with Particular Reference to the Problem of Retrolental Fibroplasia. Br. J. Ophthalmol. 1954, 38, 397–432. [Google Scholar] [CrossRef]
- McLeod, D.S.; Brownstein, R.; Lutty, G.A. Vaso-Obliteration in the Canine Model of Oxygen-Induced Retinopathy. Investig. Ophthalmol. Vis. Sci. 1996, 37, 300–311. [Google Scholar]
- Flower, R.W.; Blake, D.A. Retrolental Fibroplasia: Evidence for a Role of the Prostaglandin Cascade in the Pathogenesis of Oxygen-Induced Retinopathy in the Newborn Beagle. Pediatr. Res. 1981, 15, 1293–1302. [Google Scholar] [CrossRef]
- Patz, A.; Eastham, A.; Higginbotham, D.H.; Kleh, T. Oxygen Studies in Retrolental Fibroplasia. II. The Production of the Microscopic Changes of Retrolental Fibroplasia in Experimental Animals. Am. J. Ophthalmol. 1953, 36, 1511–1522. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-H.; Chen, J. Animal Models of Retinopathy of Prematurity. In A Quick Guide to Pediatric Retina; Wu, W.-C., Lam, W.-C., Eds.; Springer: Singapore, 2021; pp. 11–19. [Google Scholar] [CrossRef]
- Young, R.W. Cell Differentiation in the Retina of the Mouse. Anat. Rec. 1985, 212, 199–205. [Google Scholar] [CrossRef]
- Liu, C.H.; Wang, Z.; Sun, Y.; Chen, J. Animal Models of Ocular Angiogenesis: From Development to Pathologies. FASEB J. 2017, 31, 4665–4681. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, C.H.; Sapieha, P. Retinal Vascular Development. In Anti-Angiogenic Therapy in Ophthalmology, 1st ed.; Stahl, A., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 1–19. [Google Scholar] [CrossRef]
- Fruttiger, M. Development of the Retinal Vasculature. Angiogenesis 2007, 10, 77–88. [Google Scholar] [CrossRef]
- Hartnett, M.E. Pediatric Retina, 2nd ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; p. 808. [Google Scholar]
- Ito, M.; Yoshioka, M. Regression of the Hyaloid Vessels and Pupillary Membrane of the Mouse. Anat. Embryol. 1999, 200, 403–411. [Google Scholar] [CrossRef]
- Bora, K.; Kushwah, N.; Maurya, M.; Pavlovich, M.C.; Wang, Z.; Chen, J. Assessment of Inner Blood-Retinal Barrier: Animal Models and Methods. Cells 2023, 12, 2443. [Google Scholar] [CrossRef]
- Gyllensten, L.J.; Hellstrom, B.E. Retrolental Fibroplasia; Animal Experiments: The Effect of Intermittingly Administered Oxygen on the Postnatal Development of the Eyes of Fullterm Mice. Acta Paediatr. 1952, 41, 577–582. [Google Scholar] [CrossRef]
- Gerschman, R.; Nadig, P.W.; Snell, A.C., Jr.; Nye, S.W. Effect of High Oxygen Concentrations on Eyes of Newborn Mice. Am. J. Physiol. 1954, 179, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Curley, F.J.; Habegger, H.; Ingalls, T.H.; Philbrook, F.R. Retinopathy of Immaturity in the Newborn Mouse after Exposure to Oxygen Imbalances. Am. J. Ophthalmol. 1956, 42, 377–392. [Google Scholar]
- Connor, K.M.; Krah, N.M.; Dennison, R.J.; Aderman, C.M.; Chen, J.; Guerin, K.I.; Sapieha, P.; Stahl, A.; Willett, K.L.; Smith, L.E. Quantification of Oxygen-Induced Retinopathy in the Mouse: A Model of Vessel Loss, Vessel Regrowth and Pathological Angiogenesis. Nat. Protoc. 2009, 4, 1565–1573. [Google Scholar] [CrossRef]
- Lange, C.; Ehlken, C.; Stahl, A.; Martin, G.; Hansen, L.; Agostini, H.T. Kinetics of Retinal Vaso-Obliteration and Neovascularisation in the Oxygen-Induced Retinopathy (Oir) Mouse Model. Graefe’s Arch. Clin. Exp. Ophthalmol. Albrecht Von Graefes Arch. Fur Klin. Und Exp. Ophthalmol. 2009, 247, 1205–1211. [Google Scholar] [CrossRef]
- Gu, X.; Samuel, S.; El-Shabrawey, M.; Caldwell, R.B.; Bartoli, M.; Marcus, D.M.; Brooks, S.E. Effects of Sustained Hyperoxia on Revascularization in Experimental Retinopathy of Prematurity. Investig. Ophthalmol. Vis. Sci. 2002, 43, 496–502. [Google Scholar]
- Chen, J.; Connor, K.M.; Aderman, C.M.; Smith, L.E. Erythropoietin Deficiency Decreases Vascular Stability in Mice. J. Clin. Investig. 2008, 118, 526–533. [Google Scholar] [CrossRef]
- Aiello, L.P.; Pierce, E.A.; Foley, E.D.; Takagi, H.; Chen, H.; Riddle, L.; Ferrara, N.; King, G.L.; Smith, L.E. Suppression of Retinal Neovascularization in Vivo by Inhibition of Vascular Endothelial Growth Factor (Vegf) Using Soluble Vegf-Receptor Chimeric Proteins. Proc. Natl. Acad. Sci. USA 1995, 92, 10457–10461. [Google Scholar] [CrossRef]
- Pierce, E.A.; Avery, R.L.; Foley, E.D.; Aiello, L.P.; Smith, L.E. Vascular Endothelial Growth Factor/Vascular Permeability Factor Expression in a Mouse Model of Retinal Neovascularization. Proc. Natl. Acad. Sci. USA 1995, 92, 905–909. [Google Scholar] [CrossRef]
- Chen, J.; Connor, K.M.; Aderman, C.M.; Willett, K.L.; Aspegren, O.P.; Smith, L.E. Suppression of Retinal Neovascularization by Erythropoietin Sirna in a Mouse Model of Proliferative Retinopathy. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1329–1335. [Google Scholar] [CrossRef]
- Nakamura, S.; Imai, S.; Ogishima, H.; Tsuruma, K.; Shimazawa, M.; Hara, H. Morphological and Functional Changes in the Retina after Chronic Oxygen-Induced Retinopathy. PLoS ONE 2012, 7, e32167. [Google Scholar] [CrossRef]
- Boeck, M.; Thien, A.; Wolf, J.; Hagemeyer, N.; Laich, Y.; Yusuf, D.; Backofen, R.; Zhang, P.; Boneva, S.; Stahl, A.; et al. Temporospatial Distribution and Transcriptional Profile of Retinal Microglia in the Oxygen-Induced Retinopathy Mouse Model. Glia 2020, 68, 1859–1873. [Google Scholar] [CrossRef]
- Pitale, P.M.; Gorbatyuk, M.S. Diabetic Retinopathy: From Animal Models to Cellular Signaling. Int. J. Mol. Sci. 2022, 23, 1487. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.K.; Pham, L.N.; Zhou, J.; Spee, C.; Ryan, S.J.; Hinton, D.R. Differential Expression of Pro- and Antiangiogenic Factors in Mouse Strain-Dependent Hypoxia-Induced Retinal Neovascularization. Lab. Investig. 2005, 85, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Ritter, M.R.; Banin, E.; Moreno, S.K.; Aguilar, E.; Dorrell, M.I.; Friedlander, M. Myeloid Progenitors Differentiate into Microglia and Promote Vascular Repair in a Model of Ischemic Retinopathy. J. Clin. Investig. 2006, 116, 3266–3276. [Google Scholar] [CrossRef]
- Wallace, D.K.; Kylstra, J.A.; Phillips, S.J.; Hall, J.G. Poor Postnatal Weight Gain: A Risk Factor for Severe Retinopathy of Prematurity. J. AAPOS 2000, 4, 343–347. [Google Scholar] [CrossRef]
- Allegaert, K.; Vanhole, C.; Casteels, I.; Naulaers, G.; Debeer, A.; Cossey, V.; Devlieger, H. Perinatal Growth Characteristics and Associated Risk of Developing Threshold Retinopathy of Prematurity. J. AAPOS 2003, 7, 34–37. [Google Scholar] [CrossRef]
- Lofqvist, C.; Andersson, E.; Sigurdsson, J.; Engstrom, E.; Hard, A.L.; Niklasson, A.; Smith, L.E.; Hellstrom, A. Longitudinal Postnatal Weight and Insulin-Like Growth Factor I Measurements in the Prediction of Retinopathy of Prematurity. Arch. Ophthalmol. 2006, 124, 1711–1718. [Google Scholar] [CrossRef]
- Stahl, A.; Chen, J.; Sapieha, P.; Seaward, M.R.; Krah, N.M.; Dennison, R.J.; Favazza, T.; Bucher, F.; Lofqvist, C.; Ong, H.; et al. Postnatal Weight Gain Modifies Severity and Functional Outcome of Oxygen-Induced Proliferative Retinopathy. Am. J. Pathol. 2010, 177, 2715–2723. [Google Scholar] [CrossRef]
- Gole, G.A.; Browning, J.; Elts, S.M. The Mouse Model of Oxygen-Induced Retinopathy: A Suitable Animal Model for Angiogenesis Research. Doc. Ophthalmol. 1990, 74, 163–169. [Google Scholar] [CrossRef]
- Brands, K.H.; Hofmann, H.; Klees, E. Retrolental fibroplasia; an experimental study. Geburtshilfe Frauenheilkd. 1958, 18, 805–814. [Google Scholar]
- Ashton, N.; Blach, R. Studies on Developing Retinal Vessels VIII. Effect of Oxygen on the Retinal Vessels of the Ratling. Br. J. Ophthalmol. 1961, 45, 321–340. [Google Scholar] [CrossRef]
- Ricci, B.; Calogero, G. Oxygen-Induced Retinopathy in Newborn Rats: Effects of Prolonged Normobaric and Hyperbaric Oxygen Supplementation. Pediatrics 1988, 82, 193–198. [Google Scholar] [CrossRef]
- Penn, J.S.; Tolman, B.L.; Lowery, L.A.; Koutz, C.A. Oxygen-Induced Retinopathy in the Rat: Hemorrhages and Dysplasias May Lead to Retinal Detachment. Curr. Eye Res. 1992, 11, 939–953. [Google Scholar] [CrossRef]
- Ventresca, M.R.; Gonder, J.R.; Tanswell, A.K. Oxygen-Induced Proliferative Retinopathy in the Newborn Rat. Can. J. Ophthalmol. 1990, 25, 186–189. [Google Scholar] [PubMed]
- York, J.R.; Landers, S.; Kirby, R.S.; Arbogast, P.G.; Penn, J.S. Arterial Oxygen Fluctuation and Retinopathy of Prematurity in Very-Low-Birth-Weight Infants. J. Perinatol. 2004, 24, 82–87. [Google Scholar] [CrossRef]
- Cunningham, S.; Fleck, B.W.; Elton, R.A.; McIntosh, N. Transcutaneous Oxygen Levels in Retinopathy of Prematurity. Lancet 1995, 346, 1464–1465. [Google Scholar] [CrossRef]
- Madan, A.; Penn, J.S. Animal Models of Oxygen-Induced Retinopathy. Front. Biosci. A J. Virtual Libr. 2003, 8, d1030–d1043. [Google Scholar]
- Grossniklaus, H.E.; Kang, S.J.; Berglin, L. Animal Models of Choroidal and Retinal Neovascularization. Progress. Retin. Eye Res. 2010, 29, 500–519. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Favazza, T.L.; Baglieri, A.M.; Benador, I.Y.; Noonan, E.R.; Fulton, A.B.; Hansen, R.M.; Iuvone, P.M.; Akula, J.D. The Rat with Oxygen-Induced Retinopathy Is Myopic with Low Retinal Dopamine. Investig. Ophthalmol. Vis. Sci. 2013, 54, 8275–8284. [Google Scholar] [CrossRef]
- Hay, W.W., Jr.; Rozance, P.J. Neonatal Hyperglycemia-Causes, Treatments, and Cautions. J. Pediatr. 2018, 200, 6–8. [Google Scholar] [CrossRef]
- Cornblath, M.; Schwartz, R. Disorders of Carbohydrate Metabolism in Infancy. Major. Probl. Clin. Pediatr. 1976, 3, 1–483. [Google Scholar] [CrossRef] [PubMed]
- Cowett, R.M.; Rapoza, R.E.; Gelardi, N.L. The Contribution of Glucose to Neonatal Glucose Homeostasis in the Lamb. Metabolism 1998, 47, 1239–1244. [Google Scholar] [CrossRef]
- Mitanchez-Mokhtari, D.; Lahlou, N.; Kieffer, F.; Magny, J.F.; Roger, M.; Voyer, M. Both Relative Insulin Resistance and Defective Islet Beta-Cell Processing of Proinsulin Are Responsible for Transient Hyperglycemia in Extremely Preterm Infants. Pediatrics 2004, 113, 537–541. [Google Scholar] [CrossRef]
- Cowett, R.M.; Oh, W.; Schwartz, R. Persistent Glucose Production During Glucose Infusion in the Neonate. J. Clin. Investig. 1983, 71, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Sunehag, A.; Gustafsson, J.; Ewald, U. Very Immature Infants (<or = 30 Wk) Respond to Glucose Infusion with Incomplete Suppression of Glucose Production. Pediatr. Res. 1994, 36, 550–555. [Google Scholar] [CrossRef]
- Au, S.C.; Tang, S.M.; Rong, S.S.; Chen, L.J.; Yam, J.C. Association between Hyperglycemia and Retinopathy of Prematurity: A Systemic Review and Meta-Analysis. Sci. Rep. 2015, 5, 9091. [Google Scholar] [CrossRef] [PubMed]
- Mohsen, L.; Abou-Alam, M.; El-Dib, M.; Labib, M.; Elsada, M.; Aly, H. A Prospective Study on Hyperglycemia and Retinopathy of Prematurity. J. Perinatol. 2014, 34, 453–457. [Google Scholar] [CrossRef]
- Fu, Z.; Yan, W.; Chen, C.T.; Nilsson, A.K.; Bull, E.; Allen, W.; Yang, J.; Ko, M.; SanGiovanni, J.P.; Akula, J.D.; et al. Omega-3/Omega-6 Long-Chain Fatty Acid Imbalance in Phase I Retinopathy of Prematurity. Nutrients 2022, 14, 1333. [Google Scholar] [CrossRef] [PubMed]
- Ahmadpour-Kacho, M.; Motlagh, A.J.; Rasoulinejad, S.A.; Jahangir, T.; Bijani, A.; Pasha, Y.Z. Correlation between Hyperglycemia and Retinopathy of Prematurity. Pediatr. Int. 2014, 56, 726–730. [Google Scholar] [CrossRef]
- Phelps, D.L.; Rosenbaum, A.L. Effects of Marginal Hypoxemia on Recovery from Oxygen-Induced Retinopathy in the Kitten Model. Pediatrics 1984, 73, 1–6. [Google Scholar] [CrossRef]
- Kimura, T.; Chen, C.H.; Patz, A. Light and Electron Microscopic Studies of Intravitreal Proliferative Tissues in Human and Puppy Eyes (Author’s Transl). Nippon. Ganka Gakkai Zasshi 1979, 83, 255–265. [Google Scholar]
- McLeod, D.S.; Crone, S.N.; Lutty, G.A. Vasoproliferation in the Neonatal Dog Model of Oxygen-Induced Retinopathy. Investig. Ophthalmol. Vis. Sci. 1996, 37, 1322–1333. [Google Scholar]
- McLeod, D.S.; Lutty, G.A. Targeting Vegf in Canine Oxygen-Induced Retinopathy—A Model for Human Retinopathy of Prematurity. Eye Brain 2016, 8, 55–65. [Google Scholar] [CrossRef]
- Rezzola, S.; Paganini, G.; Semeraro, F.; Presta, M.; Tobia, C. Zebrafish (Danio rerio) Embryo as a Platform for the Identification of Novel Angiogenesis Inhibitors of Retinal Vascular Diseases. Biochim. Biophys. Acta 2016, 1862, 1291–1296. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, Y.; Cederlund, M.L.; Cottell, D.C.; Bill, B.R.; Ekker, S.C.; Torres-Vazquez, J.; Weinstein, B.M.; Hyde, D.R.; Vihtelic, T.S.; Kennedy, B.N. Genetic Determinants of Hyaloid and Retinal Vasculature in Zebrafish. BMC Dev. Biol. 2007, 7, 114. [Google Scholar] [CrossRef]
- Weinstein, B.M.; Stemple, D.L.; Driever, W.; Fishman, M.C. Gridlock, a Localized Heritable Vascular Patterning Defect in the Zebrafish. Nat. Med. 1995, 1, 1143–1147. [Google Scholar] [CrossRef]
- Cao, Z.; Jensen, L.D.; Rouhi, P.; Hosaka, K.; Lanne, T.; Steffensen, J.F.; Wahlberg, E.; Cao, Y. Hypoxia-Induced Retinopathy Model in Adult Zebrafish. Nat. Protoc. 2010, 5, 1903–1910. [Google Scholar] [CrossRef]
- Wu, Y.C.; Chang, C.Y.; Kao, A.; Hsi, B.; Lee, S.H.; Chen, Y.H.; Wang, I.J. Hypoxia-Induced Retinal Neovascularization in Zebrafish Embryos: A Potential Model of Retinopathy of Prematurity. PLoS ONE 2015, 10, e0126750. [Google Scholar] [CrossRef] [PubMed]
- Stahl, A.; Connor, K.M.; Sapieha, P.; Willett, K.L.; Krah, N.M.; Dennison, R.J.; Chen, J.; Guerin, K.I.; Smith, L.E. Computer-Aided Quantification of Retinal Neovascularization. Angiogenesis 2009, 12, 297–301. [Google Scholar] [CrossRef]
- Harman, J.C.; Pivodic, A.; Nilsson, A.K.; Boeck, M.; Yagi, H.; Neilsen, K.; Ko, M.; Yang, J.; Kinter, M.; Hellstrom, A.; et al. Postnatal Hyperglycemia Alters Amino Acid Profile in Retinas (Model of Phase I Rop). Iscience 2023, 26, 108021. [Google Scholar] [CrossRef]
- Kremer, I.; Kissun, R.; Nissenkorn, I.; Ben-Sira, I.; Garner, A. Oxygen-Induced Retinopathy in Newborn Kittens. A Model for Ischemic Vasoproliferative Retinopathy. Investig. Ophthalmol. Vis. Sci. 1987, 28, 126–130. [Google Scholar]
- McLeod, D.S.; D’Anna, S.A.; Lutty, G.A. Clinical and Histopathologic Features of Canine Oxygen-Induced Proliferative Retinopathy. Investig. Ophthalmol. Vis. Sci. 1998, 39, 1918–1932. [Google Scholar]
- Liegl, R.; Priglinger, C.; Ohlmann, A. Induction and Readout of Oxygen-Induced Retinopathy. Methods Mol. Biol. 2019, 1834, 179–191. [Google Scholar] [CrossRef]
- Xiao, S.; Bucher, F.; Wu, Y.; Rokem, A.; Lee, C.S.; Marra, K.V.; Fallon, R.; Diaz-Aguilar, S.; Aguilar, E.; Friedlander, M.; et al. Fully Automated, Deep Learning Segmentation of Oxygen-Induced Retinopathy Images. JCI Insight 2017, 2, e97585. [Google Scholar] [CrossRef]
- Mazzaferri, J.; Larrivée, B.; Cakir, B.; Sapieha, P.; Costantino, S. A Machine Learning Approach for Automated Assessment of Retinal Vasculature in the Oxygen Induced Retinopathy Model. Sci. Rep. 2018, 8, 3916. [Google Scholar] [CrossRef] [PubMed]
- Mezu-Ndubuisi, O.J. In Vivo Angiography Quantifies Oxygen-Induced Retinopathy Vascular Recovery. Optom. Vis. Sci. 2016, 93, 1268–1279. [Google Scholar] [CrossRef] [PubMed]
- Marra, K.V.; Chen, J.S.; Robles-Holmes, H.K.; Miller, J.; Wei, G.; Aguilar, E.; Ideguchi, Y.; Ly, K.B.; Prenner, S.; Erdogmus, D.; et al. Development of a Semi-Automated Computer-Based Tool for the Quantification of Vascular Tortuosity in the Murine Retina. Ophthalmol. Sci. 2024, 4, 100439. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Marra, K.V.; Robles-Holmes, H.K.; Ly, K.B.; Miller, J.; Wei, G.; Aguilar, E.; Bucher, F.; Ideguchi, Y.; Coyner, A.S.; et al. Applications of Deep Learning: Automated Assessment of Vascular Tortuosity in Mouse Models of Oxygen-Induced Retinopathy. Ophthalmol. Sci. 2024, 4, 100338. [Google Scholar] [CrossRef] [PubMed]
- Vahatupa, M.; Prince, S.; Vataja, S.; Mertimo, T.; Kataja, M.; Kinnunen, K.; Marjomaki, V.; Uusitalo, H.; Komatsu, M.; Jarvinen, T.A.; et al. Lack of R-Ras Leads to Increased Vascular Permeability in Ischemic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4898–4909. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Stahl, A.; Hellstrom, A.; Smith, L.E. Current Update on Retinopathy of Prematurity: Screening and Treatment. Curr. Opin. Pediatr. 2011, 23, 173–178. [Google Scholar] [CrossRef]
- Fulton, A.B.; Akula, J.D.; Mocko, J.A.; Hansen, R.M.; Benador, I.Y.; Beck, S.C.; Fahl, E.; Seeliger, M.W.; Moskowitz, A.; Harris, M.E. Retinal Degenerative and Hypoxic Ischemic Disease. Doc. Ophthalmol. 2009, 118, 55–61. [Google Scholar] [CrossRef]
- Xu, Q.; Qaum, T.; Adamis, A.P. Sensitive Blood-Retinal Barrier Breakdown Quantitation Using Evans Blue. Investig. Ophthalmol. Vis. Sci. 2001, 42, 789–794. [Google Scholar]
- Comin, C.H.; Tsirukis, D.I.; Sun, Y.; Xu, X. Quantification of Retinal Blood Leakage in Fundus Fluorescein Angiography in a Retinal Angiogenesis Model. Sci. Rep. 2021, 11, 19903. [Google Scholar] [CrossRef] [PubMed]
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and Developmental Control of O2 Homeostasis by Hypoxia-Inducible Factor 1 Alpha. Genes Dev. 1998, 12, 149–162. [Google Scholar] [CrossRef]
- Weidemann, A.; Krohne, T.U.; Aguilar, E.; Kurihara, T.; Takeda, N.; Dorrell, M.I.; Simon, M.C.; Haase, V.H.; Friedlander, M.; Johnson, R.S. Astrocyte Hypoxic Response Is Essential for Pathological but Not Developmental Angiogenesis of the Retina. Glia 2010, 58, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.T.; Scholz, C.C. The Effect of Hif on Metabolism and Immunity. Nat. Rev. Nephrol. 2022, 18, 573–587. [Google Scholar] [CrossRef]
- Hughes, J.M.; Groot, A.J.; van der Groep, P.; Sersansie, R.; Vooijs, M.; van Diest, P.J.; Van Noorden, C.J.; Schlingemann, R.O.; Klaassen, I. Active Hif-1 in the Normal Human Retina. J. Histochem. Cytochem. 2010, 58, 247–254. [Google Scholar] [CrossRef]
- Ozaki, H.; Yu, A.Y.; Della, N.; Ozaki, K.; Luna, J.D.; Yamada, H.; Hackett, S.F.; Okamoto, N.; Zack, D.J.; Semenza, G.L.; et al. Hypoxia Inducible Factor-1alpha Is Increased in Ischemic Retina: Temporal and Spatial Correlation with Vegf Expression. Investig. Ophthalmol. Vis. Sci. 1999, 40, 182–189. [Google Scholar]
- Hoppe, G.; Bolok, Y.; McCollum, L.; Zhang, J.; Sears, J.E. Rank Order of Small Molecule Induced Hypoxiamimesis to Prevent Retinopathy of Prematurity. Front. Cell Dev. Biol. 2020, 8, 488. [Google Scholar] [CrossRef]
- Sears, J.E.; Hoppe, G.; Ebrahem, Q.; Anand-Apte, B. Prolyl Hydroxylase Inhibition During Hyperoxia Prevents Oxygen-Induced Retinopathy. Proc. Natl. Acad. Sci. USA 2008, 105, 19898–19903. [Google Scholar] [CrossRef]
- Lee, D.; Miwa, Y.; Wu, J.; Shoda, C.; Jeong, H.; Kawagishi, H.; Tsubota, K.; Kurihara, T. A Fairy Chemical Suppresses Retinal Angiogenesis as a Hif Inhibitor. Biomolecules 2020, 10, 1405. [Google Scholar] [CrossRef]
- Jiang, J.; Xia, X.B.; Xu, H.Z.; Xiong, Y.; Song, W.T.; Xiong, S.Q.; Li, Y. Inhibitory Effect of Interfering RNA Targeting Hif-1alpha and Vegf on Retinal Neovascularization in the Mouse. [Zhonghua Yan Ke Za Zhi] Chin. J. Ophthalmol. 2008, 44, 921–928. [Google Scholar]
- Miwa, Y.; Hoshino, Y.; Shoda, C.; Jiang, X.; Tsubota, K.; Kurihara, T. Pharmacological Hif Inhibition Prevents Retinal Neovascularization with Improved Visual Function in a Murine Oxygen-Induced Retinopathy Model. Neurochem. Int. 2019, 128, 21–31. [Google Scholar] [CrossRef]
- Ferrara, N.; Carver-Moore, K.; Chen, H.; Dowd, M.; Lu, L.; O’Shea, K.S.; Powell-Braxton, L.; Hillan, K.J.; Moore, M.W. Heterozygous Embryonic Lethality Induced by Targeted Inactivation of the Vegf Gene. Nature 1996, 380, 439–442. [Google Scholar] [CrossRef]
- Shalaby, F.; Rossant, J.; Yamaguchi, T.P.; Gertsenstein, M.; Wu, X.F.; Breitman, M.L.; Schuh, A.C. Failure of Blood-Island Formation and Vasculogenesis in Flk-1-Deficient Mice. Nature 1995, 376, 62–66. [Google Scholar] [CrossRef]
- Alon, T.; Hemo, I.; Itin, A.; Pe’er, J.; Stone, J.; Keshet, E. Vascular Endothelial Growth Factor Acts as a Survival Factor for Newly Formed Retinal Vessels and Has Implications for Retinopathy of Prematurity. Nat. Med. 1995, 1, 1024–1028. [Google Scholar] [CrossRef]
- McCloskey, M.; Wang, H.; Jiang, Y.; Smith, G.W.; Strange, J.; Hartnett, M.E. Anti-Vegf Antibody Leads to Later Atypical Intravitreous Neovascularization and Activation of Angiogenic Pathways in a Rat Model of Retinopathy of Prematurity. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2020–2026. [Google Scholar] [CrossRef]
- Wang, H.; Smith, G.W.; Yang, Z.; Jiang, Y.; McCloskey, M.; Greenberg, K.; Geisen, P.; Culp, W.D.; Flannery, J.; Kafri, T.; et al. Short Hairpin RNA-Mediated Knockdown of Vegfa in Müller Cells Reduces Intravitreal Neovascularization in a Rat Model of Retinopathy of Prematurity. Am. J. Pathol. 2013, 183, 964–974. [Google Scholar] [CrossRef]
- Ichiyama, Y.; Matsumoto, R.; Obata, S.; Sawada, O.; Saishin, Y.; Kakinoki, M.; Sawada, T.; Ohji, M. Assessment of Mouse Vegf Neutralization by Ranibizumab and Aflibercept. PLoS ONE 2022, 17, e0278951. [Google Scholar] [CrossRef]
- Rabinowitz, R.; Priel, A.; Rosner, M.; Pri-Chen, S.; Spierer, A. Avastin Treatment Reduces Retinal Neovascularization in a Mouse Model of Retinopathy of Prematurity. Curr. Eye Res. 2012, 37, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Ristori, C.; Filippi, L.; Dal Monte, M.; Martini, D.; Cammalleri, M.; Fortunato, P.; la Marca, G.; Fiorini, P.; Bagnoli, P. Role of the Adrenergic System in a Mouse Model of Oxygen-Induced Retinopathy: Antiangiogenic Effects of Β-Adrenoreceptor Blockade. Investig. Ophthalmol. Vis. Sci. 2011, 52, 155–170. [Google Scholar] [CrossRef]
- Dal Monte, M.; Martini, D.; Latina, V.; Pavan, B.; Filippi, L.; Bagnoli, P. Beta-Adrenoreceptor Agonism Influences Retinal Responses to Hypoxia in a Model of Retinopathy of Prematurity. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2181–2192. [Google Scholar] [CrossRef]
- Chen, J.; Joyal, J.S.; Hatton, C.J.; Juan, A.M.; Pei, D.T.; Hurst, C.G.; Xu, D.; Stahl, A.; Hellstrom, A.; Smith, L.E. Propranolol Inhibition of Beta-Adrenergic Receptor Does Not Suppress Pathologic Neovascularization in Oxygen-Induced Retinopathy. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2968–2977. [Google Scholar] [CrossRef]
- Miller, J.W.; Adamis, A.P.; Shima, D.T.; D’Amore, P.A.; Moulton, R.S.; O’Reilly, M.S.; Folkman, J.; Dvorak, H.F.; Brown, L.F.; Berse, B.; et al. Vascular Endothelial Growth Factor/Vascular Permeability Factor Is Temporally and Spatially Correlated with Ocular Angiogenesis in a Primate Mo`del. Am. J. Pathol. 1994, 145, 574–584. [Google Scholar] [PubMed]
- Shima, D.T.; Gougos, A.; Miller, J.W.; Tolentino, M.; Robinson, G.; Adamis, A.P.; D’Amore, P.A. Cloning and mRNA Expression of Vascular Endothelial Growth Factor in Ischemic Retinas of Macaca Fascicularis. Investig. Ophthalmol. Vis. Sci. 1996, 37, 1334–1340. [Google Scholar]
- Adamis, A.P.; Shima, D.T.; Tolentino, M.J.; Gragoudas, E.S.; Ferrara, N.; Folkman, J.; D’Amore, P.A.; Miller, J.W. Inhibition of Vascular Endothelial Growth Factor Prevents Retinal Ischemia-Associated Iris Neovascularization in a Nonhuman Primate. Arch. Ophthalmol. 1996, 114, 66–71. [Google Scholar] [CrossRef]
- Krzystolik, M.G.; Afshari, M.A.; Adamis, A.P.; Gaudreault, J.; Gragoudas, E.S.; Michaud, N.A.; Li, W.; Connolly, E.; O’Neill, C.A.; Miller, J.W. Prevention of Experimental Choroidal Neovascularization with Intravitreal Anti-Vascular Endothelial Growth Factor Antibody Fragment. Arch. Ophthalmol. 2002, 120, 338–346. [Google Scholar] [CrossRef]
- Wallace, D.K.; Dean, T.W.; Hartnett, M.E.; Kong, L.; Smith, L.E.; Hubbard, G.B.; McGregor, M.L.; Jordan, C.O.; Mantagos, I.S.; Bell, E.F.; et al. A Dosing Study of Bevacizumab for Retinopathy of Prematurity: Late Recurrences and Additional Treatments. Ophthalmology 2018, 125, 1961–1966. [Google Scholar] [CrossRef]
- Stahl, A.; Lepore, D.; Fielder, A.; Fleck, B.; Reynolds, J.D.; Chiang, M.F.; Li, J.; Liew, M.; Maier, R.; Zhu, Q.; et al. Ranibizumab Versus Laser Therapy for the Treatment of Very Low Birthweight Infants with Retinopathy of Prematurity (Rainbow): An Open-Label Randomised Controlled Trial. Lancet 2019, 394, 1551–1559. [Google Scholar] [CrossRef]
- McColm, J.R.; Geisen, P.; Hartnett, M.E. VEGF Isoforms and Their Expression after a Single Episode of Hypoxia or Repeated Fluctuations between Hyperoxia and Hypoxia: Relevance to Clinical Rop. Mol. Vis. 2004, 10, 512–520. [Google Scholar] [PubMed]
- Budd, S.J.; Thompson, H.; Hartnett, M.E. Association of Retinal Vascular Endothelial Growth Factor with Avascular Retina in a Rat Model of Retinopathy of Prematurity. Arch. Ophthalmol. 2010, 128, 1014–1021. [Google Scholar] [CrossRef]
- Werdich, X.Q.; McCollum, G.W.; Rajaratnam, V.S.; Penn, J.S. Variable Oxygen and Retinal Vegf Levels: Correlation with Incidence and Severity of Pathology in a Rat Model of Oxygen-Induced Retinopathy. Exp. Eye Res. 2004, 79, 623–630. [Google Scholar] [CrossRef]
- Bai, Y.; Ma, J.X.; Guo, J.; Wang, J.; Zhu, M.; Chen, Y.; Le, Y.Z. Müller Cell-Derived Vegf Is a Significant Contributor to Retinal Neovascularization. J. Pathol. 2009, 219, 446–454. [Google Scholar] [CrossRef]
- Watanabe, D.; Suzuma, K.; Matsui, S.; Kurimoto, M.; Kiryu, J.; Kita, M.; Suzuma, I.; Ohashi, H.; Ojima, T.; Murakami, T.; et al. Erythropoietin as a Retinal Angiogenic Factor in Proliferative Diabetic Retinopathy. N. Engl. J. Med. 2005, 353, 782–792. [Google Scholar] [CrossRef]
- Wong-Riley, M.T. Energy Metabolism of the Visual System. Eye Brain 2010, 2, 99–116. [Google Scholar] [CrossRef]
- Gospe, S.M., III; Baker, S.A.; Arshavsky, V.Y. Facilitative Glucose Transporter Glut1 Is Actively Excluded from Rod Outer Segments. J. Cell Sci. 2010, 123, 3639–3644. [Google Scholar] [CrossRef]
- Klepper, J. Glucose Transporter Deficiency Syndrome (Glut1ds) and the Ketogenic Diet. Epilepsia 2008, 49 (Suppl. S8), 46–49. [Google Scholar] [CrossRef]
- Fu, Z.; Kern, T.S.; Hellström, A.; Smith, L.E.H. Fatty Acid Oxidation and Photoreceptor Metabolic Needs. J. Lipid Res. 2021, 62, 100035. [Google Scholar] [CrossRef]
- Joyal, J.S.; Sun, Y.; Gantner, M.L.; Shao, Z.; Evans, L.P.; Saba, N.; Fredrick, T.; Burnim, S.; Kim, J.S.; Patel, G.; et al. Retinal Lipid and Glucose Metabolism Dictates Angiogenesis through the Lipid Sensor Ffar1. Nat. Med. 2016, 22, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Tyni, T.; Paetau, A.; Strauss, A.W.; Middleton, B.; Kivelä, T. Mitochondrial Fatty Acid Beta-Oxidation in the Human Eye and Brain: Implications for the Retinopathy of Long-Chain 3-Hydroxyacyl-Coa Dehydrogenase Deficiency. Pediatr. Res. 2004, 56, 744–750. [Google Scholar] [CrossRef]
- Fletcher, A.L.; Pennesi, M.E.; Harding, C.O.; Weleber, R.G.; Gillingham, M.B. Observations Regarding Retinopathy in Mitochondrial Trifunctional Protein Deficiencies. Mol. Genet. Metab. 2012, 106, 18–24. [Google Scholar] [CrossRef]
- Gillingham, M.B.; Weleber, R.G.; Neuringer, M.; Connor, W.E.; Mills, M.; van Calcar, S.; Ver Hoeve, J.; Wolff, J.; Harding, C.O. Effect of Optimal Dietary Therapy Upon Visual Function in Children with Long-Chain 3-Hydroxyacyl Coa Dehydrogenase and Trifunctional Protein Deficiency. Mol. Genet. Metab. 2005, 86, 124–133. [Google Scholar] [CrossRef]
- Singh, C. Systems Levels Analysis of Lipid Metabolism in Oxygen-Induced Retinopathy. bioRxiv 2023. [Google Scholar] [CrossRef]
- Díaz-Coránguez, M.; Ramos, C.; Antonetti, D.A. The Inner Blood-Retinal Barrier: Cellular Basis and Development. Vision. Res. 2017, 139, 123–137. [Google Scholar] [CrossRef]
- Fu, Z.; Lofqvist, C.A.; Shao, Z.; Sun, Y.; Joyal, J.S.; Hurst, C.G.; Cui, R.Z.; Evans, L.P.; Tian, K.; SanGiovanni, J.P.; et al. Dietary Ω-3 Polyunsaturated Fatty Acids Decrease Retinal Neovascularization by Adipose-Endoplasmic Reticulum Stress Reduction to Increase Adiponectin. Am. J. Clin. Nutr. 2015, 101, 879–888. [Google Scholar] [CrossRef]
- Gong, Y.; Fu, Z.; Edin, M.L.; Liu, C.H.; Wang, Z.; Shao, Z.; Fredrick, T.W.; Saba, N.J.; Morss, P.C.; Burnim, S.B.; et al. Cytochrome P450 Oxidase 2c Inhibition Adds to Ω-3 Long-Chain Polyunsaturated Fatty Acids Protection against Retinal and Choroidal Neovascularization. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1919–1927. [Google Scholar] [CrossRef]
- Stahl, A.; Sapieha, P.; Connor, K.M.; Sangiovanni, J.P.; Chen, J.; Aderman, C.M.; Willett, K.L.; Krah, N.M.; Dennison, R.J.; Seaward, M.R.; et al. Short Communication: Ppar Gamma Mediates a Direct Antiangiogenic Effect of Omega 3-Pufas in Proliferative Retinopathy. Circ. Res. 2010, 107, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Inague, A.; Alecrim, L.C.; Monteiro, J.S.; Yoshinaga, M.Y.; Setubal, J.C.; Miyamoto, S.; Giordano, R.J. Oxygen-Induced Pathological Angiogenesis Promotes Intense Lipid Synthesis and Remodeling in the Retina. Iscience 2023, 26, 106777. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.V.; Basu, S.K.; Qaladize, B.; Grambergs, R.; Rotstein, N.P.; Mandal, N. Sphingolipids as Critical Players in Retinal Physiology and Pathology. J. Lipid Res. 2021, 62, 100037. [Google Scholar] [CrossRef]
- Lee, M.J.; Thangada, S.; Claffey, K.P.; Ancellin, N.; Liu, C.H.; Kluk, M.; Volpi, M.; Sha’afi, R.I.; Hla, T. Vascular Endothelial Cell Adherens Junction Assembly and Morphogenesis Induced by Sphingosine-1-Phosphate. Cell 1999, 99, 301–312. [Google Scholar] [CrossRef]
- Paik, J.H.; Chae, S.; Lee, M.J.; Thangada, S.; Hla, T. Sphingosine 1-Phosphate-Induced Endothelial Cell Migration Requires the Expression of Edg-1 and Edg-3 Receptors and Rho-Dependent Activation of Alpha Vbeta3- and Beta1-Containing Integrins. J. Biol. Chem. 2001, 276, 11830–11837. [Google Scholar] [CrossRef]
- Paik, J.H.; Skoura, A.; Chae, S.S.; Cowan, A.E.; Han, D.K.; Proia, R.L.; Hla, T. Sphingosine 1-Phosphate Receptor Regulation of N-Cadherin Mediates Vascular Stabilization. Genes. Dev. 2004, 18, 2392–2403. [Google Scholar] [CrossRef] [PubMed]
- Niaudet, C.; Jung, B.; Kuo, A.; Swendeman, S.; Bull, E.; Seno, T.; Crocker, R.; Fu, Z.; Smith, L.E.H.; Hla, T. Therapeutic Activation of Endothelial Sphingosine-1-Phosphate Receptor 1 by Chaperone-Bound S1p Suppresses Proliferative Retinal Neovascularization. EMBO Mol. Med. 2023, 15, e16645. [Google Scholar] [CrossRef]
- Skoura, A.; Sanchez, T.; Claffey, K.; Mandala, S.M.; Proia, R.L.; Hla, T. Essential Role of Sphingosine 1-Phosphate Receptor 2 in Pathological Angiogenesis of the Mouse Retina. J. Clin. Investig. 2007, 117, 2506–2516. [Google Scholar] [CrossRef]
- Eresch, J.; Stumpf, M.; Koch, A.; Vutukuri, R.; Ferreirós, N.; Schreiber, Y.; Schröder, K.; Devraj, K.; Popp, R.; Huwiler, A.; et al. Sphingosine Kinase 2 Modulates Retinal Neovascularization in the Mouse Model of Oxygen-Induced Retinopathy. Investig. Ophthalmol. Vis. Sci. 2018, 59, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Ancellin, N.; Colmont, C.; Su, J.; Li, Q.; Mittereder, N.; Chae, S.S.; Stefansson, S.; Liau, G.; Hla, T. Extracellular Export of Sphingosine Kinase-1 Enzyme. Sphingosine 1-Phosphate Generation and the Induction of Angiogenic Vascular Maturation. J. Biol. Chem. 2002, 277, 6667–6675. [Google Scholar] [CrossRef] [PubMed]
- Draoui, N.; de Zeeuw, P.; Carmeliet, P. Angiogenesis Revisited from a Metabolic Perspective: Role and Therapeutic Implications of Endothelial Cell Metabolism. Open Biol. 2017, 7, 170219. [Google Scholar] [CrossRef] [PubMed]
- Teuwen, L.A.; Geldhof, V.; Carmeliet, P. How Glucose, Glutamine and Fatty Acid Metabolism Shape Blood and Lymph Vessel Development. Dev. Biol. 2019, 447, 90–102. [Google Scholar] [CrossRef]
- Selvam, S.; Kumar, T.; Fruttiger, M. Retinal Vasculature Development in Health and Disease. Prog. Retin. Eye Res. 2018, 63, 1–19. [Google Scholar] [CrossRef]
- Huang, H.; Vandekeere, S.; Kalucka, J.; Bierhansl, L.; Zecchin, A.; Brüning, U.; Visnagri, A.; Yuldasheva, N.; Goveia, J.; Cruys, B.; et al. Role of Glutamine and Interlinked Asparagine Metabolism in Vessel Formation. EMBO J. 2017, 36, 2334–2352. [Google Scholar] [CrossRef]
- Amelio, I.; Cutruzzolá, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and Glycine Metabolism in Cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Fan, J.; Venneti, S.; Cross, J.R.; Takagi, T.; Bhinder, B.; Djaballah, H.; Kanai, M.; Cheng, E.H.; Judkins, A.R.; et al. Asparagine Plays a Critical Role in Regulating Cellular Adaptation to Glutamine Depletion. Mol. Cell 2014, 56, 205–218. [Google Scholar] [CrossRef]
- Kim, B.; Li, J.; Jang, C.; Arany, Z. Glutamine Fuels Proliferation but Not Migration of Endothelial Cells. EMBO J. 2017, 36, 2321–2333. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Cheng, T. Q’s Next: The Diverse Functions of Glutamine in Metabolism, Cell Biology and Cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine Supports Pancreatic Cancer Growth through a Kras-Regulated Metabolic Pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Parri, M.; Chiarugi, P. Redox Molecular Machines Involved in Tumor Progression. Antioxid. Redox Signal 2013, 19, 1828–1845. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Hui, S.; Ghergurovich, J.M.; Fan, J.; Intlekofer, A.M.; White, R.M.; Rabinowitz, J.D.; Thompson, C.B.; Zhang, J. As Extracellular Glutamine Levels Decline, Asparagine Becomes an Essential Amino Acid. Cell Metab. 2018, 27, 428–438.e425. [Google Scholar] [CrossRef]
- Zhou, Y.; Tan, W.; Zou, J.; Cao, J.; Huang, Q.; Jiang, B.; Yoshida, S.; Li, Y. Metabolomics Analyses of Mouse Retinas in Oxygen-Induced Retinopathy. Investig. Ophthalmol. Vis. Sci. 2021, 62, 9. [Google Scholar] [CrossRef]
- Lee, W.J.; Hawkins, R.A.; Viña, J.R.; Peterson, D.R. Glutamine Transport by the Blood-Brain Barrier: A Possible Mechanism for Nitrogen Removal. Am. J. Physiol. 1998, 274, C1101–C1107. [Google Scholar] [CrossRef]
- Nakanishi, T.; Sugawara, M.; Huang, W.; Martindale, R.G.; Leibach, F.H.; Ganapathy, M.E.; Prasad, P.D.; Ganapathy, V. Structure, Function, and Tissue Expression Pattern of Human Sn2, a Subtype of the Amino Acid Transport System N. Biochem. Biophys. Res. Commun. 2001, 281, 1343–1348. [Google Scholar] [CrossRef]
- Chen, J.; Stahl, A.; Krah, N.M.; Seaward, M.R.; Joyal, J.S.; Juan, A.M.; Hatton, C.J.; Aderman, C.M.; Dennison, R.J.; Willett, K.L.; et al. Retinal Expression of Wnt-Pathway Mediated Genes in Low-Density Lipoprotein Receptor-Related Protein 5 (Lrp5) Knockout Mice. PLoS ONE 2012, 7, e30203. [Google Scholar] [CrossRef]
- Xia, C.H.; Yablonka-Reuveni, Z.; Gong, X. Lrp5 Is Required for Vascular Development in Deeper Layers of the Retina. PLoS ONE 2010, 5, e11676. [Google Scholar] [CrossRef]
- Schafer, N.F.; Luhmann, U.F.; Feil, S.; Berger, W. Differential Gene Expression in Ndph-Knockout Mice in Retinal Development. Investig. Ophthalmol. Vis. Sci. 2009, 50, 906–916. [Google Scholar] [CrossRef]
- Ye, X.; Wang, Y.; Nathans, J. The Norrin/Frizzled4 Signaling Pathway in Retinal Vascular Development and Disease. Trends Mol. Med. 2010, 16, 417–425. [Google Scholar] [CrossRef]
- Benson, W.E. Familial Exudative Vitreoretinopathy. Trans. Am. Ophthalmol. Soc. 1995, 93, 473–521. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, C.H.; Sun, Y.; Gong, Y.; Favazza, T.L.; Morss, P.C.; Saba, N.J.; Fredrick, T.W.; He, X.; Akula, J.D.; et al. Pharmacologic Activation of Wnt Signaling by Lithium Normalizes Retinal Vasculature in a Murine Model of Familial Exudative Vitreoretinopathy. Am. J. Pathol. 2016, 186, 2588–2600. [Google Scholar] [CrossRef] [PubMed]
- Andersen, S.R.; Warburg, M. Norrie’s Disease: Congenital Bilateral Pseudotumor of the Retina with Recessive X-Chromosomal Inheritance; Preliminary Report. Arch. Ophthalmol. 1961, 66, 614–618. [Google Scholar] [CrossRef]
- Wang, Z.; Yemanyi, F.; Blomfield, A.K.; Bora, K.; Huang, S.; Liu, C.H.; Britton, W.R.; Cho, S.S.; Tomita, Y.; Fu, Z.; et al. Amino Acid Transporter Slc38a5 Regulates Developmental and Pathological Retinal Angiogenesis. Elife 2022, 11, e73105. [Google Scholar] [CrossRef]
- Hitzel, J.; Lee, E.; Zhang, Y.; Bibli, S.I.; Li, X.; Zukunft, S.; Pfluger, B.; Hu, J.; Schurmann, C.; Vasconez, A.E.; et al. Oxidized Phospholipids Regulate Amino Acid Metabolism through Mthfd2 to Facilitate Nucleotide Release in Endothelial Cells. Nat. Commun. 2018, 9, 2292. [Google Scholar] [CrossRef]
- Vandekeere, S.; Dubois, C.; Kalucka, J.; Sullivan, M.R.; García-Caballero, M.; Goveia, J.; Chen, R.; Diehl, F.F.; Bar-Lev, L.; Souffreau, J.; et al. Serine Synthesis Via Phgdh Is Essential for Heme Production in Endothelial Cells. Cell Metab. 2018, 28, 573–587.e513. [Google Scholar] [CrossRef]
- Eade, K.; Gantner, M.L.; Hostyk, J.A.; Nagasaki, T.; Giles, S.; Fallon, R.; Harkins-Perry, S.; Baldini, M.; Lim, E.W.; Scheppke, L.; et al. Serine Biosynthesis Defect Due to Haploinsufficiency of Phgdh Causes Retinal Disease. Nat. Metab. 2021, 3, 366–377. [Google Scholar] [CrossRef]
- Singh, C.; Hoppe, G.; Tran, V.; McCollum, L.; Bolok, Y.; Song, W.; Sharma, A.; Brunengraber, H.; Sears, J.E. Serine and 1-Carbon Metabolism Are Required for Hif-Mediated Protection Against Retinopathy of Prematurity. JCI Insight 2019, 4, e129398. [Google Scholar] [CrossRef]
- Tomita, Y.; Cagnone, G.; Fu, Z.; Cakir, B.; Kotoda, Y.; Asakage, M.; Wakabayashi, Y.; Hellström, A.; Joyal, J.S.; Talukdar, S.; et al. Vitreous Metabolomics Profiling of Proliferative Diabetic Retinopathy. Diabetologia 2021, 64, 70–82. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maurya, M.; Liu, C.-H.; Bora, K.; Kushwah, N.; Pavlovich, M.C.; Wang, Z.; Chen, J. Animal Models of Retinopathy of Prematurity: Advances and Metabolic Regulators. Biomedicines 2024, 12, 1937. https://doi.org/10.3390/biomedicines12091937
Maurya M, Liu C-H, Bora K, Kushwah N, Pavlovich MC, Wang Z, Chen J. Animal Models of Retinopathy of Prematurity: Advances and Metabolic Regulators. Biomedicines. 2024; 12(9):1937. https://doi.org/10.3390/biomedicines12091937
Chicago/Turabian StyleMaurya, Meenakshi, Chi-Hsiu Liu, Kiran Bora, Neetu Kushwah, Madeline C. Pavlovich, Zhongxiao Wang, and Jing Chen. 2024. "Animal Models of Retinopathy of Prematurity: Advances and Metabolic Regulators" Biomedicines 12, no. 9: 1937. https://doi.org/10.3390/biomedicines12091937