
Cellular Senescence: The Driving Force of Musculoskeletal Diseases
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cellular Senescence in Musculoskeletal Diseases
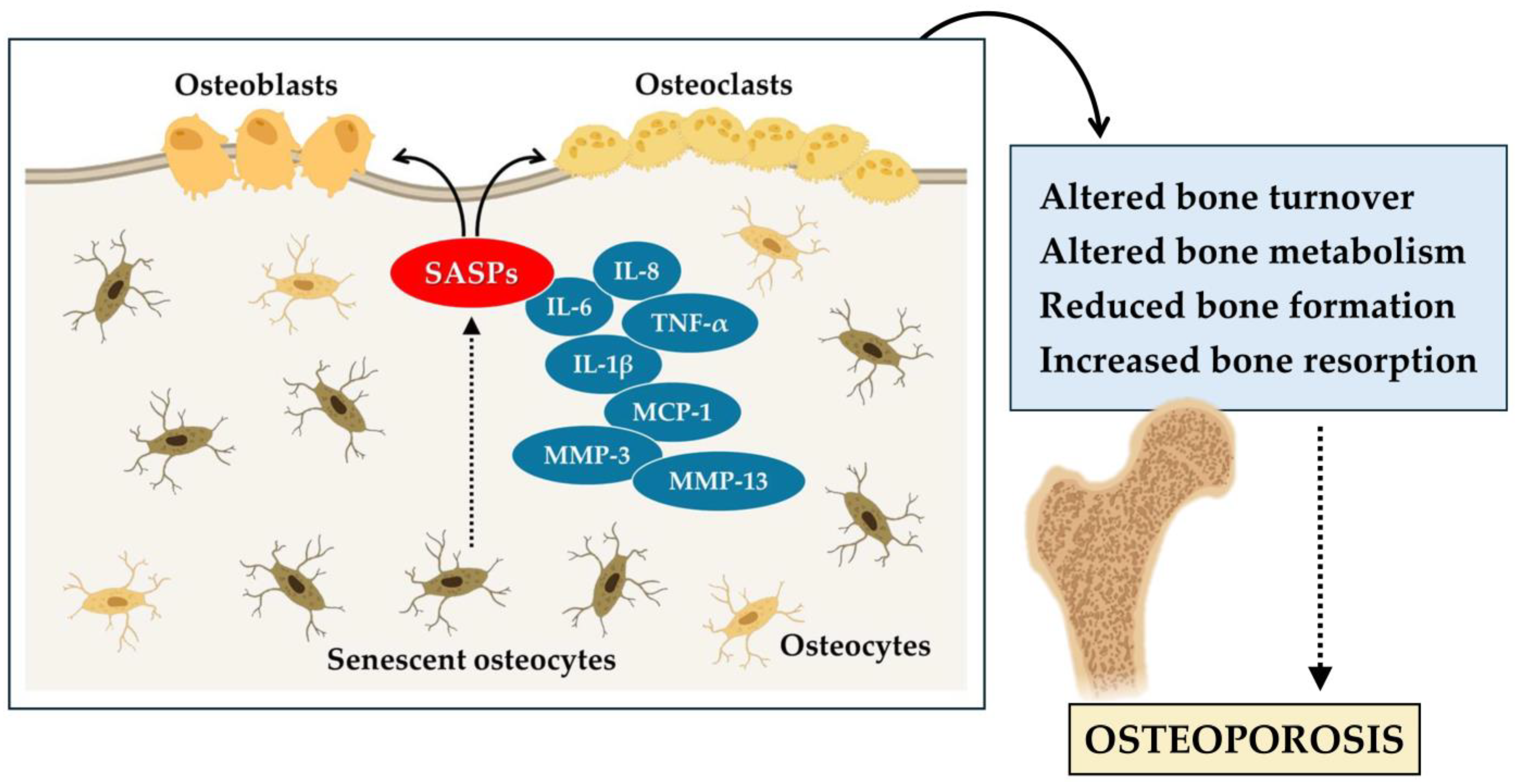
2.1. Bone Senescence in Osteoporosis
2.2. Muscle Senescence in Sarcopenia
2.3. Cartilage Senescence in Osteoarthritis
3. Anti-Senescence Strategies
3.1. Exercise
3.2. Nutrition
3.3. Senolytics and Senomorphs
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mkrtchyan, G.V.; Abdelmohsen, K.; Andreux, P.; Bagdonaite, I.; Barzilai, N.; Brunak, S.; Cabreiro, F.; de Cabo, R.; Campisi, J.; Cuervo, A.M.; et al. ARDD 2020: From aging mechanisms to interventions. Aging 2020, 12, 24484–24503. [Google Scholar] [CrossRef]
- Grote, C.; Reinhardt, D.; Zhang, M.; Wang, J. Regulatory mechanisms and clinical manifestations of musculoskeletal aging. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2019, 37, 1475–1488. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, E.; Morales-Pison, S.; Urbina, F.; Solari, A. Aging Hallmarks and the Role of Oxidative Stress. Antioxidants 2023, 12, 651. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Z.; Ren, Y.; Wang, Y.; Fang, J.; Yue, H.; Ma, S.; Guan, F. Aging and age-related diseases: From mechanisms to therapeutic strategies. Biogerontology 2021, 22, 165–187. [Google Scholar] [CrossRef] [PubMed]
- Wan, M.; Gray-Gaillard, E.F.; Elisseeff, J.H. Cellular senescence in musculoskeletal homeostasis, diseases, and regeneration. Bone Res. 2021, 9, 41. [Google Scholar] [CrossRef]
- Walston, J.D. Sarcopenia in older adults. Curr. Opin. Rheumatol. 2012, 24, 623–627. [Google Scholar] [CrossRef]
- Sözen, T.; Özışık, L.; Başaran, N.Ç. An overview and management of osteoporosis. Eur. J. Rheumatol. 2017, 4, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Huang, X.; Dou, L.; Yan, M.; Shen, T.; Tang, W.; Li, J. Aging and aging-related diseases: From molecular mechanisms to interventions and treatments. Signal Transduct. Target. Ther. 2022, 7, 391. [Google Scholar] [CrossRef]
- Englund, D.A.; Jolliffe, A.; Aversa, Z.; Zhang, X.; Sturmlechner, I.; Sakamoto, A.E.; Zeidler, J.D.; Warner, G.M.; McNinch, C.; White, T.A.; et al. p21 induces a senescence program and skeletal muscle dysfunction. Mol. Metab. 2023, 67, 101652. [Google Scholar] [CrossRef]
- Jeon, O.H.; Wilson, D.R.; Clement, C.C.; Rathod, S.; Cherry, C.; Powell, B.; Lee, Z.; Khalil, A.M.; Green, J.J.; Campisi, J.; et al. Senescence cell-associated extracellular vesicles serve as osteoarthritis disease and therapeutic markers. JCI Insight 2019, 4, e125019. [Google Scholar] [CrossRef]
- Pignolo, R.J.; Law, S.F.; Chandra, A. Bone Aging, Cellular Senescence, and Osteoporosis. JBMR Plus 2021, 5, e10488. [Google Scholar] [CrossRef]
- Chandra, A.; Rajawat, J. Skeletal Aging and Osteoporosis: Mechanisms and Therapeutics. Int. J. Mol. Sci. 2021, 22, 3553. [Google Scholar] [CrossRef] [PubMed]
- Ritschka, B.; Storer, M.; Mas, A.; Heinzmann, F.; Ortells, M.C.; Morton, J.P.; Sansom, O.J.; Zender, L.; Keyes, W.M. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017, 31, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Birch, J.; Gil, J. Senescence and the SASP: Many therapeutic avenues. Genes Dev. 2020, 34, 1565–1576. [Google Scholar] [CrossRef]
- García-Prat, L.; Martínez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodríguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef]
- Wang, T.; Huang, S.; He, C. Senescent cells: A therapeutic target for osteoporosis. Cell Prolif. 2022, 55, e13323. [Google Scholar] [CrossRef] [PubMed]
- Yılmaz, D.; Mathavan, N.; Wehrle, E.; Kuhn, G.A.; Müller, R. Mouse models of accelerated aging in musculoskeletal research for assessing frailty, sarcopenia, and osteoporosis—A review. Ageing Res. Rev. 2024, 93, 102118. [Google Scholar] [CrossRef]
- Yamakawa, H.; Kusumoto, D.; Hashimoto, H.; Yuasa, S. Stem Cell Aging in Skeletal Muscle Regeneration and Disease. Int. J. Mol. Sci. 2020, 21, 1830. [Google Scholar] [CrossRef]
- Borowik, A.K.; Lawrence, M.M.; Peelor, F.F., 3rd; Piekarz, K.M.; Crosswhite, A.; Richardson, A.; Miller, B.F.; Van Remmen, H.; Brown, J.L. Senolytic treatment does not mitigate oxidative stress-induced muscle atrophy but improves muscle force generation in CuZn superoxide dismutase knockout mice. GeroScience 2024, 46, 3219–3233. [Google Scholar] [CrossRef]
- Jeon, O.H.; Kim, C.; Laberge, R.-M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef]
- Fang, C.-L.; Liu, B.; Wan, M. “Bone-SASP” in Skeletal Aging. Calcif. Tissue Int. 2023, 113, 68–82. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Xie, W.; Li, H.; Jin, H.; Zhang, Y.; Li, Y. Cellular Senescence in Sarcopenia: Possible Mechanisms and Therapeutic Potential. Front. Cell Dev. Biol. 2021, 9, 793088. [Google Scholar] [CrossRef]
- Lotz, M.; Loeser, R.F. Effects of aging on articular cartilage homeostasis. Bone 2012, 51, 241–248. [Google Scholar] [CrossRef]
- Roberts, S.; Colombier, P.; Sowman, A.; Mennan, C.; Rölfing, J.H.D.; Guicheux, J.; Edwards, J.R. Ageing in the musculoskeletal system. Acta Orthop. 2016, 87, 15–25. [Google Scholar] [CrossRef]
- Cariati, I.; Bonanni, R.; Rinaldi, A.M.; Marini, M.; Iundusi, R.; Gasbarra, E.; Tancredi, V.; Tarantino, U. Recombinant irisin prevents cell death and mineralization defects induced by random positioning machine exposure in primary cultures of human osteoblasts: A promising strategy for the osteoporosis treatment. Front. Physiol. 2023, 14, 1107933. [Google Scholar] [CrossRef] [PubMed]
- Samsonraj, R.M.; Law, S.F.; Chandra, A.; Pignolo, R.J. An unbiased proteomics approach to identify the senescence-associated secretory phenotype of human bone marrow-derived mesenchymal stem cells. Bone Rep. 2023, 18, 101674. [Google Scholar] [CrossRef]
- Saul, D.; Khosla, S. Fracture Healing in the Setting of Endocrine Diseases, Aging, and Cellular Senescence. Endocr. Rev. 2022, 43, 984–1002. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, J.; Lin, X.; Boyce, B.F.; Zhang, H.; Xing, L. Age-associated callus senescent cells produce TGF-β1 that inhibits fracture healing in aged mice. J. Clin. Investig. 2022, 132, e148073. [Google Scholar] [CrossRef] [PubMed]
- Farr, J.N.; Kaur, J.; Doolittle, M.L.; Khosla, S. Osteocyte Cellular Senescence. Curr. Osteoporos. Rep. 2020, 18, 559–567. [Google Scholar] [CrossRef]
- Farr, J.N.; Fraser, D.G.; Wang, H.; Jaehn, K.; Ogrodnik, M.B.; Weivoda, M.M.; Drake, M.T.; Tchkonia, T.; LeBrasseur, N.K.; Kirkland, J.L.; et al. Identification of Senescent Cells in the Bone Microenvironment. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2016, 31, 1920–1929. [Google Scholar] [CrossRef]
- Ding, P.; Gao, C.; Gao, Y.; Liu, D.; Li, H.; Xu, J.; Chen, X.; Huang, Y.; Zhang, C.; Zheng, M.; et al. Osteocytes regulate senescence of bone and bone marrow. Elife 2022, 11, e81480. [Google Scholar] [CrossRef] [PubMed]
- Geng, Q.; Wang, S.; Heng, K.; Zhai, J.; Song, X.; Xia, L.; Wang, L.; Lin, Q.; Li, H.; Guo, Y. Astaxanthin attenuates irradiation-induced osteoporosis in mice by inhibiting oxidative stress, osteocyte senescence, and SASP. Food Funct. 2022, 13, 11770–11779. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Lin, B.; Deng, X.; Huang, K.; Zhang, Y.; Wang, N. VDR activation attenuates osteoblastic ferroptosis and senescence by stimulating the Nrf2/GPX4 pathway in age-related osteoporosis. Free Radic. Biol. Med. 2022, 193, 720–735. [Google Scholar] [CrossRef] [PubMed]
- Tabara, Y.; Ikezoe, T.; Yamanaka, M.; Setoh, K.; Segawa, H.; Kawaguchi, T.; Kosugi, S.; Nakayama, T.; Ichihashi, N.; Tsuboyama, T.; et al. Advanced Glycation End Product Accumulation Is Associated with Low Skeletal Muscle Mass, Weak Muscle Strength, and Reduced Bone Density: The Nagahama Study. J. Gerontol. A. Biol. Sci. Med. Sci. 2019, 74, 1446–1453. [Google Scholar] [CrossRef]
- Guo, Y.; Jia, X.; Cui, Y.; Song, Y.; Wang, S.; Geng, Y.; Li, R.; Gao, W.; Fu, D. Sirt3-mediated mitophagy regulates AGEs-induced BMSCs senescence and senile osteoporosis. Redox Biol. 2021, 41, 101915. [Google Scholar] [CrossRef] [PubMed]
- Bonanni, R.; Gino Grillo, S.; Cariati, I.; Tranquillo, L.; Iundusi, R.; Gasbarra, E.; Tancredi, V.; Tarantino, U. Osteosarcopenia and Pain: Do We Have a Way Out? Biomedicines 2023, 11, 1285. [Google Scholar] [CrossRef]
- Dungan, C.M.; Peck, B.D.; Walton, R.G.; Huang, Z.; Bamman, M.M.; Kern, P.A.; Peterson, C.A. In vivo analysis of γH2AX+ cells in skeletal muscle from aged and obese humans. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 7018–7035. [Google Scholar] [CrossRef]
- Alcalde-Estévez, E.; Asenjo-Bueno, A.; Sosa, P.; Olmos, G.; Plaza, P.; Caballero-Mora, M.Á.; Rodríguez-Puyol, D.; Ruíz-Torres, M.P.; López-Ongil, S. Endothelin-1 induces cellular senescence and fibrosis in cultured myoblasts. A potential mechanism of aging-related sarcopenia. Aging 2020, 12, 11200–11223. [Google Scholar] [CrossRef]
- Moustogiannis, A.; Philippou, A.; Taso, O.; Zevolis, E.; Pappa, M.; Chatzigeorgiou, A.; Koutsilieris, M. The Effects of Muscle Cell Aging on Myogenesis. Int. J. Mol. Sci. 2021, 22, 3721. [Google Scholar] [CrossRef]
- Yu, S.; Ren, B.; Chen, H.; Goltzman, D.; Yan, J.; Miao, D. 1,25-Dihydroxyvitamin D deficiency induces sarcopenia by inducing skeletal muscle cell senescence. Am. J. Transl. Res. 2021, 13, 12638–12649. [Google Scholar]
- Francis, T.G.; Jaka, O.; Harridge, S.D.R.; Ellison-hughes, G.M.; Lazarus, N.R. Human primary skeletal muscle-derived myoblasts and fi broblasts reveal different senescent phenotypes. JCSM Rapid Commun. 2022, 5, 226–238. [Google Scholar] [CrossRef]
- Fielding, R.A.; Atkinson, E.J.; Aversa, Z.; White, T.A.; Heeren, A.A.; Achenbach, S.J.; Mielke, M.M.; Cummings, S.R.; Pahor, M.; Leeuwenburgh, C.; et al. Associations between biomarkers of cellular senescence and physical function in humans: Observations from the lifestyle interventions for elders (LIFE) study. GeroScience 2022, 44, 2757–2770. [Google Scholar] [CrossRef]
- Fujii, Y.; Liu, L.; Yagasaki, L.; Inotsume, M.; Chiba, T.; Asahara, H. Cartilage Homeostasis and Osteoarthritis. Int. J. Mol. Sci. 2022, 23, 6316. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Z.; Li, T.; Xu, H.; Zhang, H. Senescence in osteoarthritis: From mechanism to potential treatment. Arthritis Res. Ther. 2022, 24, 174. [Google Scholar] [CrossRef] [PubMed]
- Jeon, O.H.; David, N.; Campisi, J.; Elisseeff, J.H. Senescent cells and osteoarthritis: A painful connection. J. Clin. Investig. 2018, 128, 1229–1237. [Google Scholar] [CrossRef]
- Xie, J.; Huang, Z.; Yu, X.; Zhou, L.; Pei, F. Clinical implications of macrophage dysfunction in the development of osteoarthritis of the knee. Cytokine Growth Factor Rev. 2019, 46, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, A.I.; Beekhuizen, M.; ’t Hart, M.C.; Radstake, T.R.D.J.; Dhert, W.J.A.; Saris, D.B.F.; van Osch, G.J.V.M.; Creemers, L.B. Cytokine profiles in the joint depend on pathology, but are different between synovial fluid, cartilage tissue and cultured chondrocytes. Arthritis Res. Ther. 2014, 16, 441. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Gong, W.; Shao, X.; Shi, T.; Zhang, L.; Dong, J.; Shi, Y.; Shen, S.; Qin, J.; Jiang, Q.; et al. METTL3-mediated m(6)A modification of ATG7 regulates autophagy-GATA4 axis to promote cellular senescence and osteoarthritis progression. Ann. Rheum. Dis. 2022, 81, 87–99. [Google Scholar] [CrossRef]
- Xiang, Y.; Laurent, B.; Hsu, C.-H.; Nachtergaele, S.; Lu, Z.; Sheng, W.; Xu, C.; Chen, H.; Ouyang, J.; Wang, S.; et al. RNA m(6)A methylation regulates the ultraviolet-induced DNA damage response. Nature 2017, 543, 573–576. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, L.; Wang, Y.; Li, W.; Lin, Y.; Yu, D.; Zhang, L.; Li, F.; Pan, Z. Overexpression of Sirtuin 6 suppresses cellular senescence and NF-κB mediated inflammatory responses in osteoarthritis development. Sci. Rep. 2015, 5, 17602. [Google Scholar] [CrossRef]
- Ji, M.-L.; Jiang, H.; Li, Z.; Geng, R.; Hu, J.Z.; Lin, Y.C.; Lu, J. Sirt6 attenuates chondrocyte senescence and osteoarthritis progression. Nat. Commun. 2022, 13, 7658. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Wu, B.; Hu, X.; Lu, H. Sirtuin 4 (Sirt4) downregulation contributes to chondrocyte senescence and osteoarthritis via mediating mitochondrial dysfunction. Int. J. Biol. Sci. 2024, 20, 1256–1278. [Google Scholar] [CrossRef] [PubMed]
- Zeng, N.; Yan, Z.-P.; Chen, X.-Y.; Ni, G.-X. Infrapatellar Fat Pad and Knee Osteoarthritis. Aging Dis. 2020, 11, 1317–1328. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Huebner, J.L.; Kraus, V.B.; Griffin, T.M. Effect of Aging on Adipose Tissue Inflammation in the Knee Joints of F344BN Rats. J. Gerontol. A. Biol. Sci. Med. Sci. 2016, 71, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Maleitzke, T.; Geissler, S.; Hildebrandt, A.; Fleckenstein, F.N.; Niemann, M.; Fischer, H.; Perka, C.; Duda, G.N.; Winkler, T. Source and hub of inflammation: The infrapatellar fat pad and its interactions with articular tissues during knee osteoarthritis. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2022, 40, 1492–1504. [Google Scholar] [CrossRef]
- Little-Letsinger, S.E.; Rubin, J.; Diekman, B.; Rubin, C.T.; McGrath, C.; Pagnotti, G.M.; Klett, E.L.; Styner, M. Exercise to Mend Aged-tissue Crosstalk in Bone Targeting Osteoporosis & Osteoarthritis. Semin. Cell Dev. Biol. 2022, 123, 22–35. [Google Scholar] [CrossRef]
- McGregor, N.E.; Walker, E.C.; Chan, A.S.; Poulton, I.J.; Cho, E.H.-J.; Windahl, S.H.; Sims, N.A. STAT3 Hyperactivation Due to SOCS3 Deletion in Murine Osteocytes Accentuates Responses to Exercise- and Load-Induced Bone Formation. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2022, 37, 547–558. [Google Scholar] [CrossRef]
- Sherk, V.D.; Rosen, C.J. Senescent and apoptotic osteocytes and aging: Exercise to the rescue? Bone 2019, 121, 255–258. [Google Scholar] [CrossRef]
- Jing, E.; Emanuelli, B.; Hirschey, M.D.; Boucher, J.; Lee, K.Y.; Lombard, D.; Verdin, E.M.; Kahn, C.R. Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc. Natl. Acad. Sci. USA 2011, 108, 14608–14613. [Google Scholar] [CrossRef]
- Gao, J.; Qin, A.; Liu, D.; Ruan, R.; Wang, Q.; Yuan, J.; Cheng, T.S.; Filipovska, A.; Papadimitriou, J.M.; Dai, K.; et al. Endoplasmic reticulum mediates mitochondrial transfer within the osteocyte dendritic network. Sci. Adv. 2019, 5, eaaw7215. [Google Scholar] [CrossRef]
- Gao, J.; Feng, Z.; Wang, X.; Zeng, M.; Liu, J.; Han, S.; Xu, J.; Chen, L.; Cao, K.; Long, J.; et al. SIRT3/SOD2 maintains osteoblast differentiation and bone formation by regulating mitochondrial stress. Cell Death Differ. 2018, 25, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, R.; Zhang, Z.; Wang, H.; Lu, X.; Zhang, J.; Kong, A.P.-S.; Tian, X.Y.; Chan, H.-F.; Chung, A.C.-K.; et al. Sirt3 mediates the benefits of exercise on bone in aged mice. Cell Death Differ. 2023, 30, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Cariati, I.; Bonanni, R.; Pallone, G.; Romagnoli, C.; Rinaldi, A.M.; Annino, G.; D’Arcangelo, G.; Tancredi, V. Whole Body Vibration Improves Brain and Musculoskeletal Health by Modulating the Expression of Tissue-Specific Markers: FNDC5 as a Key Regulator of Vibration Adaptations. Int. J. Mol. Sci. 2022, 23, 10388. [Google Scholar] [CrossRef] [PubMed]
- Aversa, Z.; Zhang, X.; Fielding, R.A.; Lanza, I.; LeBrasseur, N.K. The clinical impact and biological mechanisms of skeletal muscle aging. Bone 2019, 127, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.J.; Zhang, D.; Kim, S.-J.; Lee, M.-C.; Moon, H.Y. Exercise-induced AMPK activation is involved in delay of skeletal muscle senescence. Biochem. Biophys. Res. Commun. 2019, 512, 604–610. [Google Scholar] [CrossRef]
- Englund, D.A.; Sakamoto, A.E.; Fritsche, C.M.; Heeren, A.A.; Zhang, X.; Kotajarvi, B.R.; Lecy, D.R.; Yousefzadeh, M.J.; Schafer, M.J.; White, T.A.; et al. Exercise reduces circulating biomarkers of cellular senescence in humans. Aging Cell 2021, 20, e13415. [Google Scholar] [CrossRef]
- Kolasinski, S.L.; Neogi, T.; Hochberg, M.C.; Oatis, C.; Guyatt, G.; Block, J.; Callahan, L.; Copenhaver, C.; Dodge, C.; Felson, D.; et al. 2019 American College of Rheumatology/Arthritis Foundation Guideline for the Management of Osteoarthritis of the Hand, Hip, and Knee. Arthritis Care Res. 2020, 72, 149–162. [Google Scholar] [CrossRef]
- Bonanni, R.; Cariati, I.; Tancredi, V.; Iundusi, R.; Gasbarra, E.; Tarantino, U. Chronic Pain in Musculoskeletal Diseases: Do You Know Your Enemy? J. Clin. Med. 2022, 11, 2609. [Google Scholar] [CrossRef]
- Mazor, M.; Best, T.M.; Cesaro, A.; Lespessailles, E.; Toumi, H. Osteoarthritis biomarker responses and cartilage adaptation to exercise: A review of animal and human models. Scand. J. Med. Sci. Sports 2019, 29, 1072–1082. [Google Scholar] [CrossRef]
- Norimatsu, K.; Nakanishi, K.; Ijuin, T.; Otsuka, S.; Takada, S.; Tani, A.; Matsuzaki, R.; Matsuoka, T.; Sakakima, H. Effects of low-intensity exercise on spontaneously developed knee osteoarthritis in male senescence-accelerated mouse prone 8. Arthritis Res. Ther. 2023, 25, 168. [Google Scholar] [CrossRef]
- Kraus, V.B.; Sprow, K.; Powell, K.E.; Buchner, D.; Bloodgood, B.; Piercy, K.; George, S.M.; Kraus, W.E. Effects of Physical Activity in Knee and Hip Osteoarthritis: A Systematic Umbrella Review. Med. Sci. Sports Exerc. 2019, 51, 1324–1339. [Google Scholar] [CrossRef]
- Jørgensen, A.E.M.; Schjerling, P.; DellaValle, B.; Rungby, J.; Kjær, M. Acute loading has minor influence on human articular cartilage gene expression and glycosaminoglycan composition in late-stage knee osteoarthritis: A randomised controlled trial. Osteoarthr. Cartil. 2023, 31, 884–893. [Google Scholar] [CrossRef]
- Andreo-López, M.C.; Contreras-Bolívar, V.; García-Fontana, B.; García-Fontana, C.; Muñoz-Torres, M. The Influence of the Mediterranean Dietary Pattern on Osteoporosis and Sarcopenia. Nutrients 2023, 15, 3224. [Google Scholar] [CrossRef] [PubMed]
- Borghesan, M.; Hoogaars, W.M.H.; Varela-Eirin, M.; Talma, N.; Demaria, M. A Senescence-Centric View of Aging: Implications for Longevity and Disease. Trends Cell Biol. 2020, 30, 777–791. [Google Scholar] [CrossRef]
- Agostini, D.; Gervasi, M.; Ferrini, F.; Bartolacci, A.; Stranieri, A.; Piccoli, G.; Barbieri, E.; Sestili, P.; Patti, A.; Stocchi, V.; et al. An Integrated Approach to Skeletal Muscle Health in Aging. Nutrients 2023, 15, 1802. [Google Scholar] [CrossRef]
- Fontana, L.; Partridge, L.; Longo, V.D. Extending healthy life span--from yeast to humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.K.; Bhapkar, M.; Pittas, A.G.; Pieper, C.F.; Das, S.K.; Williamson, D.A.; Scott, T.; Redman, L.M.; Stein, R.; Gilhooly, C.H.; et al. Effect of Calorie Restriction on Mood, Quality of Life, Sleep, and Sexual Function in Healthy Nonobese Adults: The CALERIE 2 Randomized Clinical Trial. JAMA Intern. Med. 2016, 176, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Micheli, L.; Bertini, L.; Bonato, A.; Villanova, N.; Caruso, C.; Caruso, M.; Bernini, R.; Tirone, F. Role of Hydroxytyrosol and Oleuropein in the Prevention of Aging and Related Disorders: Focus on Neurodegeneration, Skeletal Muscle Dysfunction and Gut Microbiota. Nutrients 2023, 15, 1767. [Google Scholar] [CrossRef]
- Izadi, M.; Sadri, N.; Abdi, A.; Zadeh, M.M.R.; Jalaei, D.; Ghazimoradi, M.M.; Shouri, S.; Tahmasebi, S. Longevity and anti-aging effects of curcumin supplementation. GeroScience 2024, 46, 2933–2950. [Google Scholar] [CrossRef]
- Barbagallo, M.; Veronese, N.; Dominguez, L.J. Magnesium in Aging, Health and Diseases. Nutrients 2021, 13, 463. [Google Scholar] [CrossRef]
- Ames, B.N. Prolonging healthy aging: Longevity vitamins and proteins. Proc. Natl. Acad. Sci. USA 2018, 115, 10836–10844. [Google Scholar] [CrossRef] [PubMed]
- Maduro, A.T.; Luís, C.; Soares, R. Ageing, cellular senescence and the impact of diet: An overview. Porto Biomed. J. 2021, 6, e120. [Google Scholar] [CrossRef] [PubMed]
- Iside, C.; Scafuro, M.; Nebbioso, A.; Altucci, L. SIRT1 Activation by Natural Phytochemicals: An Overview. Front. Pharmacol. 2020, 11, 1225. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lu, C.; Chen, Y.; Wang, Q.; Bao, X.; Zhang, Z.; Huang, X. Resveratrol promotes bone mass in ovariectomized rats and the SIRT1 rs7896005 SNP is associated with bone mass in women during perimenopause and early postmenopause. Climacteric 2023, 26, 25–33. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Moore, T.W.; Sun, A.; Snyder, J.P.; Shoji, M. Novel curcumin analogue UBS109 potently stimulates osteoblastogenesis and suppresses osteoclastogenesis: Involvement in Smad activation and NF-κB inhibition. Integr. Biol. 2012, 4, 905–913. [Google Scholar] [CrossRef]
- Giordani, C.; Matacchione, G.; Giuliani, A.; Valli, D.; Scarpa, E.S.; Antonelli, A.; Sabbatinelli, J.; Giacchetti, G.; Sabatelli, S.; Olivieri, F.; et al. Pro-Osteogenic and Anti-Inflammatory Synergistic Effect of Orthosilicic Acid, Vitamin K2, Curcumin, Polydatin and Quercetin Combination in Young and Senescent Bone Marrow-Derived Mesenchymal Stromal Cells. Int. J. Mol. Sci. 2023, 24, 8820. [Google Scholar] [CrossRef]
- Sosa-Díaz, E.; Hernández-Cruz, E.Y.; Pedraza-Chaverri, J. The role of vitamin D on redox regulation and cellular senescence. Free Radic. Biol. Med. 2022, 193, 253–273. [Google Scholar] [CrossRef]
- Fantini, C.; Corinaldesi, C.; Lenzi, A.; Migliaccio, S.; Crescioli, C. Vitamin D as a Shield against Aging. Int. J. Mol. Sci. 2023, 24, 4546. [Google Scholar] [CrossRef]
- Chen, J.-R.; Lazarenko, O.P.; Wu, X.; Kang, J.; Blackburn, M.L.; Shankar, K.; Badger, T.M.; Ronis, M.J.J. Dietary-induced serum phenolic acids promote bone growth via p38 MAPK/β-catenin canonical Wnt signaling. J. bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2010, 25, 2399–2411. [Google Scholar] [CrossRef]
- Zhang, J.; Lazarenko, O.P.; Blackburn, M.L.; Shankar, K.; Badger, T.M.; Ronis, M.J.J.; Chen, J.-R. Feeding blueberry diets in early life prevent senescence of osteoblasts and bone loss in ovariectomized adult female rats. PLoS ONE 2011, 6, e24486. [Google Scholar] [CrossRef]
- Zhang, J.; Lazarenko, O.P.; Blackburn, M.L.; Badger, T.M.; Ronis, M.J.J.; Chen, J.-R. Blueberry consumption prevents loss of collagen in bone matrix and inhibits senescence pathways in osteoblastic cells. Age 2013, 35, 807–820. [Google Scholar] [CrossRef]
- Ling, Z.; Liu, X.; Cheng, Y.; Yan, X.; Wu, S. Gut microbiota and aging. Crit. Rev. Food Sci. Nutr. 2022, 62, 3509–3534. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhou, J.; Wang, L. Role and Mechanism of Gut Microbiota in Human Disease. Front. Cell. Infect. Microbiol. 2021, 11, 625913. [Google Scholar] [CrossRef]
- Kawamoto, S.; Uemura, K.; Hori, N.; Takayasu, L.; Konishi, Y.; Katoh, K.; Matsumoto, T.; Suzuki, M.; Sakai, Y.; Matsudaira, T.; et al. Bacterial induction of B cell senescence promotes age-related changes in the gut microbiota. Nat. Cell Biol. 2023, 25, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, S.; Hara, E. Crosstalk between gut microbiota and cellular senescence: A vicious cycle leading to aging gut. Trends Cell Biol. 2024, 34, 626–635. [Google Scholar] [CrossRef]
- Zhang, L.; Pitcher, L.E.; Prahalad, V.; Niedernhofer, L.J.; Robbins, P.D. Targeting cellular senescence with senotherapeutics: Senolytics and senomorphics. FEBS J. 2023, 290, 1362–1383. [Google Scholar] [CrossRef]
- He, X.; Hu, W.; Zhang, Y.; Chen, M.; Ding, Y.; Yang, H.; He, F.; Gu, Q.; Shi, Q. Cellular senescence in skeletal disease: Mechanisms and treatment. Cell. Mol. Biol. Lett. 2023, 28, 88. [Google Scholar] [CrossRef]
- Lagoumtzi, S.M.; Chondrogianni, N. Senolytics and senomorphics: Natural and synthetic therapeutics in the treatment of aging and chronic diseases. Free Radic. Biol. Med. 2021, 171, 169–190. [Google Scholar] [CrossRef] [PubMed]
- Novais, E.J.; Tran, V.A.; Johnston, S.N.; Darris, K.R.; Roupas, A.J.; Sessions, G.A.; Shapiro, I.M.; Diekman, B.O.; Risbud, M. V Long-term treatment with senolytic drugs Dasatinib and Quercetin ameliorates age-dependent intervertebral disc degeneration in mice. Nat. Commun. 2021, 12, 5213. [Google Scholar] [CrossRef]
- Farr, J.N.; Xu, M.; Weivoda, M.M.; Monroe, D.G.; Fraser, D.G.; Onken, J.L.; Negley, B.A.; Sfeir, J.G.; Ogrodnik, M.B.; Hachfeld, C.M.; et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 2017, 23, 1072–1079. [Google Scholar] [CrossRef]
- Wang, Y.; Che, L.; Chen, X.; He, Z.; Song, D.; Yuan, Y.; Liu, C. Repurpose dasatinib and quercetin: Targeting senescent cells ameliorates postmenopausal osteoporosis and rejuvenates bone regeneration. Bioact. Mater. 2023, 25, 13–28. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Zhou, J.; Lei, Z.; Yang, X. Fisetin suppresses chondrocyte senescence and attenuates osteoarthritis progression by targeting sirtuin 6. Chem. Biol. Interact. 2024, 390, 110890. [Google Scholar] [CrossRef] [PubMed]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.-M.; Marquess, D.; Dananberg, J.; van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef]
- Coryell, P.R.; Diekman, B.O.; Loeser, R.F. Mechanisms and therapeutic implications of cellular senescence in osteoarthritis. Nat. Rev. Rheumatol. 2021, 17, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Gui, Z.; Zhou, Y.; Xia, L.; Lin, K.; Xu, Y. Quercetin alleviates rat osteoarthritis by inhibiting inflammation and apoptosis of chondrocytes, modulating synovial macrophages polarization to M2 macrophages. Free Radic. Biol. Med. 2019, 145, 146–160. [Google Scholar] [CrossRef]
- Kulkarni, A.S.; Gubbi, S.; Barzilai, N. Benefits of Metformin in Attenuating the Hallmarks of Aging. Cell Metab. 2020, 32, 15–30. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falvino, A.; Gasperini, B.; Cariati, I.; Bonanni, R.; Chiavoghilefu, A.; Gasbarra, E.; Botta, A.; Tancredi, V.; Tarantino, U. Cellular Senescence: The Driving Force of Musculoskeletal Diseases. Biomedicines 2024, 12, 1948. https://doi.org/10.3390/biomedicines12091948
Falvino A, Gasperini B, Cariati I, Bonanni R, Chiavoghilefu A, Gasbarra E, Botta A, Tancredi V, Tarantino U. Cellular Senescence: The Driving Force of Musculoskeletal Diseases. Biomedicines. 2024; 12(9):1948. https://doi.org/10.3390/biomedicines12091948
Chicago/Turabian StyleFalvino, Angela, Beatrice Gasperini, Ida Cariati, Roberto Bonanni, Angela Chiavoghilefu, Elena Gasbarra, Annalisa Botta, Virginia Tancredi, and Umberto Tarantino. 2024. "Cellular Senescence: The Driving Force of Musculoskeletal Diseases" Biomedicines 12, no. 9: 1948. https://doi.org/10.3390/biomedicines12091948