
Association of Endothelial Cell Activation with Acute Kidney Injury during Coronary Angiography and the Influence of Recombinant Human C1 Inhibitor—A Secondary Analysis of a Randomized, Placebo-Controlled, Double-Blind Trial

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Description
2.2. Outcomes
2.3. Laboratory Assessment
2.4. Statistical Analysis
3. Results
3.1. Baseline Characteristics of the Study Population
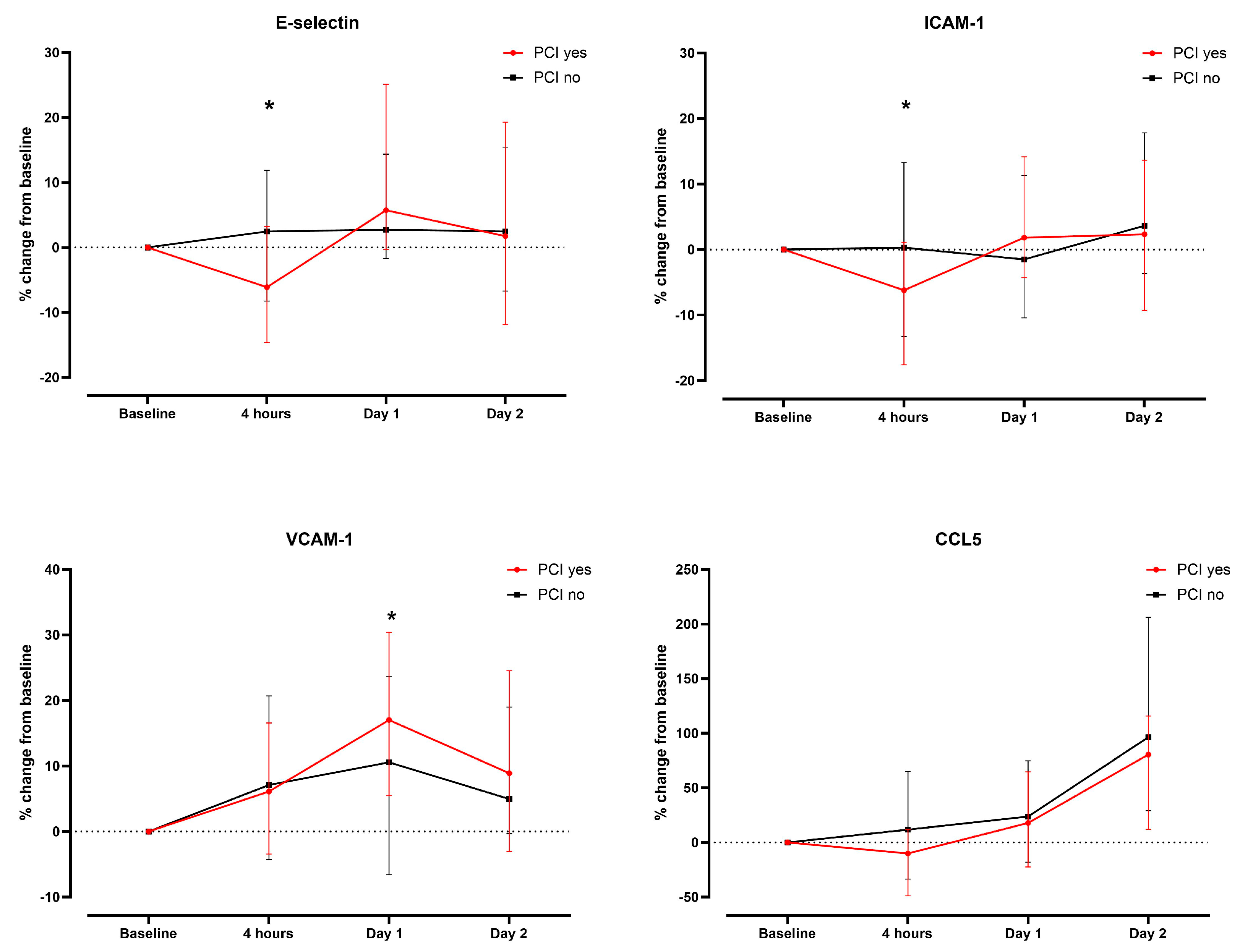
3.2. Serum Concentrations of Endothelial Cell Activation Markers/CCL5 in the Overall Study Population
3.3. Association of Endothelial Activation Marker/CCL5 with Renal Injury Markers and Renal Outcome
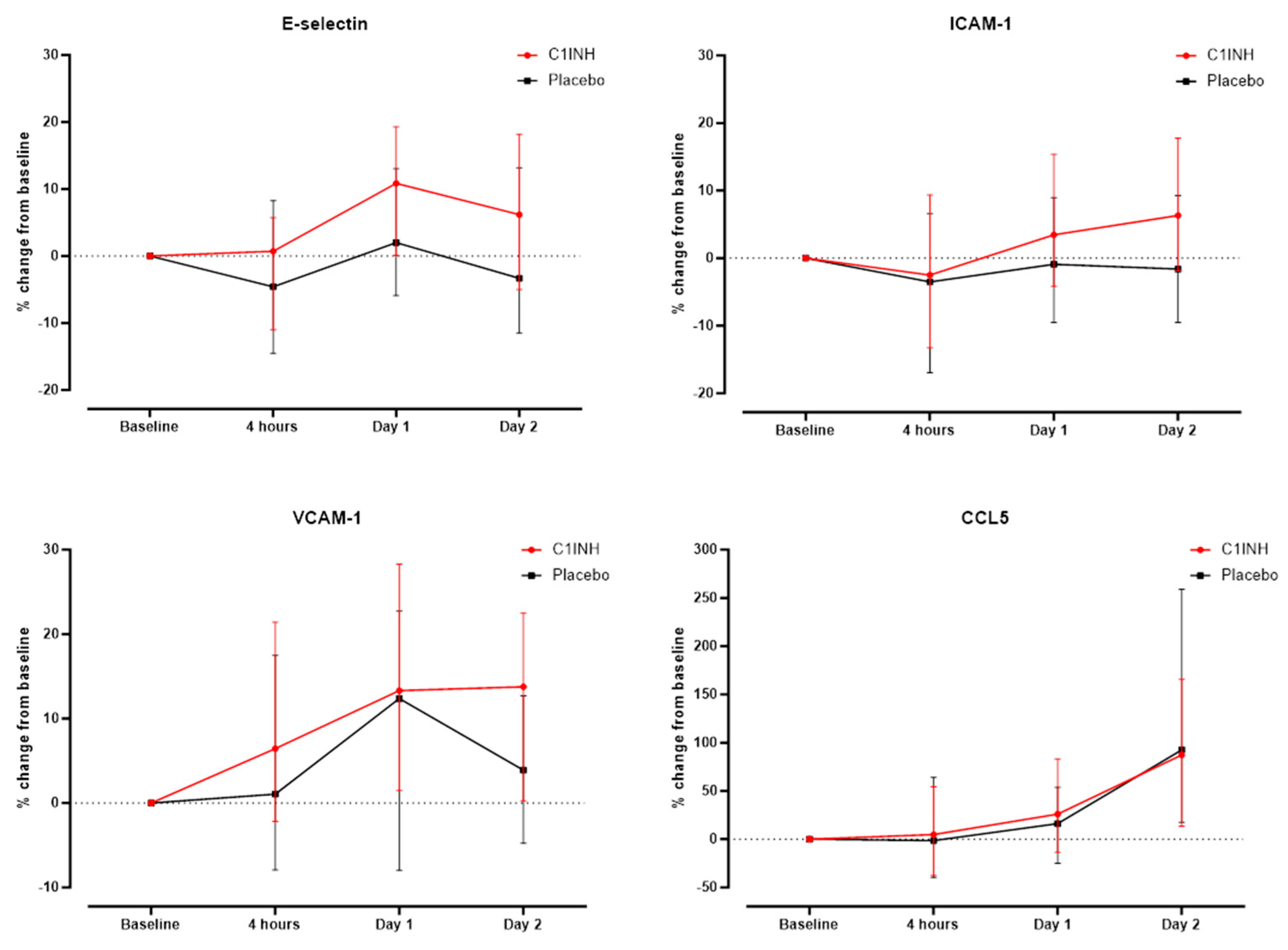
3.4. Association of Treatment with rhC1INH with Endothelial Activation Markers and CCL5
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Weisbord, S.D.; Mor, M.K.; Resnick, A.L.; Hartwig, K.C.; Sonel, A.F.; Fine, M.J.; Palevsky, P.M. Prevention, incidence, and outcomes of contrast-induced acute kidney injury. Arch. Intern. Med. 2008, 168, 1325–1332. [Google Scholar] [CrossRef]
- Seeliger, E.; Sendeski, M.; Rihal, C.S.; Persson, P.B. Contrast-induced kidney injury: Mechanisms, risk factors, and prevention. Eur. Heart J. 2012, 33, 2007–2015. [Google Scholar] [CrossRef] [PubMed]
- Nijssen, E.C.; Rennenberg, R.J.; Nelemans, P.J.; Essers, B.A.; Janssen, M.M.; Vermeeren, M.A.; van Ommen, V.; Wildberger, J.E. Prophylactic hydration to protect renal function from intravascular iodinated contrast material in patients at high risk of contrast-induced nephropathy (AMACING): A prospective, randomised, phase 3, controlled, open-label, non-inferiority trial. Ned. Tijdschr. Voor Geneeskd. 2018, 161, D1734. [Google Scholar] [CrossRef]
- Aird, W.C. Spatial and temporal dynamics of the endothelium. J. Thromb. Haemost. 2005, 3, 1392–1406. [Google Scholar] [CrossRef]
- Konukoglu, D.; Uzun, H. Endothelial Dysfunction and Hypertension. Adv. Exp. Med. Biol. 2017, 956, 511–540. [Google Scholar] [PubMed]
- Rodriguez, C.; Luque, N.; Blanco, I.; Sebastian, L.; Barbera, J.A.; Peinado, V.I.; Tura-Ceide, O. Pulmonary Endothelial Dysfunction and Thrombotic Complications in Patients with COVID-19. Am. J. Respir. Cell Mol. Biol. 2021, 64, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Videm, V.; Albrigtsen, M. Soluble ICAM-1 and VCAM-1 as markers of endothelial activation. Scand. J. Immunol. 2008, 67, 523–531. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, S.; Xu, S.; Zhu, Z.; Li, J.; Wang, Z.; Wada, Y.; Gatt, A.; Liu, J. Oxidative Stress Induces E-Selectin Expression through Repression of Endothelial Transcription Factor ERG. J. Immunol. 2023, 211, 1835–1843. [Google Scholar] [CrossRef]
- Zeng, Z.; Lan, T.; Wei, Y.; Wei, X. CCL5/CCR5 axis in human diseases and related treatments. Genes. Dis. 2022, 9, 12–27. [Google Scholar] [CrossRef]
- Ramos, T.N.; Bullard, D.C.; Barnum, S.R. ICAM-1: Isoforms and phenotypes. J. Immunol. 2014, 192, 4469–4474. [Google Scholar] [CrossRef]
- Cook-Mills, J.M.; Marchese, M.E.; Abdala-Valencia, H. Vascular cell adhesion molecule-1 expression and signaling during disease: Regulation by reactive oxygen species and antioxidants. Antioxid. Redox Signal. 2011, 15, 1607–1638. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.; Videira, P.A.; Sackstein, R. E-Selectin Ligands in the Human Mononuclear Phagocyte System: Implications for Infection, Inflammation, and Immunotherapy. Front. Immunol. 2017, 8, 1878. [Google Scholar] [CrossRef] [PubMed]
- Kjaergaard, A.G.; Dige, A.; Krog, J.; Tonnesen, E.; Wogensen, L. Soluble adhesion molecules correlate with surface expression in an in vitro model of endothelial activation. Basic. Clin. Pharmacol. Toxicol. 2013, 113, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef]
- Kerr, H.; Richards, A. Complement-mediated injury and protection of endothelium: Lessons from atypical haemolytic uraemic syndrome. Immunobiology 2012, 217, 195–203. [Google Scholar] [CrossRef]
- Roumenina, L.T.; Rayes, J.; Frimat, M.; Fremeaux-Bacchi, V. Endothelial cells: Source, barrier, and target of defensive mediators. Immunol. Rev. 2016, 274, 307–329. [Google Scholar]
- Fischetti, F.; Tedesco, F. Cross-talk between the complement system and endothelial cells in physiologic conditions and in vascular diseases. Autoimmunity 2006, 39, 417–428. [Google Scholar]
- Ricklin, D.; Lambris, J.D. Complement in immune and inflammatory disorders: Pathophysiological mechanisms. J. Immunol. 2013, 190, 3831–3838. [Google Scholar]
- Davis, A.E., III; Lu, F.; Mejia, P. C1 inhibitor, a multi-functional serine protease inhibitor. Thromb. Haemost. 2010, 104, 886–893. [Google Scholar]
- Danobeitia, J.S.; Ziemelis, M.; Ma, X.; Zitur, L.J.; Zens, T.; Chlebeck, P.J.; Van Amersfoort, E.S.; Fernandez, L.A. Complement inhibition attenuates acute kidney injury after ischemia-reperfusion and limits progression to renal fibrosis in mice. PLoS ONE 2017, 12, e0183701. [Google Scholar] [CrossRef]
- Castellano, G.; Melchiorre, R.; Loverre, A.; Ditonno, P.; Montinaro, V.; Rossini, M.; Divella, C.; Battaglia, M.; Lucarelli, G.; Annunziata, G.; et al. Therapeutic targeting of classical and lectin pathways of complement protects from ischemia-reperfusion-induced renal damage. Am. J. Pathol. 2010, 176, 1648–1659. [Google Scholar] [CrossRef]
- Panagiotou, A.; Trendelenburg, M.; Heijnen, I.; Moser, S.; Bonati, L.H.; Breidthardt, T.; Fahrni, G.; Kaiser, C.; Jeger, R.; Osthoff, M. A Randomized Trial of Recombinant Human C1-Esterase-Inhibitor in the Prevention of Contrast-Induced Kidney Injury. JACC Cardiovasc. Interv. 2020, 13, 833–842. [Google Scholar] [CrossRef]
- Hauser, I.A.; Riess, R.; Hausknecht, B.; Thuringer, H.; Sterzel, R.B. Expression of cell adhesion molecules in primary renal disease and renal allograft rejection. Nephrol. Dial. Transplant. 1997, 12, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Rickli, H.; Benou, K.; Ammann, P.; Fehr, T.; Brunner-La Rocca, H.P.; Petridis, H.; Riesen, W.; Wüthrich, R. Time course of serial cystatin C levels in comparison with serum creatinine after application of radiocontrast media. Clin. Nephrol. 2004, 61, 98–102. [Google Scholar] [CrossRef]
- Marakala, V. Neutrophil gelatinase-associated lipocalin (NGAL) in kidney injury—A systematic review. Clin. Chim. Acta 2022, 536, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Kafkas, N.; Demponeras, C.; Zoubouloglou, F.; Spanou, L.; Babalis, D.; Makris, K. Serum levels of gelatinase associated lipocalin as indicator of the inflammatory status in coronary artery disease. Int. J. Inflamm. 2012, 2012, 189797. [Google Scholar] [CrossRef]
- Li, M.; van Esch, B.; Henricks, P.A.J.; Garssen, J.; Folkerts, G. Time and Concentration Dependent Effects of Short Chain Fatty Acids on Lipopolysaccharide- or Tumor Necrosis Factor alpha-Induced Endothelial Activation. Front. Pharmacol. 2018, 9, 233. [Google Scholar]
- Scholz, D.; Devaux, B.; Hirche, A.; Potzsch, B.; Kropp, B.; Schaper, W.; Schaper, J. Expression of adhesion molecules is specific and time-dependent in cytokine-stimulated endothelial cells in culture. Cell Tissue Res. 1996, 284, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Seljeflot, I.; Tonstad, S.; Hjermann, I.; Arnesen, H. Reduced expression of endothelial cell markers after 1 year treatment with simvastatin and atorvastatin in patients with coronary heart disease. Atherosclerosis 2002, 162, 179–185. [Google Scholar] [CrossRef]
- Munk, P.S.; Breland, U.M.; Aukrust, P.; Skadberg, O.; Ueland, T.; Larsen, A.I. Inflammatory response to percutaneous coronary intervention in stable coronary artery disease. J. Thromb. Thrombolysis 2011, 31, 92–98. [Google Scholar] [CrossRef]
- Boos, C.J.; Balakrishnan, B.; Jessani, S.; Blann, A.D.; Lip, G.Y. Effects of percutaneous coronary intervention on peripheral venous blood circulating endothelial cells and plasma indices of endothelial damage/dysfunction. Chest 2007, 132, 1920–1926. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.E., III; Mejia, P.; Lu, F. Biological activities of C1 inhibitor. Mol. Immunol. 2008, 45, 4057–4063. [Google Scholar] [CrossRef]
- Tedesco, F.; Pausa, M.; Nardon, E.; Introna, M.; Mantovani, A.; Dobrina, A. The cytolytically inactive terminal complement complex activates endothelial cells to express adhesion molecules and tissue factor procoagulant activity. J. Exp. Med. 1997, 185, 1619–1627. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Bohrer, H.; Hagl, S. Rescue therapy with C1-esterase inhibitor concentrate after emergency coronary surgery for failed PTCA. Intensive Care Med. 1998, 24, 635–638. [Google Scholar] [CrossRef]
- Fattouch, K.; Bianco, G.; Speziale, G.; Sampognaro, R.; Lavalle, C.; Guccione, F.; Dioguardi, P.; Ruvolo, G. Beneficial effects of C1 esterase inhibitor in ST-elevation myocardial infarction in patients who underwent surgical reperfusion: A randomised double-blind study. Eur. J. Cardiothorac. Surg. 2007, 32, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Radke, A.; Mottaghy, K.; Goldmann, C.; Khorram-Sefat, R.; Kovacs, B.; Janssen, A.; Klosterhalfen, B.; Hafemann, B.; Pallua, N.; Kirschfink, M. C1 inhibitor prevents capillary leakage after thermal trauma. Crit. Care Med. 2000, 28, 3224–3232. [Google Scholar] [CrossRef]
- Schelzig, H.; Simon, F.; Krischer, C.; Vogel, A.; Abendroth, D. Ex-vivo hemoperfusion (eHPS) of pig-lungs with whole human blood: Effects of complement inhibition with a soluble C1-esterase-inhibitor. Ann. Transplant. 2001, 6, 34–39. [Google Scholar]
- Jansen, P.M.; Eisele, B.; de Jong, I.W.; Chang, A.; Delvos, U.; Taylor, F.B., Jr.; Hack, C.E. Effect of C1 inhibitor on inflammatory and physiologic response patterns in primates suffering from lethal septic shock. J. Immunol. 1998, 160, 475–484. [Google Scholar] [CrossRef]
- Buerke, M.; Schwertz, H.; Seitz, W.; Meyer, J.; Darius, H. Novel small molecule inhibitor of C1s exerts cardioprotective effects in ischemia-reperfusion injury in rabbits. J. Immunol. 2001, 167, 5375–5380. [Google Scholar] [CrossRef] [PubMed]
- Buerke, M.; Murohara, T.; Lefer, A.M. Cardioprotective effects of a C1 esterase inhibitor in myocardial ischemia and reperfusion. Circulation 1995, 91, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Buerke, M.; Prufer, D.; Dahm, M.; Oelert, H.; Meyer, J.; Darius, H. Blocking of classical complement pathway inhibits endothelial adhesion molecule expression and preserves ischemic myocardium from reperfusion injury. J. Pharmacol. Exp. Ther. 1998, 286, 429–438. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| All Patients n = 74 | Placebo n = 37 | rhC1INH n = 37 | |
|---|---|---|---|
| Age, years | 76.7 ± 8.1 | 77 ± 9.3 | 76.3 ± 7 |
| Male | 52 (70.3) | 27 (73) | 25 (67.6) |
| Comorbidities: | |||
| Diabetes mellitus | 30 (40.5) | 13 (35.0) | 17 (46.0) |
| Coronary artery disease | 42 (57.0) | 19 (51.0) | 23 (62.0) |
| Dyslipidemia | 47 (63.5) | 23 (62.2) | 24 (65.0) |
| PAD | 10 (13.5) | 4 (11.0) | 6 (16.0) |
| Smoking (previous and current) | 44 (59.5) | 21 (57.0) | 23 (62.0) |
| CKD stage 3 | 62 (83.8) | 31 (83.9) | 31 (83.9) |
| CKD stage 4 | 11 (14.9) | 6 (16.2) | 5 (13.5) |
| CKD stage 5 | 1 (1.4) | 0 (0) | 1 (2.7) |
| Medication: | |||
| Anticoagulation | 28 (38.0) | 16 (43.0) | 12 (32.0) |
| Antiplatelet drugs | 46 (62.0) | 21 (57.0) | 25 (67.6) |
| Statin | 49 (66.0) | 21 (57.0) | 28 (76.0) |
| Intervention: | |||
| PCI | 28 (38.0) | 14 (38.0) | 14 (38.0) |
| Laboratory parameters at baseline: | |||
| Creatinine, µmol/L | 145.2 ± 66.88 | 139.39 ± 40.48 | 149.6 ± 85.36 |
| eGFR, ml/min/1.732 | 39.81 ± 9.62 | 39.84 ± 9.12 | 39.78 ± 10.22 |
| Cystatin C, ng/mL | 1.68 ± 0.50 | 1.68 ± 0.45 | 1.68 ± 0.59 |
| Cystatin C Increase ≥ 10% within 24 h Compared to Baseline (n = 18) | No Cystatin C Increase ≥ 10% within 24 h Compared to Baseline (n = 53) | p Value | |
|---|---|---|---|
| E- selectin baseline | 31.0 ng/mL (19.4–49.6) | 28.24 ng/mL (23.4–38.2) | p = 0.94 |
| E-selectin 4 h | 27.0 ng/mL (21.5–48.2) | 28.4 ng/mL (22.7–38.6) | p = 0.849 |
| E- selectin day 1 | 28.1 ng/mL (20.1–58.9) | 30.0 ng/mL (23.5–44.4) | p = 0.98 |
| ICAM-1 baseline | 405.8 ng/mL (321.1–577.4) | 411.0 ng/mL (354.8–488.0) | p = 0.90 |
| ICAM-1 4 h | 387.8 ng/mL (372.0–638.1) | 393.6 ng/mL (342.2–471.1) | p = 0.43 |
| ICAM-1 day 1 | 411.6 ng/mL (343.7–615.2) | 416.4 ng/mL (370.6–510.4) | p = 0.69 |
| VCAM-1 baseline | 958.0 ng/mL (802.4–1284.6) | 959.0 ng/mL (778.6–1148.3) | p = 0.80 |
| VCAM-1 4 h | 1164.7 ng/mL (723.0–1604.6) | 996.0 ng/mL (834.0–1237.1) | p = 0.60 |
| VCAM-1 day 1 | 1218.6 ng/mL (845.8–1451.7) | 1084.4 ng/mL (854.4–1271.7) | p = 0.38 |
| CCL5 baseline | 17.9 ng/mL (14.6–68.3) | 17.7 ng/mL (11.1–26.0) | p = 0.30 |
| CCL5 4 h | 23.2 ng/mL (13.6–46.7) | 17.1 ng/mL (11.1–24.2) | p = 0.21 |
| CCL5 day 1 | 19.5. ng/mL (13.2–48.0) | 17.2 ng/mL (13.5–29.0) | p = 0.56 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moser, S.; Araschmid, L.; Panagiotou, A.; Bonati, L.H.; Breidthardt, T.; Fahrni, G.; Kaiser, C.; Jeger, R.; Trendelenburg, M.; Osthoff, M. Association of Endothelial Cell Activation with Acute Kidney Injury during Coronary Angiography and the Influence of Recombinant Human C1 Inhibitor—A Secondary Analysis of a Randomized, Placebo-Controlled, Double-Blind Trial. Biomedicines 2024, 12, 1956. https://doi.org/10.3390/biomedicines12091956
Moser S, Araschmid L, Panagiotou A, Bonati LH, Breidthardt T, Fahrni G, Kaiser C, Jeger R, Trendelenburg M, Osthoff M. Association of Endothelial Cell Activation with Acute Kidney Injury during Coronary Angiography and the Influence of Recombinant Human C1 Inhibitor—A Secondary Analysis of a Randomized, Placebo-Controlled, Double-Blind Trial. Biomedicines. 2024; 12(9):1956. https://doi.org/10.3390/biomedicines12091956
Chicago/Turabian StyleMoser, Stephan, Laura Araschmid, Anneza Panagiotou, Leo H. Bonati, Tobias Breidthardt, Gregor Fahrni, Christoph Kaiser, Raban Jeger, Marten Trendelenburg, and Michael Osthoff. 2024. "Association of Endothelial Cell Activation with Acute Kidney Injury during Coronary Angiography and the Influence of Recombinant Human C1 Inhibitor—A Secondary Analysis of a Randomized, Placebo-Controlled, Double-Blind Trial" Biomedicines 12, no. 9: 1956. https://doi.org/10.3390/biomedicines12091956
APA StyleMoser, S., Araschmid, L., Panagiotou, A., Bonati, L. H., Breidthardt, T., Fahrni, G., Kaiser, C., Jeger, R., Trendelenburg, M., & Osthoff, M. (2024). Association of Endothelial Cell Activation with Acute Kidney Injury during Coronary Angiography and the Influence of Recombinant Human C1 Inhibitor—A Secondary Analysis of a Randomized, Placebo-Controlled, Double-Blind Trial. Biomedicines, 12(9), 1956. https://doi.org/10.3390/biomedicines12091956