
Neuropathological mRNA Expression Changes after Single Mild Traumatic Brain Injury in Pigs

Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Subjects
2.2. Head Rotational TBI
2.3. Specimen Preparation
2.4. RNA Isolation and Nanostring Assay
2.5. Transcriptomic Analysis
3. Results
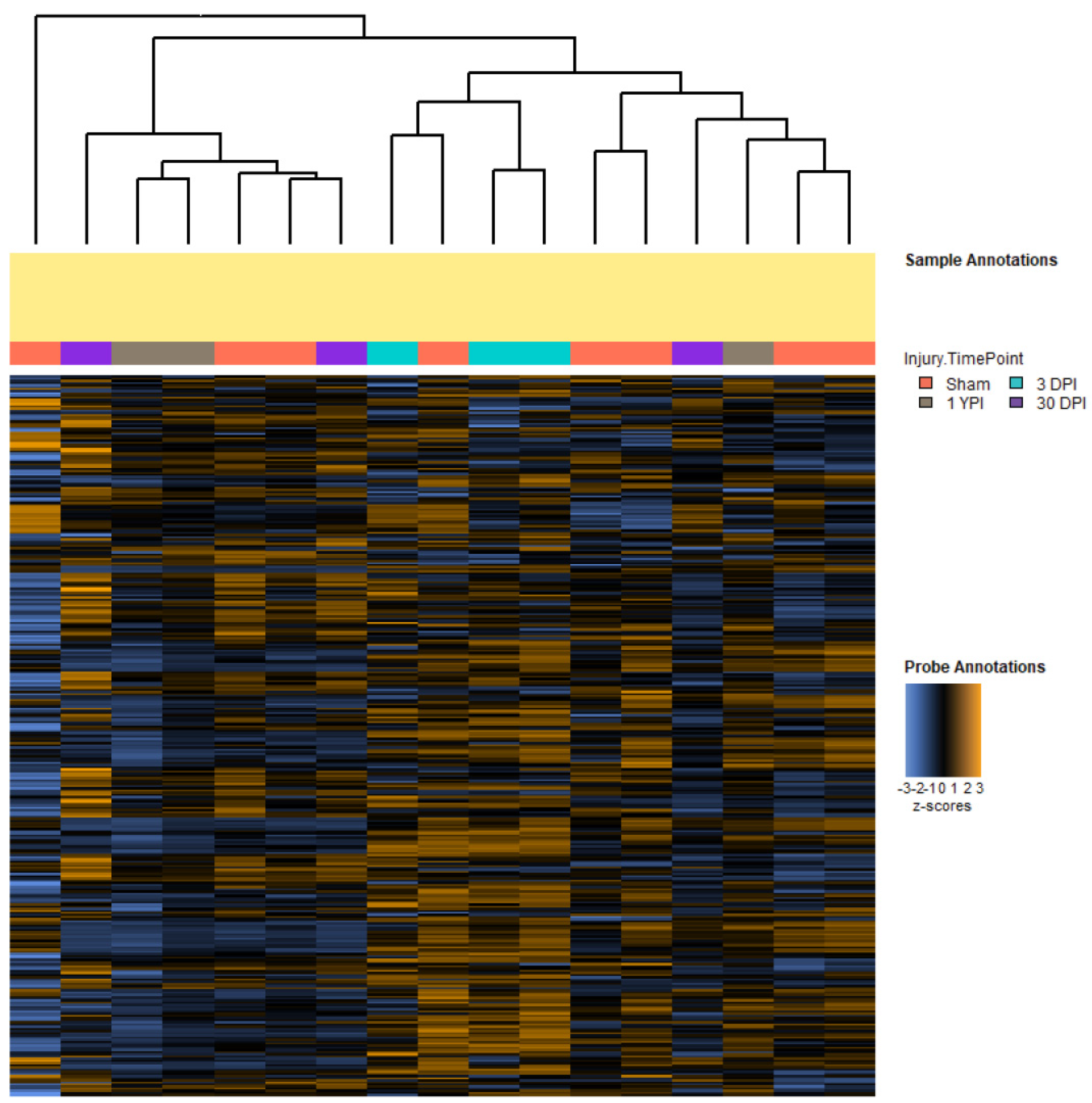
3.1. Single Mild Closed-Head TBI Influences Transcriptomic Profiles
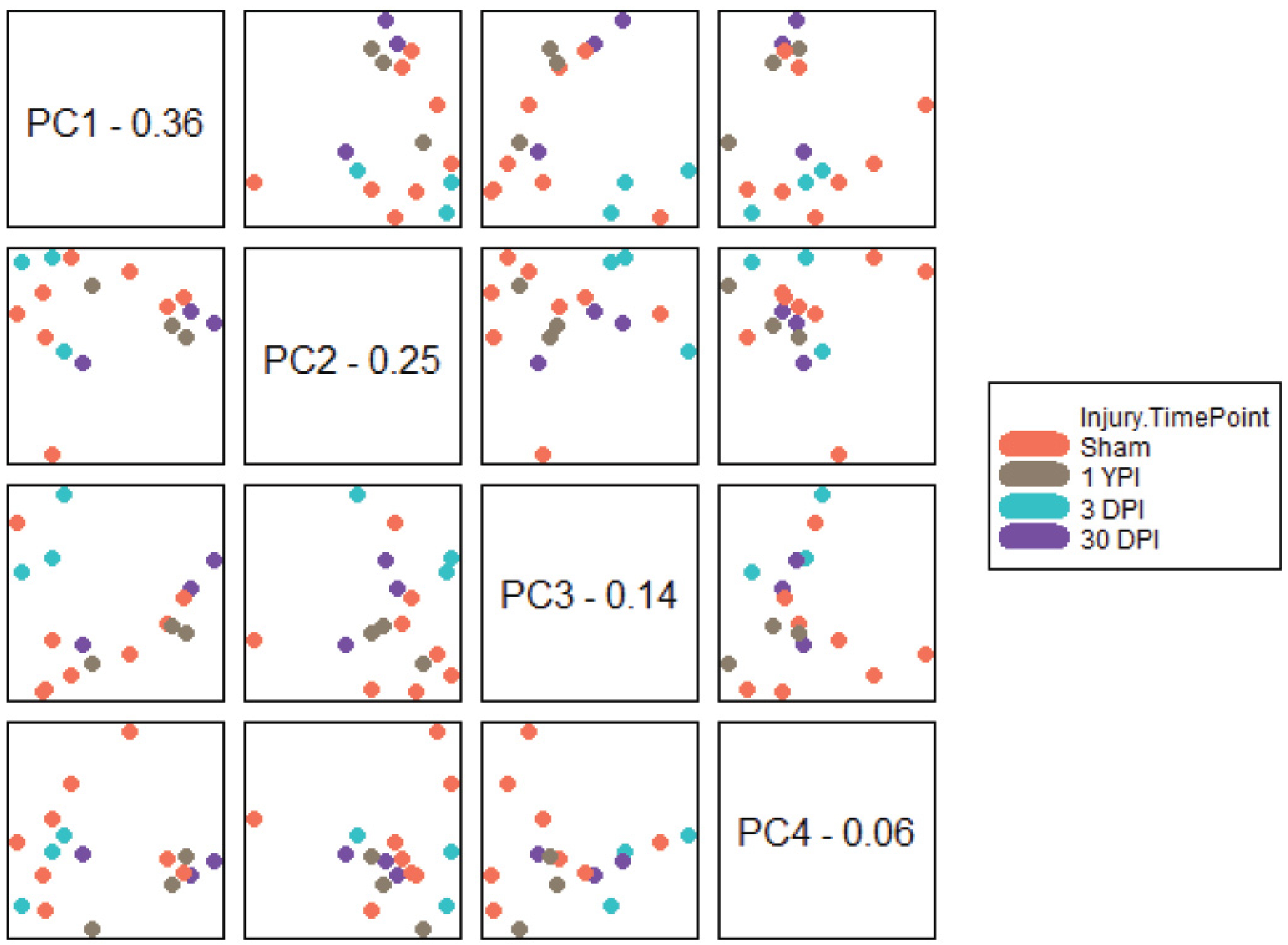
3.2. PCA Separates Transcriptomic Profiles According to Survival Timepoint
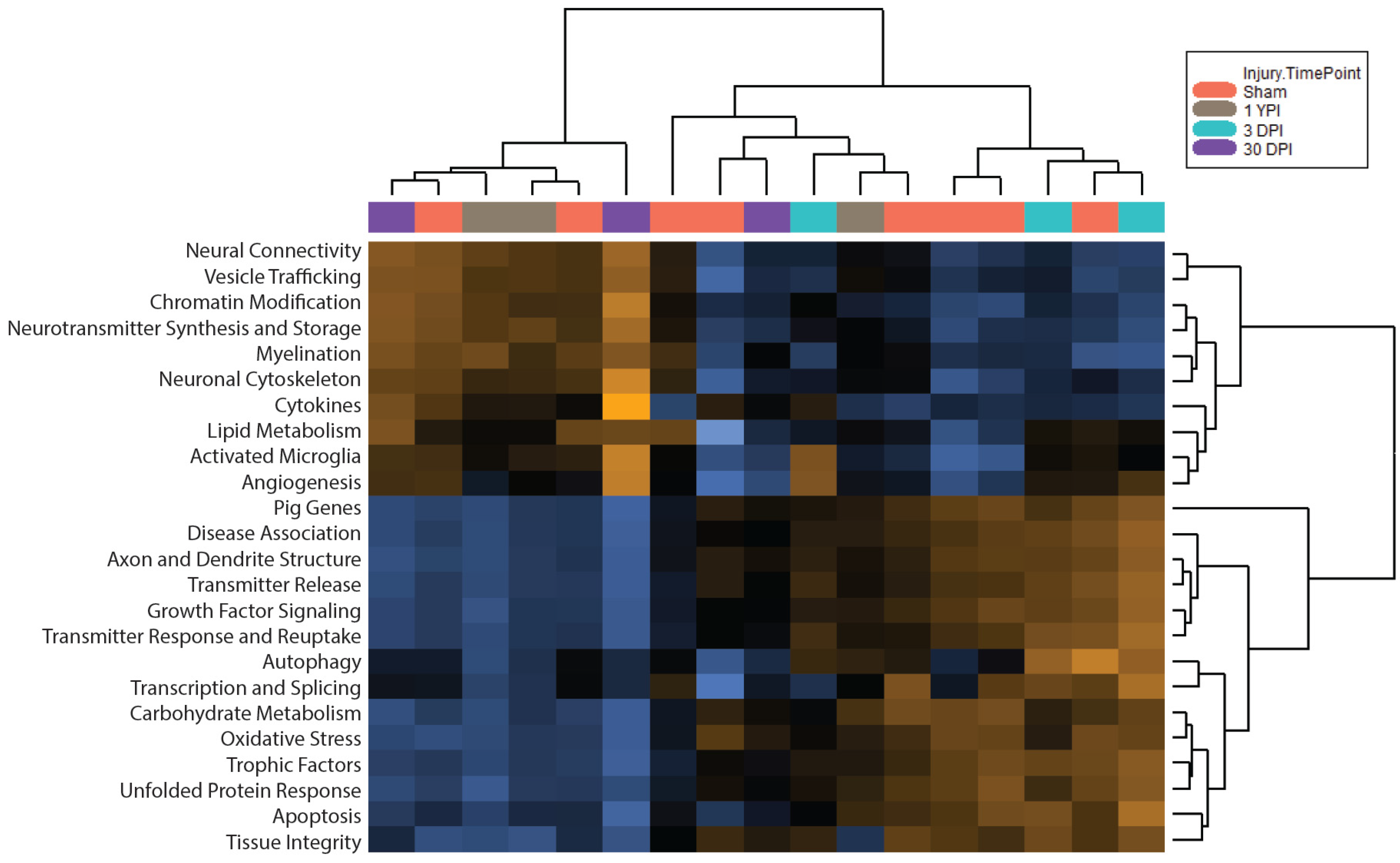
3.3. Pathway Scoring Provides a Broad Overview of Biologically Relevant Gene Sets
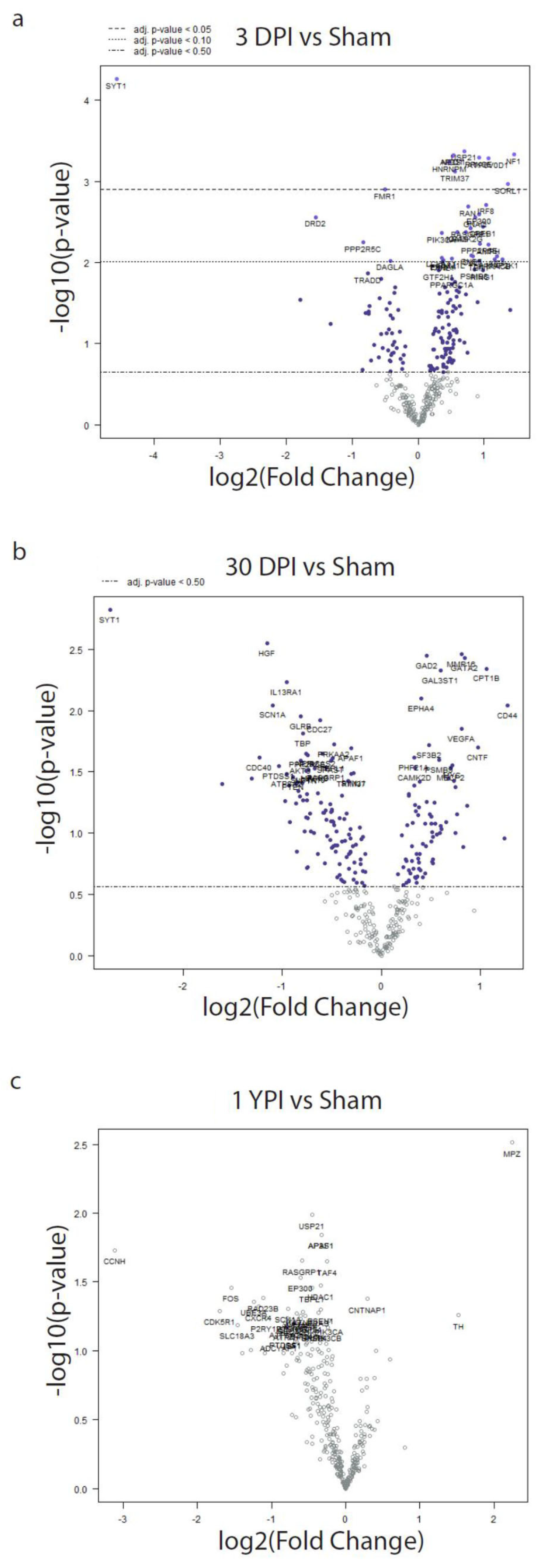
3.4. Differential Expression Analysis Reveals Significant Transcriptomic Changes at 3 DPI
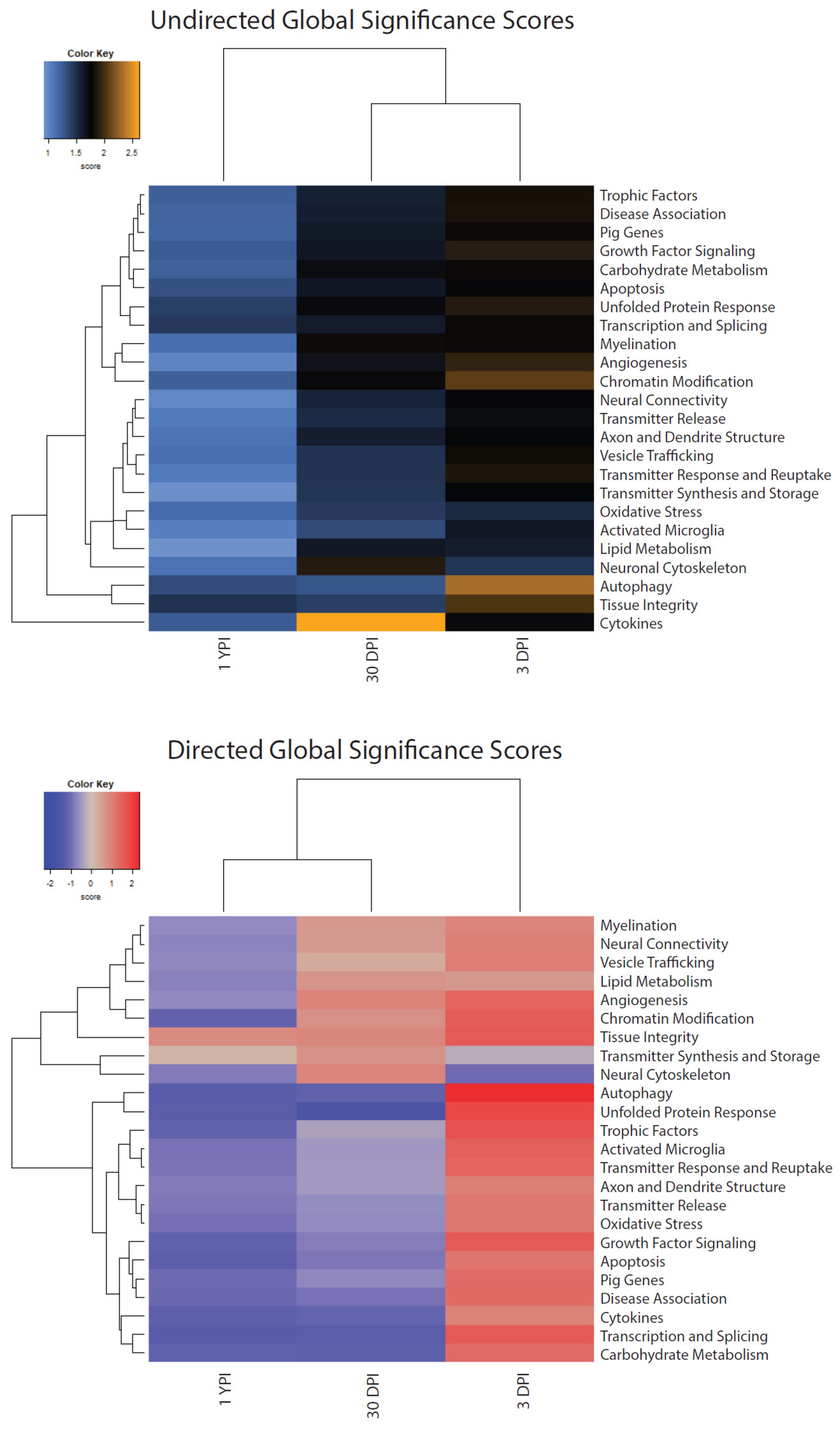
3.5. Global Significance Scores Summarize the DE Analysis by Gene Set
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Centers for Disease Control and Prevention. Surveillance Report of Traumatic Brain Injury-related Emergency Department Visits, Hospitalizations, and Deaths-United States, 2014. Centers Dis Control Prev US Dep Heal Hum Serv. 2019, 24. Available online: www.cdc.gov/TraumaticBrainInjury (accessed on 16 July 2024).
- Gardner, R.C.; Yaffe, K. Epidemiology of mild traumatic brain injury and neurodegenerative disease. Mol. Cell. Neurosci. 2015, 66, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, R.; Fiest, K.M.; McChesney, J.; Kwon, C.-S.; Jette, N.; Frolkis, A.D.; Atta, C.; Mah, S.; Dhaliwal, H.; Reid, A.; et al. The International Incidence of Traumatic Brain Injury: A Systematic Review and Meta-Analysis. Can. J. Neurol. Sci./J. Can. des Sci. Neurol. 2016, 43, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Setnik, L.; Bazarian, J.J. The characteristics of patients who do not seek medical treatment for traumatic brain injury. Brain Inj. 2007, 21, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Levin, H.S.; Diaz-Arrastia, R.R. Diagnosis, prognosis, and clinical management of mild traumatic brain injury. Lancet Neurol. 2015, 14, 506–517. [Google Scholar] [CrossRef]
- Kay, T.; Harrington, D.E.; Adams, R.; Anderson, T.; Berrol, S.; Cicerone, K.; Dahlberg, C.; Gerber, D.; Goka, R.; Harley, P.; et al. Definition of mild traumatic brain injury. J. Head Trauma Rehabil. 1993, 8, 86–87. Available online: https://acrm.org/wp-content/uploads/pdf/acrm%20mtbi%20def%20jhtr93.pdf (accessed on 16 July 2024). [CrossRef]
- Cullen, D.K.; Harris, J.P.; Browne, K.D.; Wolf, J.A.; Duda, J.E.; Meaney, D.F.; Margulies, S.S.; Smith, D.H. A porcine model of traumatic brain injury via head rotational acceleration. Methods Mol. Biol. 2016, 1462, 289–324. [Google Scholar] [CrossRef]
- O’Donnell, J.C.; Browne, K.D.; Kilbaugh, T.J.; Chen, H.I.; Whyte, J.; Cullen, D.K. Challenges and demand for modeling disorders of consciousness following traumatic brain injury. Neurosci. Biobehav. Rev. 2019, 98, 336–346. [Google Scholar] [CrossRef]
- Gennarelli, T.A.; Thibault, L.E.; Adams, J.H.; Graham, D.I.; Thompson, C.J.; Marcincin, R.P. Diffuse axonal injury and traumatic coma in the primate. Ann. Neurol. 1982, 12, 564–574. [Google Scholar] [CrossRef]
- Smith, D.H.; Meaney, D.F. Axonal Damage in Traumatic Brain Injury. Neurosci. 2000, 6, 483–495. [Google Scholar] [CrossRef]
- Gentleman, S.M.; Nash, M.J.; Sweeting, C.J.; Graham, D.I.; Roberts, G.W. Beta-amyloid precursor protein (beta APP) as a marker for axonal injury after head injury. Neurosci. Lett. 1993, 160, 139–144. [Google Scholar] [CrossRef]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Axonal pathology in traumatic brain injury. Exp. Neurol. 2013, 246, 35–43. [Google Scholar] [CrossRef]
- Grovola, M.R.; Paleologos, N.; Wofford, K.L.; Harris, J.P.; Browne, K.D.; Johnson, V.; Duda, J.E.; Wolf, J.A.; Cullen, D.K. Mossy cell hypertrophy and synaptic changes in the hilus following mild diffuse traumatic brain injury in pigs. J. Neuroinflammation 2020, 17, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Grovola, M.R.; Paleologos, N.; Brown, D.P.; Tran, N.; Wofford, K.L.; Harris, J.P.; Browne, K.D.; Shewokis, P.A.; Wolf, J.A.; Cullen, D.K.; et al. Diverse changes in microglia morphology and axonal pathology during the course of 1 year after mild traumatic brain injury in pigs. Brain Pathol. 2021, 31, e12953. [Google Scholar] [CrossRef]
- Grovola, M.R.; von Reyn, C.; Loane, D.J.; Cullen, D.K. Understanding microglial responses in large animal models of traumatic brain injury: An underutilized resource for preclinical and translational research. J. Neuroinflammation 2023, 20, 1–13. [Google Scholar] [CrossRef]
- Grovola, M.R.; Jinich, A.; Paleologos, N.; Arroyo, E.J.; Browne, K.D.; Swanson, R.L.; Duda, J.E.; Cullen, D.K. Persistence of Hyper-Ramified Microglia in Porcine Cortical Gray Matter after Mild Traumatic Brain Injury. Biomedicines 2023, 11, 1960. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.S.; Gottschalk, A.C.; Mazandi, V.M.; Kilbaugh, T.J.; Hefti, M.M. Transcriptional Profiling in a Novel Swine Model of Traumatic Brain Injury. Neurotrauma Rep. 2022, 3, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Nikolian, V.C.; Dennahy, I.S.; Higgins, G.A.; Williams, A.M.; Weykamp, M.; Georgoff, P.E.; Eidy, H.; Ghandour, M.H.; Chang, P.; Alam, H.B. Transcriptomic changes following valproic acid treatment promote neurogenesis and minimize secondary brain injury. J. Trauma Acute Care Surg. 2018, 84, 459–465. [Google Scholar] [CrossRef]
- Rai, V.; Mendoza-Mari, Y.M.; Radwan, M.M.; Brazdzionis, J.; Connett, D.A.; Miulli, D.E.; Agrawal, D.K. Transcriptional and Translational Regulation of Differentially Expressed Genes in Yucatan Miniswine Brain Tissues following Traumatic Brain Injury. J. Bioinform. Syst. Biol. 2024, 7, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.J.; Mooney, S.R.; Pearson, A.N.; McGuire, D.; Correa, D.J.; Simon, R.P.; Meller, R. Assessing the accuracy of blood RNA profiles to identify patients with post-concussion syndrome: A pilot study in a military patient population. PLoS ONE 2017, 12, e0183113. [Google Scholar] [CrossRef]
- Merchant-Borna, K.; Lee, H.; Wang, D.; Bogner, V.; van Griensven, M.; Gill, J.; Bazarian, J.J. Genome-Wide Changes in Peripheral Gene Expression following Sports-Related Concussion. J. Neurotrauma 2016, 33, 1576–1585. [Google Scholar] [CrossRef]
- Simpson, E.; Reiter, J.L.; Ren, J.; Zhang, Z.; Nudelman, K.N.; Riggen, L.D.; Menser, M.D.; Harezlak, J.; Foroud, T.M.; Saykin, A.J.; et al. Gene Expression Alterations in Peripheral Blood Following Sport-Related Concussion in a Prospective Cohort of Collegiate Athletes: A Concussion Assessment, Research and Education (CARE) Consortium Study. Sports Med. 2024, 54, 1021–1032. [Google Scholar] [CrossRef]
- Smith, D.H.; Chen, X.H.; Xu, B.N.; McIntosh, T.K.; Gennarelli, T.A.; Meaney, D.F. Characterization of diffuse axonal pathology and selective hippocampal damage following inertial brain trauma in the pig. J. Neuropathol. Exp. Neurol. 1997, 56, 822–834. [Google Scholar] [CrossRef]
- Geppert, M.; Goda, Y.; Hammer, R.E.; Li, C.; Rosahl, T.W.; Stevens, C.F.; Südhof, T.C. Synaptotagmin I: A major Ca2+ sensor for transmitter release at a central synapse. Cell 1994, 79, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Egbenya, D.L.; Lai, Y.; Dosa, Z.J.; Sørensen, J.B.; Anderson, A.E.; Davanger, S. The calcium sensor synaptotagmin 1 is expressed and regulated in hippocampal postsynaptic spines. Hippocampus 2017, 27, 1168–1177. [Google Scholar] [CrossRef]
- Xu, G.; Tan, X.; Wang, H.; Sun, W.; Shi, Y.; Burlingame, S.; Gu, X.; Cao, G.; Zhang, T.; Qin, J.; et al. Ubiquitin-specific peptidase 21 inhibits tumor necrosis factor al-pha-induced nuclear factor kappaB activation via binding to and deubiquitinating receptor-interacting protein 1. J. Biol. Chem. 2010, 285, 969–978. Available online: http://www.ncbi.nlm.nih.gov/pubmed/19910467 (accessed on 16 July 2024). [CrossRef]
- Arnaoutakis, G.J.; George, T.J.; Wang, K.K.; Wilson, M.A.; Allen, J.G.; Robinson, C.W.; Haggerty, K.A.; Weiss, E.S.; Blue, M.E.; Talbot, C.C.; et al. Serum levels of neuron-specific ubiquitin carboxyl-terminal esterase-L1 predict brain injury in a canine model of hypothermic circulatory arrest. J. Thorac. Cardiovasc. Surg. 2011, 142, 902–910.e1. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, J.; Koleske, J.P.; Chao, A.; Sauerbeck, A.D.; Chen, J.-K.; Qi, X.; Ouyang, M.; Boggs, L.G.; Idate, R.; Marquez, L.I.M.Y.; et al. Brain injury drives optic glioma formation through neuron-glia signaling. Acta Neuropathol. Commun. 2024, 12, 21. [Google Scholar] [CrossRef]
- Boone, D.K.; Weisz, H.A.; Bi, M.; Falduto, M.T.; Torres, K.E.O.; Willey, H.E.; Volsko, C.M.; Kumar, A.M.; Micci, M.-A.; Dewitt, D.S.; et al. Evidence linking microRNA suppression of essential prosurvival genes with hippocampal cell death after traumatic brain injury. Sci. Rep. 2017, 7, 6645. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Deitinghoff, L.; Davis, D.; Conrad, S.; Skutella, T.; Chedotal, A.; Mueller, B.K.; Strittmatter, S.M. Neogenin mediates the action of repulsive guidance molecule. Nat. Cell Biol. 2004, 6, 756–762. [Google Scholar] [CrossRef]
- Sun, D.; Sun, X.-D.; Zhao, L.; Lee, D.-H.; Hu, J.-X.; Tang, F.-L.; Pan, J.-X.; Mei, L.; Zhu, X.-J.; Xiong, W.-C. Neogenin, a regulator of adult hippocampal neurogenesis, prevents depressive-like behavior. Cell Death Dis. 2018, 9, 8. [Google Scholar] [CrossRef]
- Huang, Z.; Sun, D.; Hu, J.-X.; Tang, F.-L.; Lee, D.-H.; Wang, Y.; Hu, G.; Zhu, X.-J.; Zhou, J.; Mei, L.; et al. Neogenin Promotes BMP2 Activation of YAP and Smad1 and Enhances Astrocytic Differentiation in Developing Mouse Neocortex. J. Neurosci. 2016, 36, 5833–5849. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Tan, Z.-B.; Sun, X.-D.; Liu, Z.-P.; Chen, W.-B.; Milibari, L.; Ren, X.; Yao, L.-L.; Lee, D.; Shen, C.; et al. Hippocampal astrocytic neogenin regulating glutamate uptake, a critical pathway for preventing epileptic response. Proc. Natl. Acad. Sci. USA 2021, 118, e2022921118. [Google Scholar] [CrossRef] [PubMed]
- Steinhart, R.; Kazimirsky, G.; Okhrimenko, H.; Ben-Hur, T.; Brodie, C. PKCepsilon induces astrocytic differentiation of multipo-tential neural precursor cells. Glia 2007, 55, 224–232. Available online: http://www.ncbi.nlm.nih.gov/pubmed/17091491 (accessed on 16 July 2024). [CrossRef]
- Geribaldi-Doldán, N.; Gómez-Oliva, R.; Domínguez-García, S.; Nunez-Abades, P.; Castro, C. Protein Kinase C: Targets to Regen-erate Brain Injuries? Front. Cell Dev. Biol. 2019, 7, 39. [Google Scholar] [CrossRef]
- Dharshini, S.A.P.; Taguchi, Y.-H.; Gromiha, M.M. Exploring the selective vulnerability in Alzheimer disease using tissue specific variant analysis. Genomics 2019, 111, 936–949. [Google Scholar] [CrossRef]
- Meng, Q.; Zhuang, Y.; Ying, Z.; Agrawal, R.; Yang, X.; Gomez-Pinilla, F. Traumatic Brain Injury Induces Genome-Wide Transcriptomic, Methylomic, and Network Perturbations in Brain and Blood Predicting Neurological Disorders. EBioMedicine 2017, 16, 184–194. [Google Scholar] [CrossRef]
- Akinyemi, A.R.; Li, D.; Zhang, J.; Liu, Q. hnRNPM deficiency leads to cognitive deficits via disrupting synaptic plasticity. Neurosci. Lett. 2021, 751, 135824. [Google Scholar] [CrossRef]
- Ramesh, N.; Kour, S.; Anderson, E.N.; Rajasundaram, D.; Pandey, U.B. RNA-recognition motif in Matrin-3 mediates neurodegeneration through interaction with hnRNPM. Acta Neuropathol. Commun. 2020, 8, 138. [Google Scholar] [CrossRef] [PubMed]
- Bampton, A.; Gittings, L.M.; Fratta, P.; Lashley, T.; Gatt, A. The role of hnRNPs in frontotemporal dementia and amyotrophic lateral sclerosis. Acta Neuropathol. 2020, 140, 599–623. [Google Scholar] [CrossRef]
- Chang, C.; Zhu, S.; Xu, Z.; Liu, Y. Tripartite motif-containing protein 37 is overexpressed in human glioma and its downregulation inhibits human glioma cell growth in vitro. Int. J. Clin. Exp. Pathol. 2018, 11, 502–511. [Google Scholar]
- Han, C.; Xia, X.; Jiao, S.; Li, G.; Ran, Q.; Yao, S. Tripartite motif containing protein 37 involves in thrombin stimulated BV-2 microglial cell apoptosis and interleukin 1β release. Biochem. Biophys. Res. Commun. 2019, 516, 1252–1257. [Google Scholar] [CrossRef] [PubMed]
- Yin, R.-H.; Yu, J.-T.; Tan, L. The Role of SORL1 in Alzheimer’s Disease. Mol. Neurobiol. 2015, 51, 909–918. [Google Scholar] [CrossRef]
- Lamprecht, M.R.; Elkin, B.S.; Kesavabhotla, K.; Crary, J.F.; Hammers, J.L.; Huh, J.W.; Raghupathi, R.; Morrison, B. Strong Correlation of Genome-Wide Ex-pression after Traumatic Brain Injury In Vitro and In Vivo Implicates a Role for SORLA. J. Neurotrauma 2017, 34, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Bassell, G.J.; Warren, S.T. Fragile X Syndrome: Loss of Local mRNA Regulation Alters Synaptic Development and Function. Neuron 2008, 60, 201–214. [Google Scholar] [CrossRef]
- Kumari, D.; Gazy, I.; Usdin, K. Pharmacological Reactivation of the Silenced FMR1 Gene as a Targeted Therapeutic Approach for Fragile X Syndrome. Brain Sci. 2019, 9, 39. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Tsuda, M.; Yoshinaga, R.; Tozaki-Saitoh, H.; Ozato, K.; Tamura, T.; Inoue, K. IRF8 is a critical transcription factor for trans-forming microglia into a reactive phenotype. Cell Rep. 2012, 1, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Liu, K.; Sun, Y.; Cao, Y.; Yang, J. Transcriptional mechanism of IRF8 and PU.1 governs microglial activation in neuro-degenerative condition. Protein Cell 2019, 10, 87–103. Available online: http://www.ncbi.nlm.nih.gov/pubmed/30484118 (accessed on 16 July 2024). [CrossRef]
- Mastroeni, D.; Chouliaras, L.; Grover, A.; Liang, W.S.; Hauns, K.; Rogers, J.; Coleman, P.D. Reduced RAN Expression and Disrupted Transport between Cytoplasm and Nucleus; A Key Event in Alzheimer’s Disease Pathophysiology. PLoS ONE 2013, 8, e53349. [Google Scholar] [CrossRef]
- Yeo, E.J.; Shin, M.J.; Youn, G.S.; Park, J.H.; Yeo, H.J.; Kwon, H.J.; Lee, L.R.; Kim, N.Y.; Kwon, S.Y.; Kim, S.M.; et al. Tat-RAN attenuates brain ischemic injury in hippocampal HT-22 cells and ischemia animal model. Neurochem. Int. 2023, 167, 105538. [Google Scholar] [CrossRef]
- Shimizu, Y.; Kawasaki, T. Histone acetyltransferase EP300 regulates the proliferation and differentiation of neural stem cells during adult neurogenesis and regenerative neurogenesis in the zebrafish optic tectum. Neurosci. Lett. 2021, 756, 135978. [Google Scholar] [CrossRef]
- Yue, J.K.; Winkler, E.A.; Rick, J.W.; Burke, J.F.; McAllister, T.W.; Oh, S.S.; Burchard, E.G.; Hu, D.; Rosand, J.; Temkin, N.R.; et al. DRD2 C957T polymorphism is associated with improved 6-month verbal learning following traumatic brain injury. Neurogenetics 2017, 18, 29–38. [Google Scholar] [CrossRef]
- Failla, M.D.; Myrga, J.M.; Ricker, J.H.; Dixon, C.E.; Conley, Y.P.; Wagner, A.K. Posttraumatic Brain Injury Cognitive Performance Is Moderated by Variation Within ANKK1 and DRD2 Genes. J. Head Trauma Rehabilitation 2015, 30, E54–E66. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, M.E.; Bieri, G.; Kaletsky, R.; St Ange, J.; Remesal, L.; Pratt, K.J.B.; Zhou, S.; Weng, Y.; Murphy, C.T.; Villeda, S.A. Neuronal activation of Gαq EGL-30/GNAQ late in life rejuvenates cognition across species. Cell Rep. 2023, 42, 113151. [Google Scholar] [CrossRef] [PubMed]
- Nishiki, T.-I.; Augustine, G.J. Synaptotagmin I Synchronizes Transmitter Release in Mouse Hippocampal Neurons. J. Neurosci. 2004, 24, 6127–6132. [Google Scholar] [CrossRef]
- Webb, T.S.; Whitehead, C.R.; Wells, T.S.; Gore, R.K.; Otte, C.N. Neurologically-related sequelae associated with mild traumatic brain injury. Brain Inj. 2015, 29, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Hoge, C.W.; McGurk, D.; Thomas, J.L.; Cox, A.L.; Engel, C.C.; Castro, C.A. Mild Traumatic Brain Injury in U.S. Soldiers Returning from Iraq. N. Engl. J. Med. 2008, 358, 453–463. [Google Scholar] [CrossRef]
- Wolf, J.A.; Johnson, B.N.; Johnson, V.E.; Putt, M.E.; Browne, K.D.; Mietus, C.J.; Brown, D.P.; Wofford, K.L.; Smith, D.H.; Grady, M.S.; et al. Concussion Induces Hippocampal Circuitry Disruption in Swine. J. Neurotrauma 2017, 34, 2303–2314. [Google Scholar] [CrossRef]
- Bergoug, M.; Doudeau, M.; Godin, F.; Mosrin, C.; Vallée, B.; Bénédetti, H. Neurofibromin Structure, Functions and Regulation. Cells 2020, 9, 2365. Available online: http://www.ncbi.nlm.nih.gov/pubmed/33121128 (accessed on 16 July 2024). [CrossRef] [PubMed]
- Capuani, B.; Pacifici, F.; Pastore, D.; Palmirotta, R.; Donadel, G.; Arriga, R.; Bellia, A.; Di Daniele, N.; Rogliani, P.; Abete, P.; et al. The role of epsilon PKC in acute and chronic diseases: Possible pharmacological implications of its modulators. Pharmacol. Res. 2016, 111, 659–667. [Google Scholar] [CrossRef]
- Gentleman, S.M.; Roberts, G.W.; Gennarelli, T.A.; Maxwell, W.L.; Adams, J.H.; Kerr, S.; Graham, D.I. Axonal injury: A universal consequence of fatal closed head injury? Acta Neuropathol. 1995, 89, 537–543. [Google Scholar] [CrossRef]
- Browne, K.D.; Chen, X.-H.; Meaney, D.F.; Smith, D.H. Mild Traumatic Brain Injury and Diffuse Axonal Injury in Swine. J. Neurotrauma 2011, 28, 1747–1755. [Google Scholar] [CrossRef] [PubMed]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Traumatic brain injury and amyloid-β pathology: A link to Alzheimer’s disease? Nat. Rev. Neurosci. 2010, 11, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chen, Z.; Tang, Q.; Wang, Z.; Lu, J.; You, Y.; Wang, H. USP21 promotes self-renewal and tumorigenicity of mesenchymal glioblastoma stem cells by deubiquitinating and stabilizing FOXD1. Cell Death Dis. 2022, 13, 712. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yao, Y.; Ding, H.; Han, C.; Chen, Y.; Zhang, Y.; Wang, C.; Zhang, X.; Zhang, Y.; Zhai, Y.; et al. USP21 deubiquitylates Nanog to regulate protein stability and stem cell pluripotency. Signal Transduct. Target. Ther. 2016, 1, 16024. [Google Scholar] [CrossRef]
- Brouwer, J.R.; Willemsen, R.; Oostra, B.A. The FMR1 gene and fragile X-associated tremor/ataxia syndrome. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2009, 150B, 782–798. Available online: http://www.ncbi.nlm.nih.gov/pubmed/19105204 (accessed on 16 July 2024). [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Fold Change (log2) | Lower 95% Confidence Limit (log2) | Upper 95% Confidence Limit (log2) | Adjusted p-Value | Biological Function and Role |
|---|---|---|---|---|---|
| SYT1 | −4.57 | −6.09 | −3.04 | 0.0192 | |
| USP21 | 0.698 | 0.407 | 0.99 | 0.0253 | |
| NF1 | 1.45 | 0.838 | 2.07 | 0.0253 |
|
| AP3S1 | 0.53 | 0.306 | 0.755 | 0.0253 |
|
| NEO1 | 0.522 | 0.300 | 0.743 | 0.0253 | |
| PRKCE | 0.92 | 0.527 | 1.31 | 0.0253 |
|
| ATP6V0D1 | 1.07 | 0.610 | 1.52 | 0.0253 | |
| HNRNPM | 0.472 | 0.267 | 0.677 | 0.0253 |
|
| TRIM37 | 0.558 | 0.308 | 0.808 | 0.0292 |
|
| SORL1 | 1.36 | 0.723 | 2.00 | 0.0375 |
|
| FMR1 | −0.501 | −0.74 | −0.261 | 0.0396 |
|
| IRF8 | 1.03 | 0.510 | 1.56 | 0.0547 | |
| RAN | 0.765 | 0.375 | 1.15 | 0.0547 | |
| EP300 | 0.924 | 0.438 | 1.41 | 0.0610 |
|
| DRD2 | −1.55 | −2.37 | −0.723 | 0.0610 |
|
| GNAQ | 0.865 | 0.404 | 1.33 | 0.0610 |
|
| Gene Set | Undirected 3 DPI | Undirected 30 DPI | Undirected 1 YPI | Directed 3 DPI | Directed 30 DPI | Directed 1 YPI |
|---|---|---|---|---|---|---|
| Activated Microglia | 1.642 | 1.333 | 1.043 | 1.463 | −0.492 | −0.937 |
| Angiogenesis | 1.939 | 1.679 | 1.013 | 1.41 | 0.968 | −0.682 |
| Apoptosis | 1.749 | 1.649 | 1.31 | 1.173 | −0.871 | −1.255 |
| Autophagy | 2.32 | 1.287 | 1.329 | 2.32 | −1.204 | −1.316 |
| Axon and Dendrite Structure | 1.764 | 1.6 | 1.116 | 1.028 | −0.461 | −0.864 |
| Carbohydrate Metabolism | 1.791 | 1.715 | 1.233 | 1.307 | −1.216 | −1.179 |
| Chromatin Modification | 2.081 | 1.734 | 1.234 | 1.558 | 0.74 | −1.17 |
| Cytokines | 1.734 | 2.618 | 1.254 | 0.966 | −1.138 | −1.233 |
| Disease Association | 1.843 | 1.594 | 1.204 | 1.331 | −0.93 | −1.051 |
| Growth Factor Signaling | 1.899 | 1.649 | 1.248 | 1.613 | −0.801 | −1.186 |
| Lipid Metabolism | 1.61 | 1.641 | 0.922 | 0.632 | 0.678 | −0.768 |
| Myelination | 1.79 | 1.802 | 1.147 | 0.931 | 0.618 | −0.669 |
| Neural Connectivity | 1.749 | 1.566 | 0.997 | 1.015 | 0.583 | −0.726 |
| Neuronal Cytoskeleton | 1.47 | 1.89 | 1.125 | −1.006 | 0.954 | −0.87 |
| Oxidative Stress | 1.544 | 1.445 | 1.165 | 1.114 | −0.664 | −0.946 |
| Pig Genes | 1.796 | 1.626 | 1.2 | 1.309 | −0.697 | −1.018 |
| Tissue Integrity | 2.033 | 1.414 | 1.481 | 1.606 | 0.898 | 0.832 |
| Transcription and Splicing | 1.793 | 1.619 | 1.447 | 1.61 | −1.287 | −1.333 |
| Transmitter Release | 1.711 | 1.522 | 1.068 | 1.112 | −0.633 | −0.872 |
| Transmitter Response and Reuptake | 1.853 | 1.479 | 1.076 | 1.406 | −0.46 | −0.915 |
| Transmitter Synthesis and Storage | 1.776 | 1.466 | 0.929 | −0.249 | 0.739 | 0.173 |
| Trophic Factors | 1.834 | 1.589 | 1.238 | 1.724 | −0.377 | −1.179 |
| Unfolded Protein Response | 1.894 | 1.726 | 1.412 | 1.857 | −1.568 | −1.287 |
| Vesicle Trafficking | 1.804 | 1.483 | 1.144 | 1.041 | 0.31 | −0.699 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grovola, M.R.; Cullen, D.K. Neuropathological mRNA Expression Changes after Single Mild Traumatic Brain Injury in Pigs. Biomedicines 2024, 12, 2019. https://doi.org/10.3390/biomedicines12092019
Grovola MR, Cullen DK. Neuropathological mRNA Expression Changes after Single Mild Traumatic Brain Injury in Pigs. Biomedicines. 2024; 12(9):2019. https://doi.org/10.3390/biomedicines12092019
Chicago/Turabian StyleGrovola, Michael R., and D. Kacy Cullen. 2024. "Neuropathological mRNA Expression Changes after Single Mild Traumatic Brain Injury in Pigs" Biomedicines 12, no. 9: 2019. https://doi.org/10.3390/biomedicines12092019