
Succinate Dehydrogenase and Human Disease: Novel Insights into a Well-Known Enzyme

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
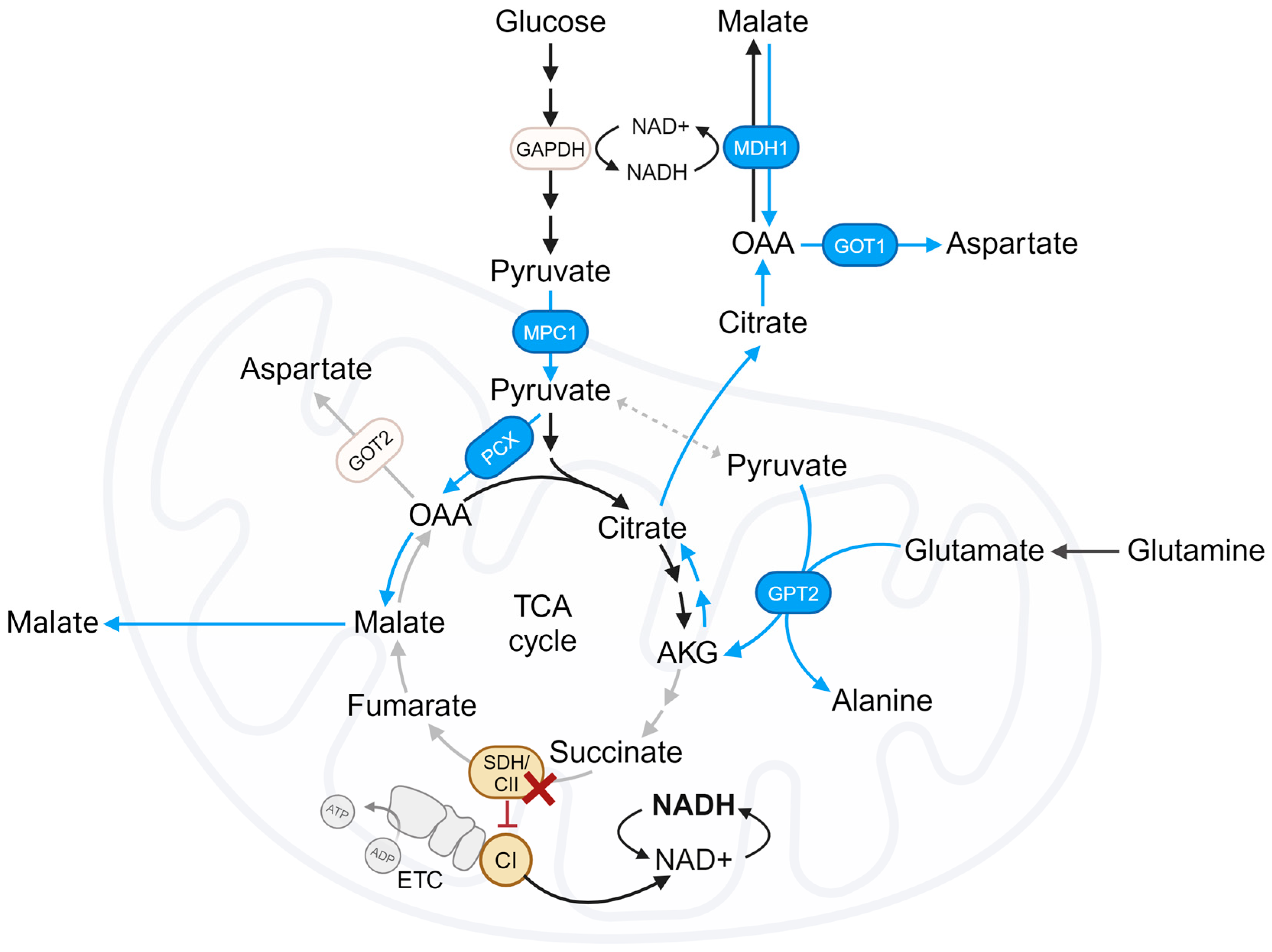
2. TCA Cycle Truncation in SDH/CII-Deficient Cells
3. The ETC and OXPHOS in SDH/CII-Deficient Cells
4. Metabolic Adaptations and Vulnerabilities Imposed by Deficiencies in SDH/CII
4.1. Reliance on Pyruvate Carboxylation
4.2. Contribution of the Reductive TCA Cycle
4.3. The Role of CII-Low
4.4. The Benefits of CI Inhibition
5. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial Electron Transport Chain: Oxidative Phosphorylation, Oxidant Production, and Methods of Measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef]
- Bezawork-Geleta, A.; Rohlena, J.; Dong, L.; Pacak, K.; Neuzil, J. Mitochondrial Complex II: At the Crossroads. Trends Biochem. Sci. 2017, 42, 312–325. [Google Scholar] [CrossRef]
- Rizwan, M.; Al Rasheed, H.; Tarjan, G. Succinate Dehydrogenase Complex An Updated Review. Arch. Pathol. Lab. Med. 2018, 142, 1564–1570. [Google Scholar]
- Rutter, J.; Winge, D.R.; Schiffman, J.D. Succinate Dehydrogenase—Assembly, Regulation and Role in Human Disease. Mitochondrion 2010, 10, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Fullerton, M.; McFarland, R.; Taylor, R.W.; Alston, C.L. The Genetic Basis of Isolated Mitochondrial Complex II Deficiency. Mol. Genet. Metab. 2020, 131, 53–65. [Google Scholar] [CrossRef]
- Cao, K.; Xu, J.; Cao, W.; Wang, X.; Lv, W.; Zeng, M.; Zou, X.; Liu, J.; Feng, Z. Assembly of Mitochondrial Succinate Dehydrogenase in Human Health and Disease. Free Radic. Biol. Med. 2023, 207, 247–259. [Google Scholar] [CrossRef]
- Hoekstra, A.S.; Bayley, J.P. The Role of Complex II in Disease. Biochim. Biophys. Acta (BBA)-Bioenerg. 2013, 1827, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Jodeiri Farshbaf, M.; Kiani-Esfahani, A. Succinate Dehydrogenase: Prospect for Neurodegenerative Diseases. Mitochondrion 2018, 42, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Eijkelenkamp, K.; Osinga, T.E.; Links, T.P.; van der Horst-Schrivers, A.N.; Anouk A van der Horst-Schrivers, C.N. Clinical Implications of the Oncometabolite Succinate in SDHx-Mutation Carriers. Clin. Genet. 2019, 97, 39–53. [Google Scholar] [CrossRef]
- Cervera, A.M.; Apostolova, N.; Crespo, F.L.; Mata, M.; McCreath, K.J. Cells Silenced for SDHB Expression Display Characteristic Features of the Tumor Phenotype. Cancer Res. 2008, 68, 4058–4067. [Google Scholar] [CrossRef]
- Letouzé, E.; Martinelli, C.; Loriot, C.; Burnichon, N.; Abermil, N.; Ottolenghi, C.; Janin, M.; Menara, M.; Nguyen, A.T.; Benit, P.; et al. SDH Mutations Establish a Hypermethylator Phenotype in Paraganglioma. Cancer Cell 2013, 23, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Morin, A.; Goncalves, J.; Moog, S.; Castro-Vega, L.J.; Job, S.; Buffet, A.; Fontenille, M.J.; Woszczyk, J.; Gimenez-Roqueplo, A.P.; Letouzé, E.; et al. TET-Mediated Hypermethylation Primes SDH-Deficient Cells for HIF2α-Driven Mesenchymal Transition. Cell Rep. 2020, 30, 4551–4566. [Google Scholar] [CrossRef] [PubMed]
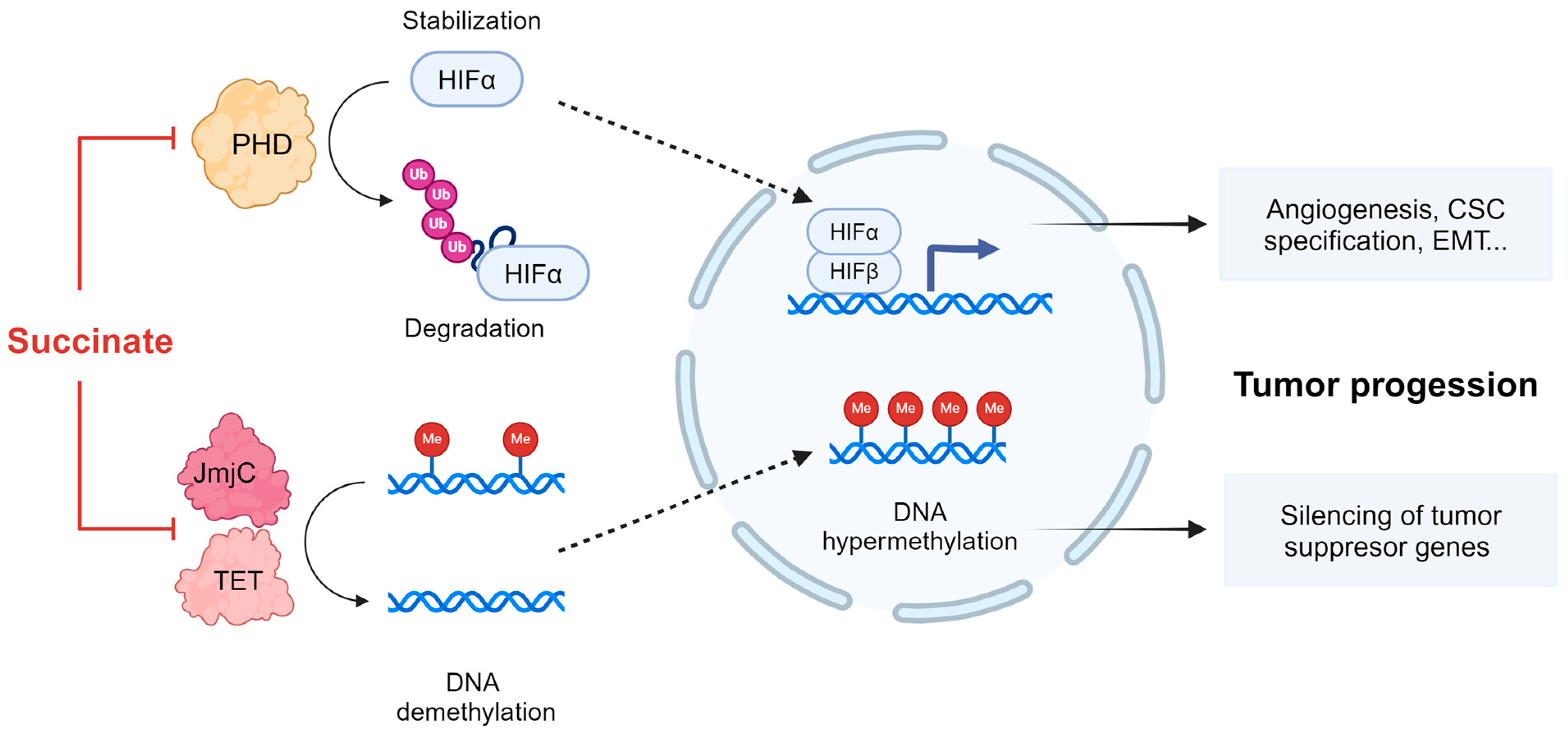
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate Links TCA Cycle Dysfunction to Oncogenesis by Inhibiting HIF-α Prolyl Hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef]
- Hirsilä, M.; Koivunen, P.; Günzler, V.; Kivirikko, K.I.; Myllyharju, J. Characterization of the Human Prolyl 4-Hydroxylases That Modify the Hypoxia-Inducible Factor. J. Biol. Chem. 2003, 278, 30772–30780. [Google Scholar] [CrossRef]
- Wicks, E.E.; Semenza, G.L. Hypoxia-Inducible Factors: Cancer Progression and Clinical Translation. J. Clin. Investig. 2022, 132, 159839. [Google Scholar] [CrossRef] [PubMed]
- Line Loriot, C.; Burnichon, N.; Mie Gadessaud, N.; Vescovo, L.; Amar, L.; Libé, R.; Bertherat, J.; Plouin, P.-F.O.; Jeunemaitre, X.; Gimenez-Roqueplo, A.-P.; et al. Epithelial to Mesenchymal Transition Is Activated in Metastatic Pheochromocytomas and Paragangliomas Caused by SDHB Gene Mutations. J. Clin. Endocrinol. Metab. 2012, 97, 954–962. [Google Scholar] [CrossRef]
- Kiss, N.B.; Muth, A.; Andreasson, A.; Juhlin, C.C.; Geli, J.; Bäckdahl, M.; Höög, A.; Wängberg, B.; Nilsson, O.; Ahlman, H.; et al. Acquired Hypermethylation of the P16 INK4A Promoter in Abdominal Paraganglioma: Relation to Adverse Tumor Phenotype and Predisposing Mutation. Endocr. Relat. Cancer 2013, 20, 65–78. [Google Scholar] [CrossRef]
- Bezawork-Geleta, A.; Wen, H.; Dong, L.; Yan, B.; Vider, J.; Boukalova, S.; Krobova, L.; Vanova, K.; Zobalova, R.; Sobol, M.; et al. Alternative Assembly of Respiratory Complex II Connects Energy Stress to Metabolic Checkpoints. Nat. Commun. 2018, 9, 2221. [Google Scholar] [CrossRef]
- Bandara, A.B.; Drake, J.C.; Brown, D.A. Complex II Subunit SDHD Is Critical for Cell Growth and Metabolism, Which Can Be Partially Restored with a Synthetic Ubiquinone Analog. BMC Mol. Cell Biol. 2021, 22, 35. [Google Scholar] [CrossRef]
- Hart, M.L.; Quon, E.; Vigil, A.-L.B.; Engstrom, I.A.; Newsom, O.J.; Davidsen, K.; Hoellerbauer, P.; Carlisle, S.M.; Sullivan, L.B. Mitochondrial Redox Adaptations Enable Alternative Aspartate Synthesis in SDH-Deficient Cells. eLife 2023, 12, 78654. [Google Scholar] [CrossRef]
- Saxena, N.; Maio, N.; Crooks, D.R.; Ricketts, C.J.; Yang, Y.; Wei, M.H.; Fan, T.W.M.; Lane, A.N.; Sourbier, C.; Singh, A.; et al. SDHB-Deficient Cancers: The Role of Mutations That Impair Iron Sulfur Cluster Delivery. J. Natl. Cancer Inst. 2016, 108, 287. [Google Scholar] [CrossRef] [PubMed]
- Gobelli, D.; Serrano-Lorenzo, P.; Esteban-Amo, M.J.; Serna, J.; Pérez-García, M.T.; Orduña, A.; Jourdain, A.A.; Martín-Casanueva, M.; de la Fuente, M.Á.; Simarro, M. The Mitochondrial Succinate Dehydrogenase Complex Controls the STAT3-IL-10 Pathway in Inflammatory Macrophages. iScience 2023, 26, 107473. [Google Scholar] [CrossRef] [PubMed]
- Cardaci, S.; Zheng, L.; Mackay, G.; Van Den Broek, N.J.F.; Mackenzie, E.D.; Nixon, C.; Stevenson, D.; Tumanov, S.; Bulusu, V.; Kamphorst, J.J.; et al. Pyruvate Carboxylation Enables Growth of SDH-Deficient Cells by Supporting Aspartate Biosynthesis. Nat. Cell Biol. 2015, 17, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Lorendeau, D.; Rinaldi, G.; Boon, R.; Spincemaille, P.; Metzger, K.; Jäger, C.; Christen, S.; Dong, X.; Kuenen, S.; Voordeckers, K.; et al. Dual Loss of Succinate Dehydrogenase (SDH) and Complex I Activity Is Necessary to Recapitulate the Metabolic Phenotype of SDH Mutant Tumors. Metab. Eng. 2017, 43, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Alston, C.L.; Davison, J.E.; Meloni, F.; van der Westhuizen, F.H.; He, L.; Hornig-Do, H.T.; Peet, A.C.; Gissen, P.; Goffrini, P.; Ferrero, I.; et al. Recessive Germline SDHA and SDHB Mutations Causing Leukodystrophy and Isolated Mitochondrial Complex II Deficiency. J. Med. Genet. 2012, 49, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Grønborg, S.; Darin, N.; Miranda, M.J.; Damgaard, B.; Cayuela, J.A.; Oldfors, A.; Kollberg, G.; Hansen, T.V.O.; Ravn, K.; Wibrand, F.; et al. Leukoencephalopathy Due to Complex II Deficiency and Bi-Allelic SDHB Mutations: Further Cases and Implications for Genetic Counselling. JIMD Rep. 2017, 33, 69–77. [Google Scholar]
- Renkema, G.H.; Wortmann, S.B.; Smeets, R.J.; Venselaar, H.; Antoine, M.; Visser, G.; Ben-Omran, T.; van den Heuvel, L.P.; Timmers, H.J.L.M.; Smeitink, J.A.; et al. SDHA Mutations Causing a Multisystem Mitochondrial Disease: Novel Mutations and Genetic Overlap with Hereditary Tumors. Eur. J. Hum. Genet. 2015, 23, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Van Coster, R.; Seneca, S.; Smet, J.; Van Hecke, R.; Gerlo, E.; Devreese, B.; Van Beeumen, J.; Leroy, J.G.; De Meirleir, L.; Lissens, W. Homozygous Gly555Glu Mutation in the Nuclear-Encoded 70 KDa Flavoprotein Gene Causes Instability of the Respiratory Chain Complex II. Am. J. Med. Genet. Part A 2003, 120A, 13–18. [Google Scholar] [CrossRef]
- Liu, Y.; Fiskum, G.; Schubert, D. Generation of Reactive Oxygen Species by the Mitochondrial Electron Transport Chain. J. Neurochem. 2002, 80, 780–787. [Google Scholar] [CrossRef]
- Robb, E.L.; Hall, A.R.; Prime, T.A.; Eaton, S.; Szibor, M.; Viscomi, C.; James, A.M.; Murphy, M.P. Control of Mitochondrial Superoxide Production by Reverse Electron Transport at Complex I. J. Biol. Chem. 2018, 293, 9869–9879. [Google Scholar] [CrossRef]
- Guzy, R.D.; Sharma, B.; Bell, E.; Chandel, N.S.; Schumacker, P.T. Loss of the SdhB, but Not the SdhA, Subunit of Complex II Triggers Reactive Oxygen Species-Dependent Hypoxia-Inducible Factor Activation and Tumorigenesis. Mol. Cell. Biol. 2008, 28, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Kluckova, K.; Sticha, M.; Cerny, J.; Mracek, T.; Dong, L.; Drahota, Z.; Gottlieb, E.; Neuzil, J.; Rohlena, J. Ubiquinone-Binding Site Mutagenesis Reveals the Role of Mitochondrial Complex II in Cell Death Initiation. Cell Death Dis. 2015, 6, 1749. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial Complex II Can Generate Reactive Oxygen Species at High Rates in Both the Forward and Reverse Reactions. J. Biol. Chem. 2012, 287, 27255–27264. [Google Scholar] [CrossRef] [PubMed]
- Siebels, I.; Dröse, S. Q-Site Inhibitor Induced ROS Production of Mitochondrial Complex II Is Attenuated by TCA Cycle Dicarboxylates. Biochim. Biophys. Acta (BBA)-Bioenerg. 2013, 1827, 1156–1164. [Google Scholar] [CrossRef] [PubMed]
- Bonke, E.; Zwicker, K.; Dröse, S. Manganese Ions Induce H 2 O 2 Generation at the Ubiquinone Binding Site of Mitochondrial Complex II. Arch. Biochem. Biophys. 2015, 580, 75–83. [Google Scholar] [CrossRef]
- Maklashina, E.; Rajagukguk, S.; Iverson, T.M.; Cecchini, G. The Unassembled Flavoprotein Subunits of Human and Bacterial Complex II Have Impaired Catalytic Activity and Generate Only Minor Amounts of ROS. J. Biol. Chem. 2018, 293, 7754–7765. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Rosen, P.C.; Sprenger, H.G.; Puszynska, A.M.; Mann, J.L.; Roessler, J.M.; Cangelosi, A.L.; Henne, A.; Condon, K.J.; Zhang, T.; et al. Fumarate Is a Terminal Electron Acceptor in the Mammalian Electron Transport Chain. Science 2021, 374, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Lussey-Lepoutre, C.; Hollinshead, K.E.R.; Ludwig, C.; Menara, M.; Morin, A.; Castro-Vega, L.-J.; Parker, S.J.; Janin, M.; Martinelli, C.; Ottolenghi, C.; et al. ARTICLE Loss of Succinate Dehydrogenase Activity Results in Dependency on Pyruvate Carboxylation for Cellular Anabolism. Nat. Commun. 2015, 6, 8784. [Google Scholar] [CrossRef]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; Deberardinis, R.J. Reductive Carboxylation Supports Growth in Tumour Cells with Defective Mitochondria. Nature 2011, 481, 385–388. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to Clinic: Glutamine Metabolism to Cancer. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Fendt, S.M.; Bell, E.L.; Keibler, M.A.; Olenchock, B.A.; Mayers, J.R.; Wasylenko, T.M.; Vokes, N.I.; Guarente, L.; Heiden, M.G.V.; Stephanopoulos, G. Reductive Glutamine Metabolism Is a Function of the α-Ketoglutarate to Citrate Ratio in Cells. Nat. Commun. 2013, 4, 2236. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond Aerobic Glycolysis: Transformed Cells Can Engage in Glutamine Metabolism That Exceeds the Requirement for Protein and Nucleotide Synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine Reliance in Cell Metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef] [PubMed]
- Ricci, L.; Stanley, F.U.; Eberhart, T.; Mainini, F.; Sumpton, D.; Cardaci, S. Pyruvate Transamination and NAD Biosynthesis Enable Proliferation of Succinate Dehydrogenase-Deficient Cells by Supporting Aerobic Glycolysis. Cell Death Dis. 2023, 14, 403. [Google Scholar] [CrossRef]
- Yang, R.Z.; Blaileanu, G.; Hansen, B.C.; Shuldiner, A.R.; Gong, D.W. CDNA Cloning, Genomic Structure, Chromosomal Mapping, and Functional Expression of a Novel Human Alanine Aminotransferase. Genomics 2002, 79, 445–450. [Google Scholar] [CrossRef]
- Kim, M.; Gwak, J.; Hwang, S.; Yang, S.; Jeong, S.M. Mitochondrial GPT2 Plays a Pivotal Role in Metabolic Adaptation to the Perturbation of Mitochondrial Glutamine Metabolism. Oncogene 2019, 38, 4729–4738. [Google Scholar] [CrossRef]
- Gaude, E.; Schmidt, C.; Gammage, P.A.; Szabadkai, G.; Minczuk, M. NADH Shuttling Couples Cytosolic Reductive Carboxylation of Glutamine with Glycolysis in Cells with Mitochondrial Dysfunction. Mol. Cell 2018, 69, 581–593. [Google Scholar] [CrossRef]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive Glutamine Metabolism by IDH1 Mediates Lipogenesis under Hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Vander Heiden, M.G. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 2015, 162, 552–563. [Google Scholar] [CrossRef]
- Jiang, L.; Shestov, A.A.; Swain, P.; Yang, C.; Parker, S.J.; Wang, Q.A.; Terada, L.S.; Adams, N.D.; McCabe, M.T.; Pietrak, B.; et al. Reductive Carboxylation Supports Redox Homeostasis during Anchorage-Independent Growth. Nature 2016, 532, 255–258. [Google Scholar] [CrossRef]
- Iverson, T.M.; Singh, P.K.; Cecchini, G. An Evolving View of Complex II-Noncanonical Complexes, Megacomplexes, Respiration, Signaling, and Beyond. J. Biol. Chem. 2023, 299, 104761. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Strange, K.; Telange, R.; Guo, A.; Hatch, H.; Sobh, A.; Elie, J.; Carter, A.M.; Totenhagen, J.; Tan, C.; et al. Genetic Impairment of Succinate Metabolism Disrupts Bioenergetic Sensing in Adrenal Neuroendocrine Cancer. Cell Rep. 2022, 40, 111218. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Maklashina, E.; Voehler, M.; Balintova, S.; Dvorakova, S.; Kraus, M.; Vanova, K.H.; Nahacka, Z.; Zobalova, R.; Boukalova, S.; et al. Disordered-to-Ordered Transitions in Assembly Factors Allow the Complex II Catalytic Subunit to Switch Binding Partners. Nat. Commun. 2024, 15, 473. [Google Scholar] [CrossRef]
- Lobo-Jarne, T.; Ugalde, C. Respiratory Chain Supercomplexes: Structures, Function and Biogenesis. Semin. Cell Dev. Biol. 2018, 76, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Scott Budigner, G.R.; et al. Metformin Inhibits Mitochondrial Complex I of Cancer Cells to Reduce Tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Canul-Tec, J.C.; Assal, R.; Cirri, E.; Legrand, P.; Brier, S.; Chamot-Rooke, J.; Reyes, N. Structure and Allosteric Inhibition of Excitatory Amino Acid Transporter 1. Nature 2017, 544, 446–451. [Google Scholar] [CrossRef]
- Titov, D.V.; Cracan, V.; Goodman, R.P.; Peng, J.; Grabarek, Z.; Mootha, V.K. Complementation of Mitochondrial Electron Transport Chain by Manipulation of the NAD+/NADH Ratio. Science 2016, 352, 231–235. [Google Scholar] [CrossRef]
- Herzig, S.; Raemy, E.; Montessuit, S.; Veuthey, J.L.; Zamboni, N.; Westermann, B.; Kunji, E.R.S.; Martinou, J.C. Identification and Functional Expression of the Mitochondrial Pyruvate Carrier. Science 2012, 336, 93–96. [Google Scholar] [CrossRef]
- Toney, M.D. Aspartate Aminotransferase: An Old Dog Teaches New Tricks. Arch. Biochem. Biophys. 2014, 544, 119–127. [Google Scholar] [CrossRef]
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef]
- Borst, P. The Malate–Aspartate Shuttle (Borst Cycle): How It Started and Developed into a Major Metabolic Pathway. IUBMB Life 2020, 72, 2241–2259. [Google Scholar] [CrossRef] [PubMed]
- Altea-Manzano, P.; Vandekeere, A.; Edwards-Hicks, J.; Fendt, S.-M.; Martin-Hernandez, M.; Finch, A.J.; Roldan, M.; Abraham, E.; Lleshi, X.; Naila Guerrieri, A.; et al. Reversal of Mitochondrial Malate Dehydrogenase 2 Enables Anaplerosis via Redox Rescue in Respiration-Deficient Cells. Mol. Cell 2022, 82, 4537–4547. [Google Scholar] [CrossRef] [PubMed]
- Bui, B.P.; Nguyen, P.L.; Lee, K.; Cho, J. Hypoxia-Inducible Factor-1: A Novel Therapeutic Target for the Management of Cancer, Drug Resistance, and Cancer-Related Pain. Cancers 2022, 14, 6054. [Google Scholar] [CrossRef]
- Peng, S.; Zhang, J.; Tan, X.; Huang, Y.; Xu, J.; Silk, N.; Zhang, D.; Liu, Q.; Jiang, J. The VHL/HIF Axis in the Development and Treatment of Pheochromocytoma/Paraganglioma. Front. Endocrinol. 2020, 11, 586857. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esteban-Amo, M.J.; Jiménez-Cuadrado, P.; Serrano-Lorenzo, P.; de la Fuente, M.Á.; Simarro, M. Succinate Dehydrogenase and Human Disease: Novel Insights into a Well-Known Enzyme. Biomedicines 2024, 12, 2050. https://doi.org/10.3390/biomedicines12092050
Esteban-Amo MJ, Jiménez-Cuadrado P, Serrano-Lorenzo P, de la Fuente MÁ, Simarro M. Succinate Dehydrogenase and Human Disease: Novel Insights into a Well-Known Enzyme. Biomedicines. 2024; 12(9):2050. https://doi.org/10.3390/biomedicines12092050
Chicago/Turabian StyleEsteban-Amo, María J., Patricia Jiménez-Cuadrado, Pablo Serrano-Lorenzo, Miguel Á. de la Fuente, and María Simarro. 2024. "Succinate Dehydrogenase and Human Disease: Novel Insights into a Well-Known Enzyme" Biomedicines 12, no. 9: 2050. https://doi.org/10.3390/biomedicines12092050
APA StyleEsteban-Amo, M. J., Jiménez-Cuadrado, P., Serrano-Lorenzo, P., de la Fuente, M. Á., & Simarro, M. (2024). Succinate Dehydrogenase and Human Disease: Novel Insights into a Well-Known Enzyme. Biomedicines, 12(9), 2050. https://doi.org/10.3390/biomedicines12092050