
Transcriptional Regulation and Function of Malic Enzyme 1 in Human Macrophage Activation

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Analysis of GEO Datasets for ME1 Expression
2.2. Isolation of Human PBMCs from Whole-Blood and Differentiation of PBMC-Derived Macrophages
2.3. RNA Interference and Treatments
2.4. Western Blot Analysis
2.5. Quantitative Real-Time PCR (RT-qPCR)
2.6. ME1 Activity
2.7. In Silico Analysis of ME1 Promoters
2.8. ChIP-qPCR
2.9. ROS and NO Detection
2.10. PGE2 and TNF-α Detection
2.11. Statistical Analysis
3. Results
3.1. ME1 Expression in Rheumatoid Arthritis and Systemic Lupus Erythematosus
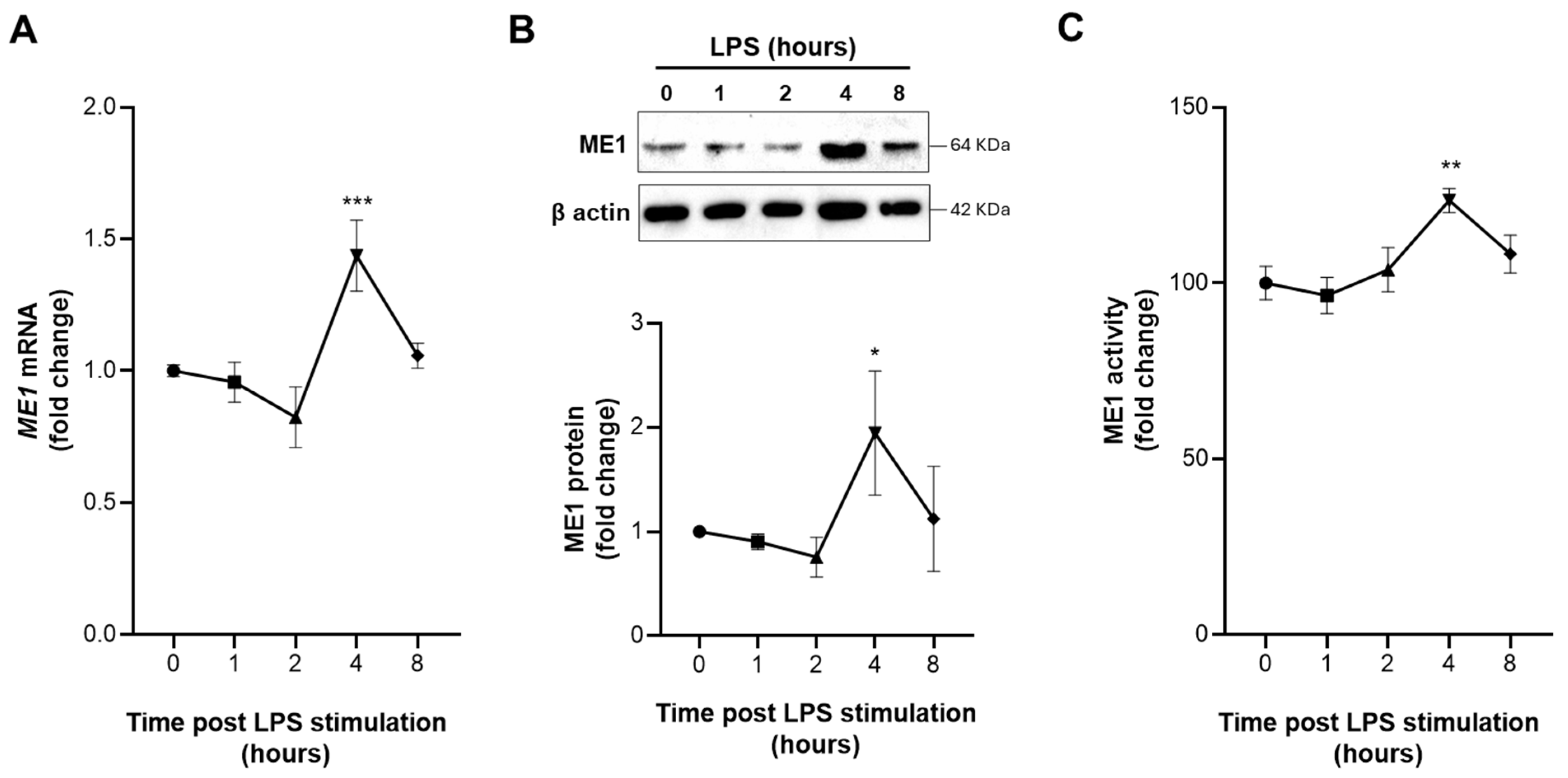
3.2. Early Activation of ME1 in LPS-Treated Human Macrophages
3.3. Identification and Functionality of the NF-κB-Responsive Element in ME1 Gene Promoter
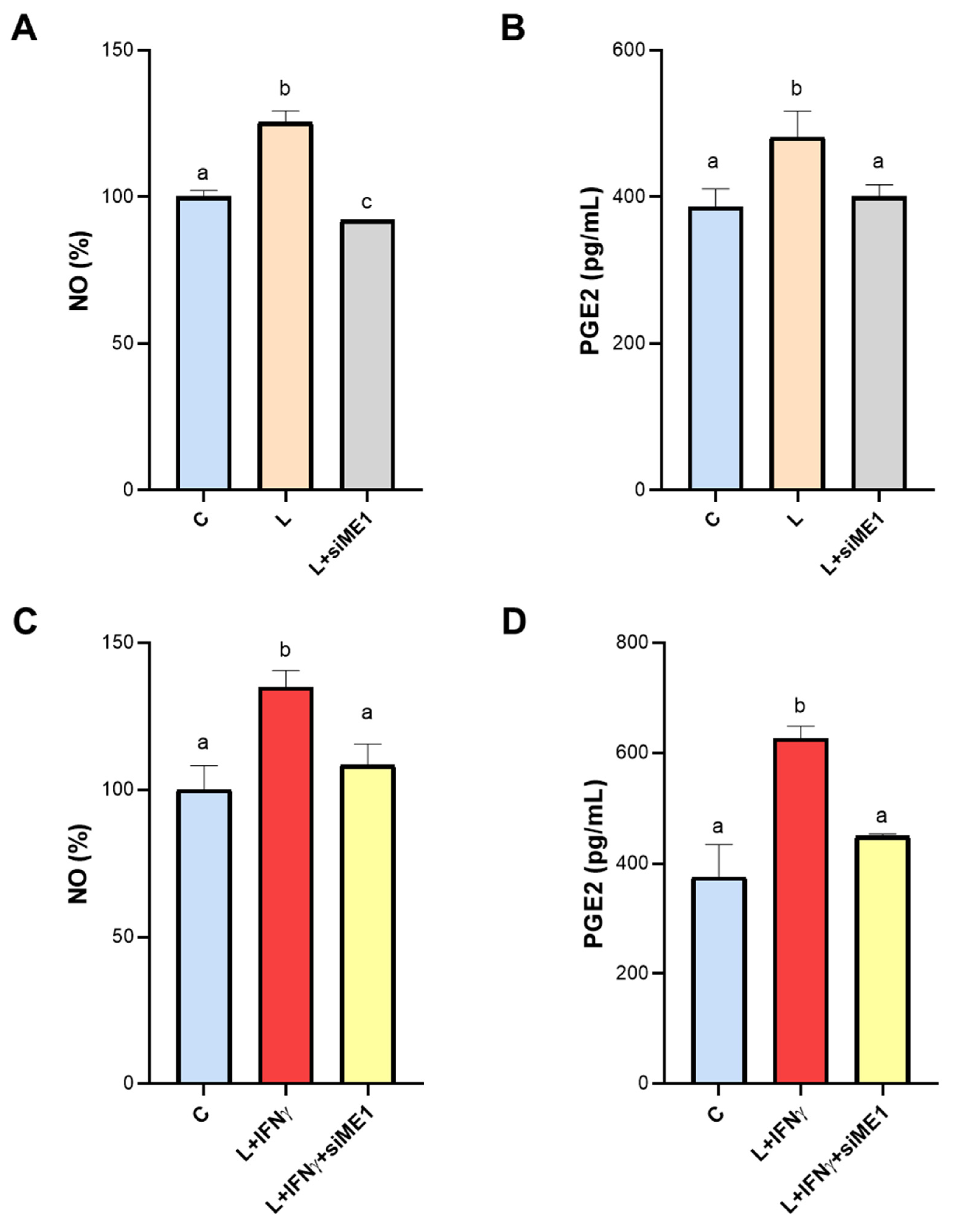
3.4. Effect of ME1 Gene Silencing on NO and PGE2 Secretion
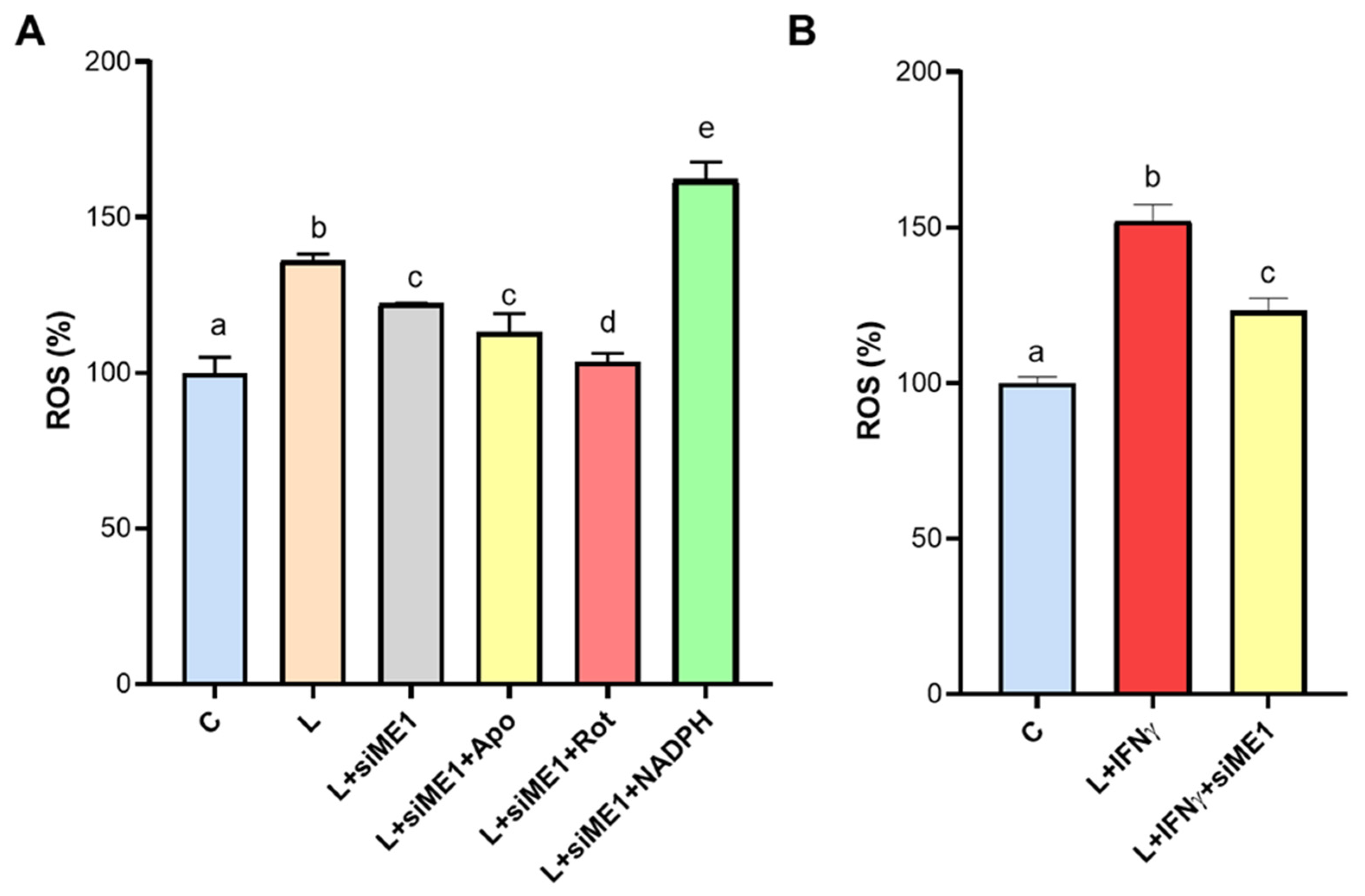
3.5. ROS Generation Mediated by ME1 in M1-like State Macrophages
3.6. Effect of ME1 Gene Silencing on TNF-α Secretion
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hsieh, J.Y.; Li, S.Y.; Chen, M.C.; Yang, P.C.; Chen, H.Y.; Chan, N.L.; Liu, J.H.; Hung, H.C. Structural characteristics of the nonallosteric human cytosolic malic enzyme. Biochim. Biophys. Acta 2014, 1844, 1773–1783. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S. NADPH-The Forgotten Reducing Equivalent. Cold Spring Harb. Perspect. Biol. 2021, 13, a040550. [Google Scholar] [CrossRef] [PubMed]
- Murai, S.; Ando, A.; Ebara, S.; Hirayama, M.; Satomi, Y.; Hara, T. Inhibition of malic enzyme 1 disrupts cellular metabolism and leads to vulnerability in cancer cells in glucose-restricted conditions. Oncogenesis 2017, 6, e329. [Google Scholar] [CrossRef] [PubMed]
- Todisco, S.; Convertini, P.; Iacobazzi, V.; Infantino, V. TCA Cycle Rewiring as Emerging Metabolic Signature of Hepatocellular Carcinoma. Cancers 2019, 12, 68. [Google Scholar] [CrossRef]
- Hu, W.C.; Yang, Y.F.; Cheng, C.F.; Tu, Y.T.; Chang, H.T.; Tsai, K.W. Overexpression of malic enzyme is involved in breast cancer growth and is correlated with poor prognosis. J. Cell Mol. Med. 2024, 28, e18163. [Google Scholar] [CrossRef]
- Simmen, F.A.; Alhallak, I.; Simmen, R.C.M. Malic enzyme 1 (ME1) in the biology of cancer: It is not just intermediary metabolism. J. Mol. Endocrinol. 2020, 65, R77–R90. [Google Scholar] [CrossRef]
- Yao, P.; Sun, H.; Xu, C.; Chen, T.; Zou, B.; Jiang, P.; Du, W. Evidence for a direct cross-talk between malic enzyme and the pentose phosphate pathway via structural interactions. J. Biol. Chem. 2017, 292, 17113–17120. [Google Scholar] [CrossRef]
- Wen, D.; Liu, D.; Tang, J.; Dong, L.; Liu, Y.; Tao, Z.; Wan, J.; Gao, D.; Wang, L.; Sun, H.; et al. Malic enzyme 1 induces epithelial-mesenchymal transition and indicates poor prognosis in hepatocellular carcinoma. Tumour Biol. 2015, 36, 6211–6221. [Google Scholar] [CrossRef]
- Nakashima, C.; Yamamoto, K.; Fujiwara-Tani, R.; Luo, Y.; Matsushima, S.; Fujii, K.; Ohmori, H.; Sasahira, T.; Sasaki, T.; Kitadai, Y.; et al. Expression of cytosolic malic enzyme (ME1) is associated with disease progression in human oral squamous cell carcinoma. Cancer Sci. 2018, 109, 2036–2045. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Locati, M. Macrophage polarization comes of age. Immunity 2005, 23, 344–346. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Fukui, S.; Iwamoto, N.; Takatani, A.; Igawa, T.; Shimizu, T.; Umeda, M.; Nishino, A.; Horai, Y.; Hirai, Y.; Koga, T.; et al. M1 and M2 Monocytes in Rheumatoid Arthritis: A Contribution of Imbalance of M1/M2 Monocytes to Osteoclastogenesis. Front. Immunol. 2017, 8, 1958. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, T.; Chen, W. Modulation of Macrophage Immunometabolism: A New Approach to Fight Infections. Front. Immunol. 2022, 13, 780839. [Google Scholar] [CrossRef]
- Wang, L.; Wang, D.; Zhang, T.; Ma, Y.; Tong, X.; Fan, H. The role of immunometabolism in macrophage polarization and its impact on acute lung injury/acute respiratory distress syndrome. Front. Immunol. 2023, 14, 1117548. [Google Scholar] [CrossRef] [PubMed]
- Van den Bossche, J.; O’Neill, L.A.; Menon, D. Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol. 2017, 38, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Infantino, V.; Pierri, C.L.; Iacobazzi, V. Metabolic Routes in Inflammation: The Citrate Pathway and its Potential as Therapeutic Target. Curr. Med. Chem. 2019, 26, 7104–7116. [Google Scholar] [CrossRef]
- Groh, L.; Keating, S.T.; Joosten, L.A.B.; Netea, M.G.; Riksen, N.P. Monocyte and macrophage immunometabolism in atherosclerosis. Semin. Immunopathol. 2018, 40, 203–214. [Google Scholar] [CrossRef]
- Makowski, L.; Chaib, M.; Rathmell, J.C. Immunometabolism: From basic mechanisms to translation. Immunol. Rev. 2020, 295, 5–14. [Google Scholar] [CrossRef]
- Santarsiero, A.; Convertini, P.; Todisco, S.; Pierri, C.L.; De Grassi, A.; Williams, N.C.; Iacobazzi, D.; De Stefano, G.; O’Neill, L.A.J.; Infantino, V. ACLY Nuclear Translocation in Human Macrophages Drives Proinflammatory Gene Expression by NF-κB Acetylation. Cells 2021, 10, 2962. [Google Scholar] [CrossRef]
- Lauterbach, M.A.; Hanke, J.E.; Serefidou, M.; Mangan, M.S.J.; Kolbe, C.C.; Hess, T.; Rothe, M.; Kaiser, R.; Hoss, F.; Gehlen, J.; et al. Toll-like Receptor Signaling Rewires Macrophage Metabolism and Promotes Histone Acetylation via ATP-Citrate Lyase. Immunity 2019, 51, 997–1011.e7. [Google Scholar] [CrossRef]
- Baardman, J.; Verberk, S.G.S.; van der Velden, S.; Gijbels, M.J.J.; van Roomen, C.P.P.A.; Sluimer, J.C.; Broos, J.Y.; Griffith, G.R.; Prange, K.H.M.; van Weeghel, M.; et al. Macrophage ATP citrate lyase deficiency stabilizes atherosclerotic plaques. Nat. Commun. 2020, 11, 6296. [Google Scholar] [CrossRef] [PubMed]
- Convertini, P.; Menga, A.; Andria, G.; Scala, I.; Santarsiero, A.; Castiglione Morelli, M.A.; Iacobazzi, V.; Infantino, V. The contribution of the citrate pathway to oxidative stress in Down syndrome. Immunology 2016, 149, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Convertini, P.; Santarsiero, A.; Todisco, S.; Gilio, M.; Palazzo, D.; Pappalardo, I.; Iacobazzi, D.; Frontuto, M.; Infantino, V. ACLY as a modulator of liver cell functions and its role in Metabolic Dysfunction-Associated Steatohepatitis. J. Transl. Med. 2023, 21, 568. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Santarsiero, A.; Convertini, P.; Vassallo, A.; Santoro, V.; Todisco, S.; Iacobazzi, D.; Fondufe-Mittendorf, Y.; Martelli, G.; de Oliveira, M.R.; Montanaro, R.; et al. Phenolic Compounds of Red Wine. Oxid. Med. Cell Longev. 2021, 2021, 5533793. [Google Scholar] [CrossRef]
- Convertini, P.; Todisco, S.; De Santis, F.; Pappalardo, I.; Iacobazzi, D.; Castiglione Morelli, M.A.; Fondufe-Mittendorf, Y.N.; Martelli, G.; Palmieri, F.; Infantino, V. Transcriptional Regulation Factors of the Human Mitochondrial Aspartate/Glutamate Carrier Gene, Isoform 2. Int. J. Mol. Sci. 2019, 20, 1888. [Google Scholar] [CrossRef]
- Zhou, Z.; Luo, G.; Li, C.; Zhang, P.; Chen, W.; Li, X.; Tang, J.; Qing, L. Metformin induces M2 polarization via AMPK/PGC-1α/PPAR-γ pathway to improve peripheral nerve regeneration. Am. J. Transl. Res. 2023, 15, 3778–3792. [Google Scholar]
- Abdalla, H.B.; Napimoga, M.H.; Lopes, A.H.; de Macedo Maganin, A.G.; Cunha, T.M.; Van Dyke, T.E.; Clemente Napimoga, J.T. Activation of PPAR-γ induces macrophage polarization and reduces neutrophil migration mediated by heme oxygenase 1. Int. Immunopharmacol. 2020, 84, 106565. [Google Scholar] [CrossRef]
- Ghosh, S.; Hayden, M.S. New regulators of NF-kappaB in inflammation. Nat. Rev. Immunol. 2008, 8, 837–848. [Google Scholar] [CrossRef]
- Kang, K.; Bachu, M.; Park, S.H.; Kang, K.; Bae, S.; Park-Min, K.H.; Ivashkiv, L.B. IFN-γ selectively suppresses a subset of TLR4-activated genes and enhancers to potentiate macrophage activation. Nat. Commun. 2019, 10, 3320. [Google Scholar] [CrossRef]
- Liu, H.; Sidiropoulos, P.; Song, G.; Pagliari, L.J.; Birrer, M.J.; Stein, B.; Anrather, J.; Pope, R.M. TNF-alpha gene expression in macrophages: Regulation by NF-kappa B is independent of c-Jun or C/EBP beta. J. Immunol. 2000, 164, 4277–4285. [Google Scholar] [CrossRef] [PubMed]
- Jang, D.I.; Lee, A.H.; Shin, H.Y.; Song, H.R.; Park, J.H.; Kang, T.B.; Lee, S.R.; Yang, S.H. The Role of Tumor Necrosis Factor Alpha (TNF-α) in Autoimmune Disease and Current TNF-α Inhibitors in Therapeutics. Int. J. Mol. Sci. 2021, 22, 2719. [Google Scholar] [CrossRef] [PubMed]
- Ghorbaninezhad, F.; Leone, P.; Alemohammad, H.; Najafzadeh, B.; Nourbakhsh, N.S.; Prete, M.; Malerba, E.; Saeedi, H.; Tabrizi, N.J.; Racanelli, V.; et al. Tumor necrosis factor-α in systemic lupus erythematosus: Structure, function and therapeutic implications. Int. J. Mol. Med. 2022, 49, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Xie, J.; Zhao, L.; Fei, X.; Zhang, H.; Tan, Y.; Nie, X.; Zhou, L.; Liu, Z.; Ren, Y.; et al. Alveolar macrophage dysfunction and cytokine storm in the pathogenesis of two severe COVID-19 patients. EBioMedicine 2020, 57, 102833. [Google Scholar] [CrossRef]
- Belchamber, K.B.R.; Donnelly, L.E. Macrophage Dysfunction in Respiratory Disease. Results Probl. Cell Differ. 2017, 62, 299–313. [Google Scholar]
- Iacobazzi, D.; Convertini, P.; Todisco, S.; Santarsiero, A.; Iacobazzi, V.; Infantino, V. New Insights into NF-κB Signaling in Innate Immunity: Focus on Immunometabolic Crosstalks. Biology 2023, 12, 776. [Google Scholar] [CrossRef]
- Todisco, S.; Santarsiero, A.; Convertini, P.; De Stefano, G.; Gilio, M.; Iacobazzi, V.; Infantino, V. PPAR Alpha as a Metabolic Modulator of the Liver: Role in the Pathogenesis of Nonalcoholic Steatohepatitis (NASH). Biology 2022, 11, 792. [Google Scholar] [CrossRef]
- Taylor, J.P.; Tse, H.M. The role of NADPH oxidases in infectious and inflammatory diseases. Redox Biol. 2021, 48, 102159. [Google Scholar] [CrossRef]
- Idelchik, M.D.P.S.; Begley, U.; Begley, T.J.; Melendez, J.A. Mitochondrial ROS control of cancer. Semin. Cancer Biol. 2017, 47, 57–66. [Google Scholar] [CrossRef]
- Mills, E.L.; O’Neill, L.A. Reprogramming mitochondrial metabolism in macrophages as an anti-inflammatory signal. Eur. J. Immunol. 2016, 46, 13–21. [Google Scholar] [CrossRef]
- Chen, S.; Saeed, A.F.U.H.; Liu, Q.; Jiang, Q.; Xu, H.; Xiao, G.G.; Rao, L.; Duo, Y. Macrophages in immunoregulation and therapeutics. Signal Transduct. Target. Ther. 2023, 8, 207. [Google Scholar] [CrossRef] [PubMed]
- Dweik, R.A.; Boggs, P.B.; Erzurum, S.C.; Irvin, C.G.; Leigh, M.W.; Lundberg, J.O.; Olin, A.C.; Plummer, A.L.; Taylor, D.R.; American Thoracic Society Committee on Interpretation of Exhaled Nitric Oxide Levels (FENO) for Clinical Applications. An official ATS clinical practice guideline: Interpretation of exhaled nitric oxide levels (FENO) for clinical applications. Am. J. Respir. Crit. Care Med. 2011, 184, 602–615. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, J.O.; Weitzberg, E. Nitric oxide signaling in health and disease. Cell 2022, 185, 2853–2878. [Google Scholar] [CrossRef] [PubMed]
- Ting, K.K.Y.; Jongstra-Bilen, J.; Cybulsky, M.I. The multi-faceted role of NADPH in regulating inflammation in activated myeloid cells. Front. Immunol. 2023, 14, 1328484. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HC (n = 19) | RA (n = 9) | |
|---|---|---|
| Age, mean ± SD (years) | 42.0 ± 13.3 | 50.8 ± 11.1 |
| Male sex, num (%) | 6 (32) | 1 (11) |
| Female sex, num (%) | 13 (68) | 8 (89) |
| HC (n = 25) | SLE (n = 15) | |
|---|---|---|
| Age, mean ± SD (years) | 32.2 ± 9.0 | 28.0 ± 8.7 |
| Male sex, num (%) | 5 (20) | 2 (13) |
| Female sex, num (%) | 20 (80) | 13 (87) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santarsiero, A.; Todisco, S.; Convertini, P.; De Leonibus, C.; Infantino, V. Transcriptional Regulation and Function of Malic Enzyme 1 in Human Macrophage Activation. Biomedicines 2024, 12, 2089. https://doi.org/10.3390/biomedicines12092089
Santarsiero A, Todisco S, Convertini P, De Leonibus C, Infantino V. Transcriptional Regulation and Function of Malic Enzyme 1 in Human Macrophage Activation. Biomedicines. 2024; 12(9):2089. https://doi.org/10.3390/biomedicines12092089
Chicago/Turabian StyleSantarsiero, Anna, Simona Todisco, Paolo Convertini, Chiara De Leonibus, and Vittoria Infantino. 2024. "Transcriptional Regulation and Function of Malic Enzyme 1 in Human Macrophage Activation" Biomedicines 12, no. 9: 2089. https://doi.org/10.3390/biomedicines12092089
APA StyleSantarsiero, A., Todisco, S., Convertini, P., De Leonibus, C., & Infantino, V. (2024). Transcriptional Regulation and Function of Malic Enzyme 1 in Human Macrophage Activation. Biomedicines, 12(9), 2089. https://doi.org/10.3390/biomedicines12092089