Abstract
Diabetes is a chronic metabolic disorder whose prevalence increases every year, affecting more than 530 million adults worldwide. Type 1 (T1D) and type 2 diabetes (T2D), the most common forms of diabetes, are characterized by the loss of functional pancreatic β-cells, mostly due to apoptosis. B-cell leukemia/lymphoma 2 (Bcl-2) and B-cell lymphoma-extra large (Bcl-xL), two anti-apoptotic proteins belonging to the Bcl-2 family, are crucial for regulating the intrinsic pathway of apoptosis. However, over the years, they have been implicated in many other cellular processes, including intracellular Ca2+ homeostasis and the regulation of mitochondrial metabolism. Thus, understanding the biological processes in which these proteins are involved may be crucial to designing new therapeutic targets. This review summarizes the roles of Bcl-2 and Bcl-xL in apoptosis and metabolic homeostasis. It focuses on how the dysregulation of Bcl-2 and Bcl-xL affects pancreatic β-cell function and survival, and the consequences for diabetes development.
1. Introduction
Diabetes is a chronic metabolic disorder characterized by elevated blood glucose levels, also known as hyperglycemia, which stems from a deficiency in insulin secretion, insulin action, or both. According to the 2021 International Diabetes Federation report, diabetes currently affects 537 million adults worldwide, and it has been projected that approximately 783 million will be living with diabetes by 2045 [1].
Type 1 (T1D) and type 2 diabetes (T2D), the most common forms of diabetes, are characterized by the loss of functional pancreatic β-cells, which are responsible for the synthesis and release of insulin, the main hormone involved in the regulation of blood glucose levels. Regarding their pathogenesis, T1D is the result of an autoimmune assault that culminates in islet inflammation and β-cell death, whereas T2D derives from mild-to-moderate β-cell loss due to metabolic stress (e.g., glucolipotoxicity) [2,3,4]. Despite their fundamentally different etiologies, a growing body of data suggests that T1D and T2D share common features concerning their development and progression. The onset of both diseases seems to mainly arise from a complex combination of a predisposing genetic background and environmental triggers [5,6]. While in T1D viral and bacterial infections, as well as dietarian immunogens, have been reported to induce or potentiate autoimmunity [7], T2D is primarily promoted by an obesogenic diet and lifestyle changes that can be worsened by exposure to environmental pollutants [8,9].
In T1D and T2D, pancreatic β-cell mass dysfunction and apoptosis are the aftermaths of stress responses to several insults, such as viral infections, proinflammatory cytokines, and free fatty acids [3]. These triggers activate stress-responsive pathways, including endoplasmic reticulum (ER) stress, oxidative stress response, autophagy, and cellular senescence, which are regulated by cell-specific and context-dependent transcription factors and gene/protein networks [10]. For example, the transcription factor nuclear factor-κB (NF-κB) is pro-apoptotic in β-cells [11,12] but has protective effects in other cell types [13].
The B-cell leukemia/lymphoma 2 (Bcl-2) family of proteins is crucial for the survival of pancreatic β-cells. The balance between pro- and anti-apoptotic members of the family in response to stress is a tightly regulated and dynamic process that determines whether cells undergo apoptosis via the intrinsic mitochondrial pathway or survive [14]. The crucial role of the Bcl-2 family of proteins in the cell fate has made them interesting actors in the search for pro-death therapies in cancer [15,16], and pro-survival therapies in diseases affecting cell types with low proliferation rates, such as neuronal pathologies [17,18] and diabetes [19]. The purpose of this review is to analyze the cell-specific and context-dependent roles of two anti-apoptotic members of the Bcl-2 family of proteins, namely, Bcl-2 and B-cell lymphoma-extra large (Bcl-xL). Moreover, we aim to shed light on recent research regarding their roles in β-cells. Finally, we discuss some limitations, gaps in the current knowledge, and potential therapeutic opportunities related to these pro-survival proteins in the context of β-cells and diabetes.
2. Function and Structure of Bcl-2 and Bcl-xL
2.1. Homology of Bcl-2 Proteins
The proteins belonging to the Bcl-2 family are crucial regulators of the cell cycle and survival. The anti-apoptotic protein Bcl-2 was the first member to be described in cancerous lymphocytic B-cells, where its exacerbated steady expression was related to increased cell viability and acute lymphocytic B-cell leukemia [20].
Since its discovery, 25 homologues of Bcl-2 have been described [21]. These homologues are divided into three categories, depending on the Bcl-2 homology (BH) domains present in their structure and their function: (1) anti-apoptotic proteins (Bcl-2, Bcl-xL, Bcl-W, Bcl-B, Bfl-1, and Mcl-1L), all harboring the four highly conserved BH regions (BH1, BH2, BH3, and BH4); (2) pro-apoptotic proteins (Bax, Bak, Bok, and Bcl-xS), which also share the four conserved BH1-4 domains; and (3) pro-apoptotic proteins exclusively presenting a single BH3 domain, also known as BH3-only proteins. This last category can be subdivided into BH3-only sensitizers (Bad, Bik, Bmf, Bnip3, Hrk/DP5, Beclin-1, and Noxa) and BH3-only activators (Bim, tBid, Mule, and Puma). Of note, several anti-apoptotic members of the family lack the BH4 domain, such as Mcl-1, Bfl-1/A1, and Bcl2L121 [21,22]. Additionally, other BH-3-only isoforms, namely, Bcl-Rambo, Bcl-G, Mcl-1S, and Mcl-1ES, have been proposed as potentially pro-apoptotic [23,24,25].
The BCL2 gene, located on chromosome 18, renders three mRNA variants that translate into two splicing alternatives: Bcl-2α, which consists of 239 amino acids and encodes the active, membrane-bound isoform; and Bcl-2β, which has only 205 residues due to a truncation of the hydrophobic tail at the C-terminus and lack the transmembrane domain [26,27]. Although the anti-apoptotic activity of the Bcl-2β isoform is still controversial [21], recent evidence suggests that it might be involved in promoting tumor angiogenesis [28].
Bcl-xL is one of the two isoforms yielded by alternative splicing of exon 2 of the BCL2L1 gene (Bcl-2 like 1), located on chromosome 20q11.21. The long, anti-apoptotic isoform Bcl-xL, which contains 233 amino acids, results from the selection of the proximal 5′ site on exon 2. In contrast, the short, pro-apoptotic isoform Bcl-xS is translated from the distal 5′ splicing site and contains 170 amino acids [29]. Due to its role as an antagonist of the anti-apoptotic function of Bcl-xL and Bcl-2, the alternative splicing of Bcl-xS is tightly regulated to favor a high Bcl-xL/Bcl-xS ratio under normal conditions [30].
2.2. Structure of Bcl-2 and Bcl-xL
Curiously, the crystallographic and solution structures of Bcl-xL [31] were established before those of Bcl-2 [32]. Briefly, Bcl-xL consists of two central hydrophobic α-helices, surrounded by amphipathic helices. Helices α1 and α2 are connected to a flexible loop of 60 residues, which is not essential for Bcl-xL anti-apoptotic activity. The BH1, BH2, and BH3 regions are spatially close, forming an elongated hydrophobic groove that may act as the binding site for interacting members of the Bcl-2 family [31].
Regarding the two Bcl-2 isoforms, their structures consist of six α-helices with a surface hydrophobic pocket similarly to Bcl-xL. Comparisons between Bcl-2 and Bcl-xL structures show that, although the overall fold is the same, their structural topology and electrostatic potential of the binding groove present some differences [32]. In Bcl-2α, the 22 amino acids in the COOH-terminal act as a signal-anchor sequence responsible for its integration into the outer mitochondrial membrane [33], but it is not essential for its anti-apoptotic effects [34].
2.3. Preferential Localization of Bcl-2 and Bcl-xL
In the early 1990s, a series of studies established that Bcl-2 was localized to the inner mitochondrial membrane, more specifically, at the interacting points between the external and internal mitochondrial membranes [33,35,36]. Later research, however, indicated that it is situated on the outer mitochondrial membrane [37,38,39,40,41]. Additionally, subsequent studies revealed the presence of Bcl-2 not only in the mitochondria, but also in the nuclear outer membrane, the nucleus, and the ER [42]. Similarly to Bcl-2, Bcl-xL is primarily localized to the outer mitochondrial membrane [14,43] but can also be found in the inner mitochondrial membrane, in the cytosol, or bound to the ER [44,45]. Many of these studies were performed in B-cell lymphoma cell lines [35,37,38,39,40,41,43,45]. However, this localization seems to be conserved among cell types, including heart [33], liver [36] and pancreatic β-cells [46,47,48]. Specifically, Luciani et al. described a strong correlation between Bcl-xL and mitochondrial membranes in β-cells, which was higher than its ER localization and Bcl-2 mitochondrial localization [47] in MIN6 cells. Later, Aharoni and colleagues confirmed Bcl-2 localization in the inner mitochondrial membrane of MIN6 cells and primary β-cells from C57/BL6 mice [46], in line with human islet reports that indicated a microsomal localization of the Bcl-2 anti-apoptotic protein [48].
The anti-apoptotic role of Bcl-2 might exhibit spatial dependence, varying according to the specific apoptotic pathway involved. For instance, ER-bound Bcl-2 is particularly effective in inhibiting starvation/myc-induced apoptosis, where a loss in mitochondrial membrane potential precedes the release of cytochrome c from mitochondria. In contrast, only Bcl-2 localized to the outer mitochondrial membrane can counteract etoposide-induced apoptosis, characterized by an early release of cytochrome c before the loss of mitochondrial membrane potential [49].
2.4. Interactions Within the Family of Bcl-2 Proteins
The complex interactions between pro- and anti-apoptotic members of the Bcl-2 family play a pivotal role in determining whether cells survive or undergo cell death through mitochondrial-mediated apoptosis, also known as the intrinsic pathway of apoptosis, in response to pathophysiological challenges. Classically, the role played by Bcl-2 and Bcl-xL in the regulation of the intrinsic pathway of apoptosis is known as their canonical function. As this canonical role has been previously reviewed elsewhere [50,51,52], here we will focus on how these proteins interact with other members of the Bcl-2 family involved in the intrinsic pathway of apoptosis.
Under basal conditions, BH3-only activators are sequestered in a heterodimer structure by pro-survival members of the Bcl-2 family, such as Bcl-2 and Bcl-xL. This inhibition occurs by direct interaction between the BH3 domain of BH3-only sensitizers and the highly conserved BH3 domain-binding pocket in both pro- and anti-apoptotic members of the family [53]. In the presence of apoptotic signals, these BH3-only activators are released from the anti-apoptotic proteins upon displacement by BH3-only sensitizers and are free to bind to pro-apoptotic partners of the family (Figure 1).
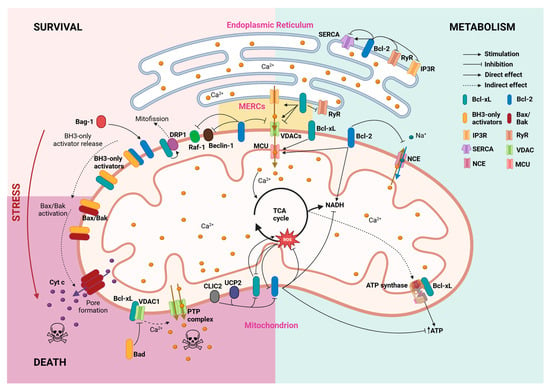
Figure 1.
Survival and metabolic homeostasis maintenance by Bcl-2 and Bcl-xL proteins. Through multiple interactions with mitochondrial and ER proteins, Bcl-2 (blue) and Bcl-xL (green) establish a network of processes that connect survival promotion with Ca2+ homeostasis and fluxes between both organelles. Arrows depict direct interactions, while dashed arrows refer to indirect effects reported in the literature. MERCs: mitochondria-endoplasmic reticulum contact sites; MCU: mitochondrial calcium uniporter; NCE: Na+-Ca2+ exchanger; PTP: permeability transition pore.
The interactions between Bcl-2 family proteins are determined by their abundance in each locus of action and the affinity between partner proteins [14]. Different models describe this process, among which the “embedded together” model emphasizes the importance of the intracellular membrane where these proteins are attached [54].
The interaction with activated BH3-only proteins induces a conformational change in the apoptotic effectors Bax/Bak, that disfavors their binding to anti-apoptotic Bcl-2 proteins, and which results in their homo-oligomerization and pore-formation in the outer membrane of the mitochondria.
Despite their high homology, Bcl-xL and Bcl-2 present different interactions with their associated pro-apoptotic antagonists, such as Bcl-2-associated death promoter (Bad) and Bcl-2-associated X protein (Bax) [55]. Computational interaction entropy analyses performed by Duan and colleagues found a higher affinity between Bcl-xL and Bad than Bcl-xL and Bax, which was also the case for Bcl-2. These researchers also identified the key residues in Bcl-xL complexes (arginine 104, tyrosine 105, leucine 116, and leucine 134) and Bcl-2 complexes (arginine 107, tyrosine 108, phenylalanine 112, glutamine 118, leucine 137, arginine 146, and tyrosine 202) with their pro-apoptotic counterparts [55].
In spite of the fact that the regulation of anti- and pro-apoptotic members of the Bcl-2 family occurs through direct interaction between their BH3 domains [56], the transmembrane domain of Bcl-2 proteins can mediate these interactions. In this line, Beigl and collaborators recently found that Bcl-2 regulates Bok-induced apoptosis through the direct interaction of their C-terminal transmembrane domains at the ER membrane. In fact, the Bcl-2 inhibition of Bok specifically depends on this interaction [57]. Furthermore, the functionality of the Bcl-xL transmembrane domain is also a novel matter of research. A recent study by Wu and colleagues found that Bcl-xL transmembrane domain shows strong anti-apoptotic activity on its own, comparable to the full-length protein, being able to directly inhibit Bad and Bax at the mitochondrial membrane of living HeLa cells [58].
Furthermore, the phosphorylation status seems to have a differential role in Bcl-xL and Bcl-2 proteins. While the phosphorylation of the serine residue at position 70 is crucial for the Bcl-2 full pro-survival phenotype [59], multiple-site phosphorylation can have a pro-apoptotic effect. Serum-starved cells presenting multiple Bcl-2 phosphorylations—including serine 70 among others—lose the Bcl-2 autophagy inhibition as phosphorylated Bcl-2 is unable to interact with the major autophagy regulator Beclin-1 [60]. On the other hand, the phosphorylation of the Bcl-xL serine-14 residue has an inhibitory effect, favoring its detachment from Bax [61]. Moreover, the phosphorylation of the serine-62 residue in Bcl-xL has been shown to reduce its ability to bind and retain Bax [62], instead causing it to interact with the cyclin-dependent kinase 1 to arrest the cell cycle during the G2 checkpoint [63].
2.5. Non-Canonical Functions of Bcl-2 and Bcl-xL
Besides their canonical roles in the hierarchical regulation of mitochondrion-dependent apoptosis, Bcl-2 proteins are also involved in other cellular processes, mainly through the regulation of Ca2+ homeostasis [64,65]. In this review, we will refer to these apoptosis-independent functions as the “non-canonical” roles of Bcl-2 and Bcl-xL.
2.5.1. ER Ca2+ Homeostasis
Many studies report that Bcl-2 and Bcl-xL can control intracellular Ca2+ homeostasis through different and complex pathways [66,67]. Both proteins are present at the surface of the two main cell Ca2+ reservoirs: the ER and the mitochondria. Moreover, as will be further discussed in this review, their interaction with both ER and mitochondrial Ca2+ machinery, together with their localization at the mitochondrial-associated ER membranes [68], suggests that these proteins could participate in the modulation of the ER–mitochondrial Ca2+ trafficking (Figure 1). In this line, a study from Williams and colleagues showed that Bcl-xL interacts with 1,4,5-trisphosphate receptor 3 (IP3R) type 3 at the mitochondrial-associated ER membrane to enhance transient ER-to-mitochondrial Ca2+ trafficking, dynamically modulating cell metabolism in CHO cells [69].
In contrast, ER-localized IP3R regulation by both Bcl-2 and Bcl-xL has been extensively studied [70,71,72]. Bcl-2 interaction with IP3R promotes survival by diminishing ER Ca2+ trafficking towards the cytosol and mitochondria through a BH4-domain dependent mechanism [70,71,72]. This effect has been narrowed to the Bcl-2α isoform [73]. Even though the ER Ca2+ lowering effects of Bcl-2 might seem contradictory to its anti-apoptotic nature, several hypotheses have arisen to explain this effect. One hypothesis suggests that modest Ca2+ intraluminal concentrations lead to reduced ER stress-induced Ca2+ mobilization and decreased mitochondrial Ca2+ uptake [74,75]. Supporting this hypothesis, Pinton and colleagues found that Bcl-2-mediated ER Ca2+ depletion prevented the ceramide-induced increase in cytoplasmic Ca2+ and mitochondrial damage, which protected HeLa cells from apoptosis [76]. On the other hand, the Bcl-2-induced reduction of Ca2+ release might not be related to a decrease in the ER Ca2+ pool but to the inhibition of IP3R and, therefore, reduction in Ca2+ efflux. In 1997, He and colleagues demonstrated that Bcl-2 prevented the depletion of the ER Ca2+ pool in mouse lymphoma cells [77]. Despite their differences, both hypotheses emphasize that the anti-apoptotic effect of Bcl-2 resides in its control of the ER-induced cytosolic Ca2+ oscillations.
The Bcl-xL interaction with IP3R has shown both functional and survival outcomes [70,78,79,80]. On the one hand, Bcl-xL interacts with the C-terminus of IP3R, sensitizing the channel to lower IP3 concentrations and thus enhancing both Ca2+- and IP3-dependent regulation of the channel. This effect is accompanied by a reduction in the ER Ca2+ content, the consequent sensitization of ER Ca2+ oscillations to extracellular signals, and increased mitochondrial bioenergetics, suggesting that Bcl-xL mediates ER–mitochondrial Ca2+ flux [79]. Recently, a study by Nakamura and colleagues deepened this issue and demonstrated that the ability of Bcl-xL to modulate the IP3R function depends on its phosphorylation status [61]. Alternatively, the Bcl-xL inhibitory effect on IP3R activity has a protective effect against Ca2+-driven apoptosis [81]. Furthermore, the inhibitory effect of Bcl-xL on IP3R extends to the regulation of the receptor expression [82]. An analysis of Bcl-xL effects on each of the three isoforms of IP3R, performed by Li and colleagues, demonstrated that while Bcl-xL only reduced ER Ca2+ storage in IP3R3-expressing cells, its enhancement of Ca2+ signaling was isoform-independent. Therefore, the pro-survival effects of Bcl-xL on IP3R are conferred through increasing the spontaneous intracellular Ca2+ oscillations rather than ER Ca2+ content dampening [83].
Ryanodine receptors (RyRs) are the other co-protagonists of receptor-mediated ER Ca2+ dynamics [84]. As with IP3R binding, the BH4 domain of Bcl-2 is essential for its interaction with RyRs [85]. In contrast to Bcl-2, Bcl-xL requires the combined contribution of its BH4 and BH3 domains to fully bind to RyR channels [86]. Importantly, regardless of the specific binding mechanism, Bcl-2 and Bcl-xL binding to RyRs leads to a decrease in RyR activity, thereby inhibiting RyR-mediated ER Ca2+ release [85,86] (Figure 1).
The Bcl-2 effect on the Ca2+ levels inside the lumen of the ER has also been hypothesized to act through the Bcl-2 regulation of store-operated Ca2+ entry [87] or through direct effects of Bcl-2 on the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA). The ability of Bcl-2 to control ER Ca2+ is directly related with its cell survival function, as phosphorylated forms of Bcl-2 are unable to lower ER Ca2+ content or interact with BH3-only members of the family [88]. To our knowledge, no relationship between Bcl-xL and SERCA has been reported. However, the relationship between Bcl-2 and the sarcoplasmic Ca2+ pump has been a matter of study since the late 1990s. In 1998, Kuo and colleagues reported that clonal Bcl-2 expression increased SERCA2 expression, promoting increased ER Ca2+ uptake [89]. Nonetheless, a truncated form of Bcl-2 directly inhibits SERCA activity in rat skeletal muscle. This inhibition was associated with a partial unfolding of SERCA, yet it did not involve proteolytic degradation or an increased sensitivity of SERCA to oxidation [90]. Together, these results show that differing functions of ER-related Bcl-2 may occur depending on cell context.
2.5.2. Mitochondrial Homeostasis
Along with the ER Ca2+ storages, the mitochondrial Ca2+ content is crucial for regulating cell survival and maintaining the Ca2+ reservoir [91,92]. The Ca2+ flux across the outer mitochondrial membrane occurs mainly through the voltage-dependent anion channels (VDACs), whose isoforms can transfer Ca2+ with different functional implications [93]. Bcl-xL was the first Bcl-2 protein to be described as able to interfere with VDACs by Craig Thompson and colleagues. These researchers provided evidence that Bcl-xL sustained coupled respiration and ATP production in growth factor-deprived cells by maintaining VDACs in an open state [94,95]. When the BH3-only protein Bad is dephosphorylated, it displaces Bcl-xL from VDAC1. Once liberated, VDAC1 forms the mitochondrial permeability transition pore, leading to the depletion of mitochondrial Ca2+ [96,97]. Furthermore, it has been stated that Bcl-xL direct interaction with VDCA1 and the less studied VDCA3 promotes mitochondrial Ca2+ intake [98]. In contrast, the BH4 domain from Bcl-xL, but not from Bcl-2, limits VDAC1 Ca2+ permeability, thus restricting the Ca2+ uptake by the organelle and inhibiting cell death [99,100]. For its part, Bcl-2 interaction with the N-terminal α-helix of VDAC1 inhibits channel function and has a cytoprotective effect [101].
The disparity of effects of Bcl-2 and Bcl-xL on mitochondrial Ca2+ status has been hypothesized to be a consequence of their implications in cell survival. In this model, under physiological conditions, Bcl-xL and Bcl-2 would promote mitochondrial Ca2+ uptake to stimulate bioenergetics, whereas under pro-apoptotic stimuli, they might inhibit the VDAC to prevent lethal Ca2+ overload in the mitochondria [93]. Following this hypothesis, it would be argued that Bcl-2 anti-apoptotic members could interfere in other mitochondrial Ca2+ exchange pathways. This extension of the model is supported by the results of Zhu and colleagues, who demonstrated that Bcl-2 overexpression increases mitochondrial Ca2+ matrix content by reducing the mitochondrial Na+-Ca2+ exchange in cardiomyocytes [102] (Figure 1).
Another crucial process in mitochondria homeostasis is the regulation of mitochondrial dynamics (i.e., mitochondrial fusion and fission) and mitophagy, of special relevance for neuronal physiology [103].
The protein dynamin-related protein 1 (Drp1) plays a crucial part in mitochondrial fission. In hippocampal neurons, Bcl-xL ensures neuronal survival and function by increasing the synaptic localization of mitochondria and acts through Drp1 to promote mitochondrial fission and the consequent mitophagy of damaged mitochondria [104]. Similarly to its role in the regulation of VDAC activity, Bcl-xL shows a dual effect on mitochondrial fission, as its counteraction of Bax and Bak inhibits mitochondrial-like membrane pore formation, budding, and fission in in vitro models [105]. Even though the Bcl-xL inhibition of Bax and Bak abolishes cytochrome c release, it is unable to counteract Bax- and Bak-initiated mitochondrial network remodeling in HeLa cells [106]. These findings suggest that Bcl-xL effects on mitochondrial network dynamics are the results of multiple factors.
Along with the promotion of the open state of VDACs discussed above, Bcl-xL control over mitochondrial dynamics might indirectly promote reactive oxygen species (ROS) production [107] and ATP production, which has already been proposed to contribute to neural plasticity [108]. For instance, Alavian and colleagues found out that Bcl-xL directly interacts with the mitochondrial F1FO-ATP synthase β-subunit, increasing metabolic efficiency by reducing ion leak and thus improving net H+ transport [109]. These findings were later confirmed by Chen and colleagues [110]. Furthermore, a decreased expression of Bcl-xL in hippocampal neurons reduces mitochondrial motility and ATP retainment, enhancing their vulnerability to excitotoxic insults [111]. Finally, and consistent with the previously mentioned results, the pro-metastatic effects of Bcl-xL and Bcl-2 in breast cancer cells can be at least partially attributed to their pathological promotion of VDAC activity, which is accompanied by mitochondrial Ca2+ uniporter increased activity and consequent ATP production [112].
Recent research has shown that the Bcl-xL regulatory effect on ATP synthase is shared with the anti-apoptotic Bcl-2 family member Mcl-1 but not with Bcl-2 itself [113]. This seems contradictory considering Bcl-2 ability to arrest both ATP and ROS production to delay cell cycle progression to S phase entry [114] (Figure 1). Later proteomics research by the same authors showed that the Bcl-2 arrest of the cell cycle progression from G0/G1 to S phase entry occurs via the downregulation of ribosomal dynamics and oxidative phosphorylation. These authors found that in serum-deprived fibroblasts, Bcl-2 decreased the expression of transcripts related to mitochondrial Ca2+ homeostasis, while the electron transport chain and response to nutrients pathways were upregulated [115]. The authors observed changes in NADH dehydrogenase, cytochrome c reductase/oxidase, and mitochondrial ATPase expression in Bcl-2-overexpressing fibroblasts. Curiously, in the case of ATPase, Bcl-2 expression upregulated the F subunit, while downregulating the B, H, and D subunits. These results indicate that Bcl-2 takes control of oxidative phosphorylation pathways, regulating ATP synthesis to counteract energy deficiency during the cell cycle arrest. Interestingly, while Bcl-2 proteins can regulate ROS production, that is a two-way street in which ROS increases have been shown to reduce the expression of Bcl-2, and both Bcl-2 and Bcl-xL phosphorylation in squamous cell carcinoma cells [116].
While Bcl-xL promotes the mitophagy of damaged mitochondria, Bcl-2 ensures that autophagy levels remain within the physiological range. It does so by inhibiting Beclin-1 [117]. Additionally, the Bcl-2 protein participates in a pathway that ensures mitochondrial integrity via Raf-1 sequestering to the outer mitochondrial membrane [118]. Interestingly, the modulation of Raf-1 expression via both Bcl-2 and Bcl-xL has a regulatory effect on cell differentiation, conducting hematopoietic differentiation towards myeloid or erythroid lineage [119].
In conclusion, Bcl-2 and Bcl-xL effects on mitochondrial homeostasis are not limited to the counteraction of pro-apoptotic signals. Contrarily, these members of the Bcl-2 family exert control on mitochondrial ATP production and Ca2+ homeostasis, ensuring a bioenergetic balance compatible with life.
3. Bcl-2 and Bcl-xL in Pancreatic Islets
Both in T1D and T2D, the clinical presentation of the disease is a consequence of the reduction in functional pancreatic β-cell mass below the threshold necessary for maintaining glucose homeostasis. As previously reviewed by Gurzov and Eizirik, the Bcl-2 family of proteins plays an important role in the β-cell demise in both T1D and T2D [52]. The pro-apoptotic members of this family Bax, Bad, Bak, DP5 and Puma have become classical markers of programmed cell death activation by proinflammatory cytokines and free fatty acids in β-cells [120,121,122,123]. In addition, the anti-apoptotic members of the family have also been studied in the context of β-cell survival. Lipotoxicity, a classical T2D insult, has been shown to downregulate Bcl-2, Bcl-xL, Mcl-1, and Bcl-w [124,125] in β-cells. Mirroring the establishment of Bax, Bad, Bak, DP5, and Puma as cell death standards, Mcl-1, Bcl-w, Bcl-2, and Bcl-xL have been used as cell survival markers in β-cell studies [126]. In this section, we will summarize what is currently known about the canonical (apoptosis-dependent) and non-canonical (apoptosis-independent) roles of Bcl-2 and Bcl-xL in different pancreatic cells.
3.1. Bcl-2 and Bcl-xL Effects in β-Cell Survival
There is no doubt that strong survival regulators such as Bcl-2 and Bcl-xL need a tight expression and function regulation, and deficiencies in this control are related to the development of different diseases, namely, different types of cancer [127,128], bone deficiencies [129], asthma complications [130], anemia [131,132,133], and neuronal abnormalities [134,135].
The protective effect of a high expression of Bcl-xL on pancreatic β-cell survival has been demonstrated by us and others. This was firstly reported by Zhou and colleagues, who found that the overexpression of human Bcl-xL in transgenic mouse islets was protective against thapsigargin-induced apoptosis [136]. Subsequent studies demonstrated that Bcl-xL protected β-cells from different insults, as described next. Klein and colleagues demonstrated that the exogenous overexpression of Bcl-xL or its anti-apoptotic BH4 domain protects the NIT-1 mouse β-cell line, as well as human and non-human primate islets, from starvation or staurosporine-induced cell death [137]. Similarly, Bcl-xL-overexpressing RIN-r β-cells were protected against different proinflammatory cytokines [138]. Furthermore, Cunha et al. demonstrated that palmitate-induced lipotoxic death in rat INS-1E β-cells is at least partially due to Bcl-2 and Bcl-xL depletion, and that Bcl-2 knock-down sensitizes INS-1E cells to free fatty acids [121]. Finally, we have recently shown that the effects of Bcl-xL overexpression on β-cell physiology seem to be species-dependent. Bcl-xL overexpression in both INS-1E and human EndoC-βH1 β-cells protects them from both cytokine- and palmitate-induced apoptosis, proving its ability to counteract classical death signals from T1D (i.e., proinflammatory cytokines) and T2D (i.e., palmitate). While Bcl-xL overexpression in rat INS-1E β-cells conferred a 40% protection against proinflammatory cytokines- and palmitate-induced apoptosis, INS-1E cells also showed impaired glucose-induced Ca2+ oscillations and insulin secretion. In contrast, Bcl-xL-overexpressing human EndoC-βH1 β-cells conserved their physiological functions while showing an 80% increased protection against proinflammatory cytokines and a complete protection against palmitate-induced apoptosis. Notably, our findings suggest that Bcl-xL protection on human β-cells acts, at least partially, through ER stress alleviation, as Bcl-xL-overexpressing EndoC-βH1 showed a decreased expression of the ER stress markers Bip and XBP1s upon treatment with both insults (i.e., cytokines and palmitate), and reduced Chop expression in response to metabolic stress [19]. Together, these studies present Bcl-xL overexpression as a potential strategy to increase β-cell survival. However, this might not be the case for Bcl-2 overexpression. Allison and colleagues demonstrated that the overexpression of human Bcl-2 was insufficient to prevent or reduce cytotoxic or autoimmune β-cell damage in transgenic mice expressing human Bcl-2 in their islet β-cells. As a result, the onset and incidence of diabetes in three mouse models of T1D—multiple low-dose streptozotocin-induced diabetes, RIP-B7-1 mice, and non-obese diabetic (NOD) mice—were similar between wild-type and transgenic mice overexpressing Bcl-2 [139].
Due to their classical role in ensuring cell survival, many studies have investigated the role of Bcl-2 and Bcl-xL in β-cell endurance against different pro-apoptotic stimuli, such as proinflammatory cytokines [19,140,141,142,143,144], palmitate [19,121,145], and cyclopiazonic acid-induced ER stress [146]. While Bcl-xL-deficient β-cell mice develop even with abnormally high sensitivity to apoptotic insults, including thapsigargin, cytokines, and Fas ligand [141], the lack of this protein seems to be extremely detrimental in human β-cells. A recent study by Loo and colleagues revealed the fundamental role of Bcl-xL in the survival and identity establishment of human pluripotent stem cells during pancreatic specification. Bcl-xL inhibition not only increased the apoptosis rate but also downregulated the expression of pancreatic identity and metabolic genes, preventing the formation of insulin-positive β-like cells and disturbing glycolysis and oxidative phosphorylation [147].
The protection against apoptosis conferred by these proteins can also be detrimental to β-cell survival in the context of diabetes. In NOD mice, Bcl-2 plays an important role in ensuring the survival of activated T lymphocytes against apoptosis induced by IL-2 deprivation. This effect contributes to the autoimmune positive feedback and worsens insulitis and β-cell destruction [148]. Regarding the relationship between the autoimmune attack on β-cells and the expression of Bcl-2 proteins, a recent study from Brozzi and colleagues demonstrated that activated CD4+ T lymphocytes promote apoptosis in targeted β-cells not only by secreting proinflammatory cytokines, but also through the secretion of extracellular vesicles containing Bcl-xL-inhibiting non-coding tRNA fragments [149].
Polymorphisms/mutations in the diabetes-linked paired/homeodomain transcription factor Pax4 have been associated with several forms of diabetes, including T1D, T2D, and monogenic diabetes [150,151]. Pax4 plays a crucial role in β-cell development, proliferation, survival, and function [150,151]. For instance, it promotes β-cell proliferation by regulating the expression of genes involved in cell cycle (e.g., cyclin-dependent kinase inhibitor 2A and c-Myc) [152,153,154]. Moreover, Pax4 enhances β-cell survival by regulating genes involved in ER integrity in response to stress [154] and genes involved in anti-apoptotic pathways, such as Bcl-2 and Bcl-xL [153,155,156]. Studies led by Brun and colleagues reported that Pax4 upregulated Bcl-xL expression in the rat β-cell line INS-1E in comparison with rat islets [153,156]. This 25-fold higher expression of Pax4 is not related to cell proliferation. Nevertheless, its higher content in Bcl-xL directly translates to increased cell viability both in basal conditions and in the presence of the proinflammatory cytokines tumor necrosis α (TNFα), interferon gamma (IFNγ), and interleukin-1β (IL-1β). Of note, adenoviral-mediated Pax4 overexpression in human islets induces a small increase in Bcl-xL expression, showing mechanistic differences in the regulation of Bcl-xL expression by Pax4 between humans and rodents [157]. This regulatory effect has also been reported for Bcl-2 in transgenic mice overexpressing Pax4 in β-cells [155]. Interestingly, Hu He and collaborators compared the overexpression of Pax4 with the equivalent overexpression of the diabetes-linked mutant variant of Pax4, Pax4R129W. These researchers found that Pax4 overexpression, but not Pax4R129W, protected animals from streptozotocin-induced hyperglycemia as well as mouse islets from cytokine-induced apoptosis; these protective effects were at least partially due to a three-fold increase in the expression of Bcl-2 [155]. Of note, the overexpression of Pax4 or its mutant version decreased insulin expression, with a long-term effect of insulin content reduction [155]. This is consistent with the suppressing effect of Bcl-2 in glucose tolerance found by Luciani and colleagues [47].
Pro-apoptotic insults, such as proinflammatory cytokines and palmitate, can either directly or indirectly regulate Bcl-2 and Bcl-xL expression in β-cells [52]. Different combinations of proinflammatory cytokines activate distinct pathways that regulate Bcl-2 proteins in β-cells. The combination of IL-1β + IFNγ activates signal transducer and activator of transcription 1 (STAT1), activator protein 1 (AP-1), and NF-κB pathways in β-cells [158,159]. These pathways converge to promote the pro-survival induction of the transcription factor Jun-B and the Bcl-2-related protein A1 [160,161]. Simultaneously, exposure to these cytokines activates the c-Jun N-terminal kinase (JNK) pathway, which upregulates the BH3-only sensitizer DP5 [52]. The pro-survival JunB sequestering of DP5 is surpassed due to Jun-B degradation after JNK activation [161] and the consequential phosphorylation of the E3 ligase Itch [162]. JNK activation also downregulates Mcl-1 [163,164] which, along with Bad activation by IL-1β + IFNγ [165], accelerates DP5-induced β-cell apoptosis. DP5, which is also transcriptionally activated by STAT1 and ER stress [159,166], selectively binds to Bcl-xL, releasing the BH3-only activator Puma, after which this pro-apoptotic Bcl-2 protein binds to Bax and initiates the intrinsic pathway of apoptosis [167]. The combination of TNFα with IFNγ is also regulated by STAT1, which transcriptionally activates Bim, DP5, and Puma, while inactivating Bcl-xL and Mcl-1 [52,164].
Upon treatment with TNFα + IFNγ, Bcl-2 expression remained unaltered, whereas Bcl-xL expression, both at mRNA and protein levels, was diminished in rat INS-1E cells [140]. In contrast, in the mouse NIT-1 cell line, the protein levels of Bcl-2 were enhanced and Bcl-xL expression was not affected by treatment with TNFα alone during 24 h [168]. Mehmeti and colleagues reported that IL-1β alone was sufficient to drastically reduce both the gene and protein expression of Bcl-2 in rat RINm5F cells and rat islets, while Bcl-xL gene expression was upregulated by this proinflammatory cytokine in rat islets. The combination of IL-1β, TNFα, and IFNγ also increased Bcl-xL gene and protein expression in RINm5F cells. Of note, IL-1β-induced Bcl-2 downregulation depended on mitochondrial hydrogen peroxide accumulation [169]. In INS-1 832/13 cells and human islets, treatment with IL-1β, TNFα, and IFNγ—but not glucotoxicity or ER stress—stimulated the expression of microRNA miR-21. This microRNA, in turn, increased β-cell death by degrading BCL2/Bcl2 transcripts and inhibiting Bcl-2 translation [170].
The palmitate-induced activation of the intrinsic apoptotic pathway begins with JNK pathway activation and ATF3 transcription factor induction via the PERK arm of ER stress. PERK activation downregulates Bcl-2, Bcl-xL, and Mcl-1, while simultaneously inducing ATF3 and FoxO3A to upregulate Puma. These events collectively lead to Bax activation and the permeabilization of the mitochondrial outer membrane [52,158]. Although not yet demonstrated in β-cells, it is noteworthy that JNK can directly phosphorylate Bcl-2 and Bcl-xL at their BH3-binding domains [171,172]. As pointed out by Stanley and colleagues [168], this phosphorylation either prevents binding to BH3-only apoptotic proteins or marks Bcl-2 and Bcl-xL for degradation, ultimately releasing BH3-only proteins and increasing the susceptibility to apoptosis. Finally, it has been shown that palmitate blocks the ubiquitin–proteasome system, resulting in ER stress and the downregulation of Bcl-2 and Bcl-xL in MIN6 cells [145].
Bcl-xL is also the object of indirect regulation by other agents. As stated above, the BH3-only sensitizer DP5 binds to and inhibits Bcl-xL in response to IFNγ and IL-1β [159,173] or palmitate [173], favoring apoptosis. On the other hand, exposure to a high concentration of 16.7 mM glucose reduces Bcl-xL expression while leaving Bcl-2 unaffected and increasing the expression of pro-apoptotic members of the family Bad, Bid, and Bik in human islets [174]. This evidence suggests a modulatory effect of high glucose on Bcl-2 proteins towards apoptosis, which could be of special relevance in the T2D context. Bcl-xL downregulation as a vehicular contributor to β-cell damage has also been reported in chemically induced diabetes models. Streptozotocin, a classical inductor of β-cell destruction in murine models of diabetes, exerts its effects by promoting the binding of the BH3-only sensitizer Bad with Bcl-xL while decreasing the interaction with its pro-survival phosphorylated form, P-Bad, thus increasing apoptosis in rat β-cells [175].
An analysis of Bcl-xL expression in α- and β-like cells derived from human induced pluripotent stem cells exposed to IFNα showed that α-like cells present a higher expression of the anti-apoptotic protein Bcl-xL when compared to their β-like counterparts [176]. These findings are consistent with previous results from our group with primary rat α- and β-cells, where we found that the higher basal expression of Bcl-xL in α-cells conferred them resistance to palmitate-induced ER stress compared to β-cells [177]. These findings suggest that the increased expression of Bcl-xL in α-cells both in basal conditions and in response to IFNα could be a crucial feature of α-cell survival in T1D.
Interestingly, transcriptomic data from T1D donor β-cells from the nPOD consortium show a significant increase in Bcl-2 expression in comparison to non-diabetic donors, without changes in Bcl-xL expression [178]. This upregulation might be due to a late but insufficient effort to reestablish a functional β-cell mass during the autoimmune attack, which could be influenced by the already low expression of Bcl-2 in human islets [179]. In fact, previous results from Allagnat and colleagues evidenced that the cytotoxic effectors characteristic of T1D, such as proinflammatory cytokines, affect the expression of Bcl-2 and Bcl-xL as a late event of β-cell apoptosis [164].
The synergistic effect of proinflammatory cytokines towards β-cell death can be aggravated by other regulators of the intrinsic pathway of apoptosis. In brain-dead donors, which are usual candidates for islet transplantation, the increase in expression of the uncoupling protein-2 (UCP2) in islets contributes to accelerating the loss of islet yield and quality by downregulating Bcl-2 both at protein and mRNA levels [180] (Figure 1). Similarly, the cytokine-induced gene Clic4 sensitizes primary mouse β-cells to apoptosis by reducing the steady state levels of Bcl-2 and Bcl-xL [181] (Figure 1). Well-known non-cytokine regulators of Bcl-2 expression are glucagon-like peptide 1 (GLP-1) agonists such as exedin-4, that has been shown to protect β-cells from ER stress [182]. In fact, exedin-4 has been observed to counteract TNFα-induced apoptosis in INS-1 and MIN6 cells and isolated pancreatic human islets at least partially due to Bcl-2 expression level restoration [183]. Bcl-2 can also be regulated by Bag-1 [184], an anti-cell death protein that binds to and upregulates Bcl-2 activity in hippocampal neurons [185] and cardiomyocytes [186] in the context of diabetes. However, little is known about its effects on pancreatic β-cell survival.
Furthermore, paracrine signals have been proved to regulate Bcl-2 expression. The pancreatic-derived cytokine-like protein PANDER, also known as FAM3B, is secreted by α- and β-cells and induces apoptosis in these cells [187,188,189]. PANDER knockdown has been shown to reduce Bcl-2 expression in colon carcinoma cells, and to decrease cell viability in the β-cell MIN6 cell line, in an arguably similar mechanism [190]. However, these results remain controversial, as a subsequent study by Cao and colleagues showed that PANDER overexpression induces apoptosis without changes in Bcl-2 protein levels in βTC3 cells and mouse islets [191].
In β-cells, cellular stress responses can promote a senescent cell state in the form of replicative senescence activated by a cyclin-dependent kinase inhibitor-induced cell cycle arrest. Although senescent β-cells are a normal feature of adult islets and present enhanced GSIS compared with young pre-senescent β-cells [192,193], they have been found to display DNA double-strand breaks and the activation of DNA damage response in islets from T1D cadaveric donors [194]. The survival of these cells has been reported to act through the upregulation of Bcl-2 expression and to increase during T1D progression in islets from both NOD mice and T1D patients. Interestingly, the pharmacological inhibition of Bcl-2 by the ABT-737 Bcl-2/Bcl-xL inhibitor or the Bcl-2-specific inhibitor ABT-199 eliminated senescent β-cells in NOD mice, preventing diabetes development in this model without affecting the immune cell population or non-senescent β-cells [195]. In this line, a recent study by Rampazzo Morelli and colleagues found that a major senescence-associated secretory phenotype factor of pancreatic β-cells, the growth and differentiation factor 15 (GDF15), is directly linked to the expression of the BCL2L1 gene in senescent human β-cells. While the antibody neutralization of GDF15 in EndoC-βH5 cells did not affect cell viability, it reduced the expression of the Bcl-xL transcript, among other key features of the senescence-associated secretory phenotype [196].
As described above, we demonstrated that α-cells express higher levels of Bcl-xL than β-cells [177]. In this study, α-cells exhibited a higher mRNA expression of both Bcl-2 and Bcl-xL compared to β-cells, both at baseline and after palmitate exposure. Notably, silencing Bcl-xL significantly increased α-cell sensitivity to palmitate, whereas Bcl-2 knockdown did not enhance susceptibility to palmitate-induced apoptosis [177]. These results suggest that the protective effect in α-cells primarily arises from elevated Bcl-xL expression and may explain the greater resistance of α-cells to metabolic stress in T2D.
Taken together, these findings emphasize the intertwined relationship between Bcl-2 and Bcl-xL protein expression and the pathophysiology of diabetes. In pancreatic endocrine cells, these proteins have a complex role where their expression can be regulated by many different factors, including proinflammatory cytokines and ER stress, but also transcription factors and hormones (e.g., GLP-1).
3.2. Bcl-2 and Bcl-xL Effects on β-Cell Function
While Bcl-2 and Bcl-xL overexpression might have beneficial effects on β-cell survival, these anti-apoptotic proteins have also been reported to interfere with insulin secretion and metabolic regulation in murine models. Bcl-xL preserves mitochondrial transcriptional, morphological, and functional integrity under non-apoptotic levels of chronic glucose stress in β-cells [197]. Bcl-2 has been shown to control the fine-tuning of redox balance and ROS signaling in mitochondria [46] and mitochondrial morphology [121]. While these beneficial effects on mitochondrial homeostasis would identify a good starting point for hormone secretion regulation, mice overexpressing high levels of Bcl-xL (i.e., >10-fold increase) presented impaired intracellular Ca2+ signaling and mitochondrial ATP production. These changes led to reduced glucose-stimulated insulin secretion (GSIS) [136].
Later, it was reported that the adenoviral-induced overexpression of Pax4 increased the expression of Bcl-xL in rat islets. This effect was also concomitant to decreased mitochondrial Ca2+ concentration and therefore ATP production and GSIS [153]. Curiously, the overexpression of Pax4 did not alter insulin or glucagon expression in the rat islets. Conversely, transgenic mice overexpressing Pax4 exhibited a significative increase in Bcl-2 mRNA but not in Bcl-xL, and they were protected from streptozotocin-induced hyperglycemia [155].
Nevertheless, most evidence points towards an inhibitory activity of Bcl-2 and Bcl-xL in β-cell metabolism. In a very thorough study, Luciani and colleagues used knock-out mouse models of both Bcl-2 and Bcl-xL to demonstrate that the lack of these proteins significantly increased glucose-stimulated intracellular Ca2+ and metabolic responses, concomitant to increased glucose-induced insulin secretion. Furthermore, these effects could also be obtained by the small-molecule antagonism of Bcl-2 and Bcl-xL, and through the conditional ablation of Bcl-xL on adult β-cells [47,198] (Figure 1). We have recently confirmed this effect on Bcl-xL-overexpressing rat INS-1E β-cells, which showed dampened intracellular Ca2+ oscillations and insulin secretion in response to glucose and forskolin. However, the overexpression of Bcl-xL did not affect the Ca2+ response to glucose and GSIS in human EndoC-βH1 β-cells, suggesting species-specific mechanisms in the regulation of cell function by Bcl-2 proteins [19]. The observed lack of physiological effects of Bcl-xL overexpression in human β-cells might also suggest an existing threshold of Bcl-xL overexpression for Ca2+ homeostasis alterations. As reported by Zhou and collaborators, an over 10-fold overexpression of Bcl-xL might be enough to induce such Ca2+ homeostasis impairment, but a 2- to 3-fold increase has no effects on glucose tolerance [136]. Similarly, our results show that a 7-fold increase in Bcl-xL expression affects intracellular Ca2+ and insulin secretory response to glucose in rat INS-1E cells. In contrast, a 2.5- to 5-fold induction of Bcl-xL is enough to significantly protect human β-cells from diabetogenic insults without reaching the physiologically disturbing threshold. This highlights the importance of fine-tuning the dose–response of Bcl-xL overexpression for future therapeutic approaches.
Discrepancies in outcomes could be due to intrinsic differences between animal models. For example, human Pax4 DNA binding activity has been found to be lower than that of the mouse homologue in human islets, as can be seen in the fact that the overexpression of mouse Pax4 induces human islet cell proliferation, while the human homologue lacks this effect [157]. Additionally, it is difficult to compare effects between models when Bcl-2/Bcl-xL overexpression levels have great disparity: Zhou and colleagues reached a 10-fold increase in Bcl-xL expression [136], much higher than the increased expression of Bcl-xL by 2.7-fold in rat islets overexpressing Pax4 found by Brun and colleagues [153] and the 3-fold increase in Bcl-2 obtained in transgenic mice by Hu He and coworkers [155]. Conversely, Allison and colleagues observed that human Bcl-2 overexpression in mice had no effects on β-cell function or resistance to β-cell apoptosis [139]; of note, the level of overexpression was not clearly established in this study. The results obtained by our group corresponded to a 7-fold increase in Bcl-xL expression in rat INS-1E cells and a 5-fold increase in human EndoC-βH1 cells [19]. Our results in the rat cell line concur with those obtained by Brun and colleagues, but this is a field in which more research is needed, as it is possible that a certain level of Bcl-2/Bcl-xL hyperexpression is needed to interfere with Ca2+ and mitochondrial homeostasis, and this threshold might be different for each animal or human model.
4. Therapeutic Opportunities
Due to their role in β-cell survival, a growing body of evidence suggests that Bcl-2 and Bcl-xL represent exciting therapeutic candidates to enhance β-cell robustness in the context of T1D and T2D. As discussed by Singh and colleagues, enhancing β-cell robustness may protect individuals predisposed to T1D or prolong the honeymoon phase in recent-onset T1D patients. In T2D patients, it could help prevent insulin dependency, thereby reducing complications and improving long-term outcomes [199].
Based on the literature, we discuss two possible therapeutic approaches that might be used for future clinical studies.
4.1. Gene Therapy
Adeno-associated virus (AAV) vectors have emerged as the preferred choice in clinical trials due to their broad tissue tropism and safety. In addition, their non-pathogenic nature, minimal genome integration, and long-term transgene expression contribute to their effectiveness in enhancing cellular entry and transduction [200,201,202]. Therefore, the use of an AAV-based gene delivery system to increase Bcl-xL expression in β-cells could be considered. More specifically, we could use a pancreas-selective adeno-associated virus (serotype 8) encoding for BCL2L1 under the control of the insulin promoter to enhance Bcl-xL expression in β-cells, as previously shown for other proteins [199,203].
Despite the positive safety profile, patients receiving AAV-based gene therapy would need continuous monitoring. This should focus on the potential long-term risks related to the expression of an anti-apoptotic protein, which includes the risk of tumor development [201]. Current AAV-based gene therapies undergoing clinical trials demonstrate favorable safety profiles, with no significant evidence of tumorigenesis. Additionally, the predominantly epichromosomal nature of AAV-delivered DNA [204] reduces the likelihood of genomic integration, further mitigating oncogenic risks. It is worth mentioning that studies using transgenic mice overexpressing Bcl-xL in islets/β-cells have not reported tumor growth, suggesting that these models remained cancer-free during experiments [136,205].
Beyond in vivo gene therapy, an alternative use for Bcl-xL could be to prolong graft survival in pancreatic islets for transplantation. For this, either AAV-based gene therapy [206] or CRISPR/dCas9-based enhancer activation [207] could be employed to induce Bcl-xL overexpression in human islets, which could subsequently be transplanted into individuals with T1D.
4.2. Bcl-2/Bcl-xL Inhibitors
The inhibition of specific Bcl-2 proteins has been described as a promising therapeutic strategy for cancer. In recent years, researchers have combined nuclear magnetic resonance (NMR)-based screening, fragment chemistry, and structure-based drug design to generate a class of compounds known as BH3 mimetics [15,16]. BH3 mimetics are small molecules that mimic the binding of the BH3-only initiator proteins to the pro-survival members, effectively displacing native BH3-only proteins. Interestingly, it has been reported that these inhibitors can induce apoptosis with some specificity in cells exhibiting high levels of Bcl-2 or related members such as Bcl-xL and Bcl-w [51].
Most studies in β-cells centered on the use of ABT-737 (a Bcl-2, Bcl-xL, and Bcl-w inhibitor), ABT-263 (a Bcl-2 and Bcl-xL dual antagonist), and ABT-199 (a highly selective Bcl-2 inhibitor) (Table 1). Initial studies showed that ABT-737 induced apoptosis in human islet cells, primary rat β-cells, and INS-1E cells. Moreover, Puma silencing partially reduced ABT-737-induced apoptosis, indicating that Bcl-2/Bcl-xL inhibition triggers Puma-mediated β-cell death [142]. Several years later, it was reported that the specific inhibition of Bcl-2 by ABT-199 did not induce β-cell death after treatment for up to 24 h. This suggests that, compared to other anti-apoptotic members of the Bcl-2 family, Bcl-2 plays a minimal role in preventing apoptosis in β-cells under oxidative stress [46].

Table 1.
Preclinical studies using Bcl-2/Bcl-xL inhibitors in the context of β-cells/diabetes.
The therapeutic potential of Bcl-2/Bcl-xL inhibitors in diabetes emerged in 2019, when two complementary studies introduced the use of BH3 mimetics as senolytic compounds, which are drugs that selectively induce apoptosis in senescent cells [195,208]. First, Thompson and colleagues demonstrated that ABT-737 and ABT-199 mainly targeted and eliminated senescent β-cells in mouse islets without altering the abundance of the immune cell types (e.g., macrophages, effector T cells, and B cells). In addition, compared to control animals, treatment with ABT-199 in normoglycemic NOD mice suppressed diabetes development [195]. A couple of months later, a study by Aguayo-Mazzucato et al. demonstrated that oral treatment with ABT-263 alleviated hyperglycemia and enhanced the β-cell gene expression profile in animals treated with an insulin receptor antagonist (a drug-induced insulin-resistant mouse model). In human islets, the senescent cell subpopulation showed increased cell death after ABT-263 treatment [208]. Altogether, these results suggest that in T1D and T2D, β-cells become senescent, develop a senescence-associated secretory profile, and resist apoptosis through the upregulation of Bcl-2. Consequently, targeting the elimination of senescent β-cells with BH3 mimetics arrests the immune- and metabolic-mediated loss of functional β-cells and inhibits the progression of T1D and T2D by preserving β-cell mass.
Regarding their use in humans, Aguayo-Mazzucato and colleagues recognize that the primary limitation of senolytic therapies is their broad, non-specific targeting of cells and tissues, as they act on pathways that can be upregulated across various cell types. Additionally, senolytic drugs with greater potency and oral efficacy with manageable side effects must be developed. Finally, continuous understanding of senescence biology will aid in the development of effective therapeutics, paving the way for optimal and personalized interventions [208,209].
5. Limitations/Gaps in Knowledge and Future Perspectives
Although significant progress has been made in recent years, several questions remain unanswered concerning the roles of Bcl-2 and Bcl-xL in β-cell survival and physiology. Here, we highlight two of the major gaps in our current knowledge and areas where further research is needed to bridge these gaps.
5.1. Limited Research on Human Models
It is well known that rodent and human β-cells differ in several aspects, including susceptibility to cytotoxic agents, electrophysiological properties, and the regulation of insulin secretion [210,211]. Despite these differences, the evidence discussed herein suggests that Bcl-2 and Bcl-xL play a conserved role in β-cell survival across species. However, some differences can be seen regarding protection against some stressors (e.g., cytokines and palmitate). For instance, while Bcl-xL overexpression fully prevented palmitate-induced apoptosis in human EndoC-βH1 β-cells, rodent β-cells (INS-1E and mouse islets) were only partially protected by Bcl-2/Bcl-xL overexpression [19,145]. This suggests that palmitate-induced apoptosis may have a component that does not depend on the role of Bcl-2 proteins in rodents. Despite the absence of data from human islets, the gold standard in β-cell research, it is reasonable to state that Bcl-xL plays a protective role in both rodent and human β-cells against different pro-apoptotic stimuli.
Regarding β-cell function, the literature lacks an in-depth comparative analysis of how Bcl-2 and Bcl-xL influence β-cell electrophysiology, exocytosis dynamics, mitochondrial metabolism, and insulin secretion in rodents and humans. To our knowledge, only two studies have investigated the physiological effects of Bcl-2 and Bcl-xL in human β-cells and both only analyzed intracellular Ca2+ signaling [19,47]. Luciani et al. showed that Bcl-2/Bcl-xL inhibition rapidly induced significant Ca2+ fluctuations in mouse and human islet cells [47]. We, on the other hand, showed that Bcl-xL overexpression in rat INS-1E cells slightly reduced intracellular Ca2+ responses and glucose-stimulated insulin secretion, whereas these effects were not observed in the human EndoC-βH1 cells [19]. As discussed above, it remains unclear whether this lack of effect in human cells is due to the level of Bcl-xL expression or to differences between humans and rodents.
To overcome one of the main limitations of human studies, namely, the acquisition of human islets, we believe that future research should focus on alternative approaches, such as organoids and β-cells derived from human pluripotent stem cells [212], as well as β-cell lines that closely resemble primary human cells (e.g., EndoC-βH5 cells) [213].
5.2. Limited Preclinical Studies in Mouse Models of T1D and T2D
In this review we described several studies in which Bcl-2 or Bcl-xL overexpression in islets/β-cells conferred β-cell protection against pro-apoptotic stressors. Nonetheless, none of these studies investigated whether Bcl-2/Bcl-xL overexpression was effective in preventing diabetes development in animal models for T1D (e.g., NOD or RIP-B7.1 mice) or T2D (e.g., high-fat diet-induced diabetes or db/db mice). These preclinical studies are crucial to provide evidence of the feasibility of using Bcl-2/Bcl-xL overexpression as a therapeutic approach (as discussed in Section 4). Additionally, we would have the opportunity to further investigate the threshold of Bcl-2/Bcl-xL expression that protects against a diabetogenic environment without disturbing β-cell physiology. Lastly, these studies will be essential to evaluate whether targeting these proteins therapeutically will eventually trigger off-target effects.
As discussed above, excessive Bcl-xL expression (>10-fold) disrupts intracellular Ca2+ homeostasis, impairing insulin secretion in rodent β-cells [136]. Furthermore, excessive Bcl-2/Bcl-xL levels may hinder the natural apoptosis process, allowing dysfunctional β-cells to persist, ultimately impairing glucose-stimulated insulin secretion. Finally, by interacting with Beclin-1, Bcl-2 inhibits autophagy, a process essential for β-cell survival and function [214]. Hence, prolonged Bcl-2 overexpression may lead to autophagy imbalance, worsening β-cell health and promoting apoptosis. To mitigate these potential risks, we suggest the use of molecular tools that allow the fine-tuning of Bcl-2/Bcl-xL expression, such as inducible expression systems (e.g., Tet-on/Tet-off) and CRISPR-based approaches. Ideally, Bcl-2/Bcl-xL levels should match those observed in human α-cells, which are approximately 3–4 times higher than basal levels (HPAP database: https://hpap.pmacs.upenn.edu/).
6. Conclusions
In the present review, we summarize the different roles of Bcl-xL and Bcl-2 in survival and function, emphasizing their roles in β-cells, where they serve a dual impact on physiological processes, such as Ca2+ homeostasis and insulin secretion, as well as cell survival. The lack of studies on human models represents a significant gap, as human β-cells respond differently to stress compared to rodent cells. Addressing this gap is essential for translating findings into therapeutic approaches targeting human β-cell preservation under proinflammatory or metabolic conditions. Recent evidence, however, shows the ability of Bcl-2 and Bcl-xL to protect human β-cells without impairing insulin release, introducing a promising avenue for β-cell-specific therapies aimed at preventing β-cell loss during diabetes progression. Altogether, these findings suggest that targeting Bcl-2 and Bcl-xL could represent a viable strategy to mitigate β-cell dysfunction and death, contributing to advancements in diabetes treatment.
Author Contributions
A.A.P.-S.: Conceptualization, Visualization, and Writing—original draft and editing; D.G.-L.: Writing—review and editing; R.S.D.S.: Conceptualization, Supervision, and Writing—original draft and editing; L.M.: Conceptualization, Supervision, Writing—review and editing, Funding acquisition, and Project administration. All authors have read and agreed to the published version of the manuscript.
Funding
L.M. is funded by grants PID2020-117569RA-I00, PID2023-147823OB-I00, and CNS2022-135505 funded by MCIN/AEI/10.13039/501100011033 and, as appropriate, by “ERDF A way of making Europe”, by the “European Union”, or by the “European Union NextGenerationEU/PRTR”. RSdS is a recipient of a Miguel Servet contract from the Instituto de Salud Carlos III (ISCIII), CP23/00026, co-funded by the European Union. This research was supported by the CIBER-Consorcio Centro de Investigación Biomédica en Red (CB07/08/0002), Instituto de Salud Carlos III, Ministerio de Ciencia e Innovación.
Acknowledgments
Figure 1 was created with BioRender. While preparing this work, the authors utilized ChatGPT GPT-4o mini (https://chatgpt.com/; accessed on 7 January 2025) to structure and refine the flow of the manuscript. Following the use of this tool, the authors thoroughly reviewed and revised the content as necessary, taking full accountability for the final publication.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, Regional and Country-Level Diabetes Prevalence Estimates for 2021 and Projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef] [PubMed]
- Dludla, P.V.; Mabhida, S.E.; Ziqubu, K.; Nkambule, B.B.; Mazibuko-Mbeje, S.E.; Hanser, S.; Basson, A.K.; Pheiffer, C.; Kengne, A.P. Pancreatic β-Cell Dysfunction in Type 2 Diabetes: Implications of Inflammation and Oxidative Stress. World J. Diabetes 2023, 14, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Eizirik, D.L.; Pasquali, L.; Cnop, M. Pancreatic β-Cells in Type 1 and Type 2 Diabetes Mellitus: Different Pathways to Failure. Nat. Rev. Endocrinol. 2020, 16, 349–362. [Google Scholar] [CrossRef]
- Herold, K.C.; Delong, T.; Perdigoto, A.L.; Biru, N.; Brusko, T.M.; Walker, L.S.K. The Immunology of Type 1 Diabetes. Nat. Rev. Immunol. 2024, 24, 435–451. [Google Scholar] [CrossRef] [PubMed]
- Grarup, N.; Sandholt, C.H.; Hansen, T.; Pedersen, O. Genetic Susceptibility to Type 2 Diabetes and Obesity: From Genome-Wide Association Studies to Rare Variants and Beyond. Diabetologia 2014, 57, 1528–1541. [Google Scholar] [CrossRef]
- Noble, J.A.; Valdes, A.M.; Varney, M.D.; Carlson, J.A.; Moonsamy, P.; Fear, A.L.; Lane, J.A.; Lavant, E.; Rappner, R.; Louey, A.; et al. HLA Class I and Genetic Susceptibility to Type 1 Diabetes: Results from the Type 1 Diabetes Genetics Consortium. Diabetes 2010, 59, 2972–2979. [Google Scholar] [CrossRef] [PubMed]
- Knip, M.; Simell, O. Environmental Triggers of Type 1 Diabetes. Cold Spring Harb. Perspect. Med. 2012, 2, a007690. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Magdalena, P.; Quesada, I.; Nadal, A. Endocrine Disruptors in the Etiology of Type 2 Diabetes Mellitus. Nat. Rev. Endocrinol. 2011, 7, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Janssen, J.A.M.J.L. Hyperinsulinemia and Its Pivotal Role in Aging, Obesity, Type 2 Diabetes, Cardiovascular Disease and Cancer. Int. J. Mol. Sci. 2021, 22, 7797. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.; Muralidharan, C.; May, S.C.; Tersey, S.A.; Mirmira, R.G. Inside the β Cell: Molecular Stress Response Pathways in Diabetes Pathogenesis. Endocrinology 2023, 164, bqac184. [Google Scholar] [CrossRef] [PubMed]
- Meyerovich, K.; Fukaya, M.; Terra, L.F.; Ortis, F.; Eizirik, D.L.; Cardozo, A.K. The Non-Canonical NF-κB Pathway Is Induced by Cytokines in Pancreatic Beta Cells and Contributes to Cell Death and Proinflammatory Responses in Vitro. Diabetologia 2016, 59, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Ortis, F.; Pirot, P.; Naamane, N.; Kreins, A.Y.; Rasschaert, J.; Moore, F.; Théâtre, E.; Verhaeghe, C.; Magnusson, N.E.; Chariot, A.; et al. Induction of Nuclear Factor-kappaB and Its Downstream Genes by TNF-Alpha and IL-1beta Has a pro-Apoptotic Role in Pancreatic Beta Cells. Diabetologia 2008, 51, 1213–1225. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 Family Proteins: Changing Partners in the Dance towards Death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef]
- Kapoor, I.; Bodo, J.; Hill, B.T.; Hsi, E.D.; Almasan, A. Targeting BCL-2 in B-Cell Malignancies and Overcoming Therapeutic Resistance. Cell Death Dis. 2020, 11, 941. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Fairbrother, W.J.; Leverson, J.D.; Souers, A.J. From Basic Apoptosis Discoveries to Advanced Selective BCL-2 Family Inhibitors. Nat. Rev. Drug Discov. 2017, 16, 273–284. [Google Scholar] [CrossRef]
- Wang, J.; Wang, X.; Ren, J.; Lin, J.; Yu, Z.; Huang, S.; Hu, Y.; Fu, J.; Wang, M.; Zhang, Y.; et al. S-9-PAHSA’s Neuroprotective Effect Mediated by CAIII Suppresses Apoptosis and Oxidative Stress in a Mouse Model of Type 2 Diabetes. CNS Neurosci. Ther. 2024, 30, e14594. [Google Scholar] [CrossRef]
- Wu, Y.; Peng, X.; Ang, S.; Gao, Y.; Chi, Y.; Wang, J.; Tang, C.; Zhou, X.; Feng, Y.; Zhang, K.; et al. Bcl-xL Promotes the Survival of Motor Neurons Derived from Neural Stem Cells. Biology 2023, 12, 132. [Google Scholar] [CrossRef]
- Perez-Serna, A.A.; Dos Santos, R.S.; Ripoll, C.; Nadal, A.; Eizirik, D.L.; Marroqui, L. BCL-XL Overexpression Protects Pancreatic β-Cells against Cytokine- and Palmitate-Induced Apoptosis. Int. J. Mol. Sci. 2023, 24, 5657. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Finger, L.R.; Yunis, J.; Nowell, P.C.; Croce, C.M. Cloning of the Chromosome Breakpoint of Neoplastic B Cells with the t(14;18) Chromosome Translocation. Science 1984, 226, 1097–1099. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.F.A.; Wong-Brown, M.W.; Bowden, N.A. BCL-2 Family Isoforms in Apoptosis and Cancer. Cell Death Dis. 2019, 10, 177. [Google Scholar] [CrossRef]
- Scorilas, A.; Kyriakopoulou, L.; Yousef, G.M.; Ashworth, L.K.; Kwamie, A.; Diamandis, E.P. Molecular Cloning, Physical Mapping, and Expression Analysis of a Novel Gene, BCL2L12, Encoding a Proline-Rich Protein with a Highly Conserved BH2 Domain of the Bcl-2 Family. Genomics 2001, 72, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Godzik, A.; Reed, J.C. Bcl-G, a Novel pro-Apoptotic Member of the Bcl-2 Family. J. Biol. Chem. 2001, 276, 2780–2785. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, T.; Holler, N.; Micheau, O.; Martinon, F.; Tinel, A.; Hofmann, K.; Tschopp, J. Bcl-Rambo, a Novel Bcl-2 Homologue That Induces Apoptosis via Its Unique C-Terminal Extension. J. Biol. Chem. 2001, 276, 19548–19554. [Google Scholar] [CrossRef]
- Kim, J.-H.; Sim, S.-H.; Ha, H.-J.; Ko, J.-J.; Lee, K.; Bae, J. MCL-1ES, a Novel Variant of MCL-1, Associates with MCL-1L and Induces Mitochondrial Cell Death. FEBS Lett. 2009, 583, 2758–2764. [Google Scholar] [CrossRef] [PubMed]
- Haldar, S.; Beatty, C.; Tsujimoto, Y.; Croce, C.M. The Bcl-2 Gene Encodes a Novel G Protein. Nature 1989, 342, 195–198. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Croce, C.M. Analysis of the Structure, Transcripts, and Protein Products of Bcl-2, the Gene Involved in Human Follicular Lymphoma. Proc. Natl. Acad. Sci. USA 1986, 83, 5214–5218. [Google Scholar] [CrossRef] [PubMed]
- Hang, X.; Zhao, L.; Wu, B.; Li, S.; Liu, P.; Xu, J.; Wang, X.; Chi, P.; Chen, C.; Niu, T.; et al. BCL-2 Isoform β Promotes Angiogenesis by TRiC-Mediated Upregulation of VEGF-A in Lymphoma. Oncogene 2022, 41, 3655–3663. [Google Scholar] [CrossRef] [PubMed]
- Boise, L.H.; González-García, M.; Postema, C.E.; Ding, L.; Lindsten, T.; Turka, L.A.; Mao, X.; Nuñez, G.; Thompson, C.B. Bcl-x, a Bcl-2-Related Gene That Functions as a Dominant Regulator of Apoptotic Cell Death. Cell 1993, 74, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Michels, J.; Kepp, O.; Senovilla, L.; Lissa, D.; Castedo, M.; Kroemer, G.; Galluzzi, L. Functions of BCL-XL at the Interface between Cell Death and Metabolism. Int. J. Cell Biol. 2013, 2013, 705294. [Google Scholar] [CrossRef] [PubMed]
- Muchmore, S.W.; Sattler, M.; Liang, H.; Meadows, R.P.; Harlan, J.E.; Yoon, H.S.; Nettesheim, D.; Chang, B.S.; Thompson, C.B.; Wong, S.L.; et al. X-Ray and NMR Structure of Human Bcl-xL, an Inhibitor of Programmed Cell Death. Nature 1996, 381, 335–341. [Google Scholar] [CrossRef]
- Petros, A.M.; Medek, A.; Nettesheim, D.G.; Kim, D.H.; Yoon, H.S.; Swift, K.; Matayoshi, E.D.; Oltersdorf, T.; Fesik, S.W. Solution Structure of the Antiapoptotic Protein Bcl-2. Proc. Natl. Acad. Sci. USA 2001, 98, 3012–3017. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.; Millar, D.G.; Yong, V.W.; Korsmeyer, S.J.; Shore, G.C. Targeting of Bcl-2 to the Mitochondrial Outer Membrane by a COOH-Terminal Signal Anchor Sequence. J. Biol. Chem. 1993, 268, 25265–25268. [Google Scholar] [CrossRef]
- Borner, C.; Martinou, I.; Mattmann, C.; Irmler, M.; Schaerer, E.; Martinou, J.C.; Tschopp, J. The Protein Bcl-2 Alpha Does Not Require Membrane Attachment, but Two Conserved Domains to Suppress Apoptosis. J. Cell Biol. 1994, 126, 1059–1068. [Google Scholar] [CrossRef]
- Hockenbery, D.; Nuñez, G.; Milliman, C.; Schreiber, R.D.; Korsmeyer, S.J. Bcl-2 Is an Inner Mitochondrial Membrane Protein That Blocks Programmed Cell Death. Nature 1990, 348, 334–336. [Google Scholar] [CrossRef] [PubMed]
- Nakai, M.; Takeda, A.; Cleary, M.L.; Endo, T. The Bcl-2 Protein Is Inserted into the Outer Membrane but Not into the Inner Membrane of Rat Liver Mitochondria in Vitro. Biochem. Biophys. Res. Commun. 1993, 196, 233–239. [Google Scholar] [CrossRef]
- Akao, Y.; Otsuki, Y.; Kataoka, S.; Ito, Y.; Tsujimoto, Y. Multiple Subcellular Localization of Bcl-2: Detection in Nuclear Outer Membrane, Endoplasmic Reticulum Membrane, and Mitochondrial Membranes. Cancer Res. 1994, 54, 2468–2471. [Google Scholar] [PubMed]
- de Jong, D.; Prins, F.A.; Mason, D.Y.; Reed, J.C.; van Ommen, G.B.; Kluin, P.M. Subcellular Localization of the Bcl-2 Protein in Malignant and Normal Lymphoid Cells. Cancer Res. 1994, 54, 256–260. [Google Scholar] [PubMed]
- Krajewski, S.; Tanaka, S.; Takayama, S.; Schibler, M.J.; Fenton, W.; Reed, J.C. Investigation of the Subcellular Distribution of the Bcl-2 Oncoprotein: Residence in the Nuclear Envelope, Endoplasmic Reticulum, and Outer Mitochondrial Membranes. Cancer Res. 1993, 53, 4701–4714. [Google Scholar]
- Lithgow, T.; van Driel, R.; Bertram, J.F.; Strasser, A. The Protein Product of the Oncogene Bcl-2 Is a Component of the Nuclear Envelope, the Endoplasmic Reticulum, and the Outer Mitochondrial Membrane. Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 1994, 5, 411–417. [Google Scholar]
- Monaghan, P.; Robertson, D.; Amos, T.A.; Dyer, M.J.; Mason, D.Y.; Greaves, M.F. Ultrastructural Localization of Bcl-2 Protein. J. Histochem. Cytochem. 1992, 40, 1819–1825. [Google Scholar] [CrossRef]
- Popgeorgiev, N.; Jabbour, L.; Gillet, G. Subcellular Localization and Dynamics of the Bcl-2 Family of Proteins. Front. Cell Dev. Biol. 2018, 6, 13. [Google Scholar] [CrossRef]
- González-García, M.; Pérez-Ballestero, R.; Ding, L.; Duan, L.; Boise, L.H.; Thompson, C.B.; Núñez, G. Bcl-XL Is the Major Bcl-x mRNA Form Expressed during Murine Development and Its Product Localizes to Mitochondria. Development 1994, 120, 3033–3042. [Google Scholar] [CrossRef]
- Eno, C.O.; Eckenrode, E.F.; Olberding, K.E.; Zhao, G.; White, C.; Li, C. Distinct Roles of Mitochondria- and ER-Localized Bcl-xL in Apoptosis Resistance and Ca2+ Homeostasis. Mol. Biol. Cell 2012, 23, 2605–2618. [Google Scholar] [CrossRef]
- Hsu, Y.-T.; Wolter, K.G.; Youle, R.J. Cytosol-to-Membrane Redistribution of Bax and Bcl-XL during Apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 3668–3672. [Google Scholar] [CrossRef] [PubMed]
- Aharoni-Simon, M.; Shumiatcher, R.; Yeung, A.; Shih, A.Z.L.; Dolinsky, V.W.; Doucette, C.A.; Luciani, D.S. Bcl-2 Regulates Reactive Oxygen Species Signaling and a Redox-Sensitive Mitochondrial Proton Leak in Mouse Pancreatic β-Cells. Endocrinology 2016, 157, 2270–2281. [Google Scholar] [CrossRef] [PubMed]
- Luciani, D.S.; White, S.A.; Widenmaier, S.B.; Saran, V.V.; Taghizadeh, F.; Hu, X.; Allard, M.F.; Johnson, J.D. Bcl-2 and Bcl-xL Suppress Glucose Signaling in Pancreatic β-Cells. Diabetes 2013, 62, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Contreras, J.L.; Smyth, C.A.; Bilbao, G.; Eckstein, C.; Young, C.J.; Thompson, J.A.; Curiel, D.T.; Eckhoff, D.E. Coupling Endoplasmic Reticulum Stress to Cell Death Program in Isolated Human Pancreatic Islets: Effects of Gene Transfer of Bcl-2. Transpl. Int. Off. J. Eur. Soc. Organ Transplant. 2003, 16, 537–542. [Google Scholar] [CrossRef]
- Annis, M.G.; Zamzami, N.; Zhu, W.; Penn, L.Z.; Kroemer, G.; Leber, B.; Andrews, D.W. Endoplasmic Reticulum Localized Bcl-2 Prevents Apoptosis When Redistribution of Cytochrome c Is a Late Event. Oncogene 2001, 20, 1939–1952. [Google Scholar] [CrossRef] [PubMed]
- Banjara, S.; Suraweera, C.D.; Hinds, M.G.; Kvansakul, M. The Bcl-2 Family: Ancient Origins, Conserved Structures, and Divergent Mechanisms. Biomolecules 2020, 10, 128. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of Apoptosis by the BCL-2 Protein Family: Implications for Physiology and Therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Gurzov, E.N.; Eizirik, D.L. Bcl-2 Proteins in Diabetes: Mitochondrial Pathways of β-Cell Death and Dysfunction. Trends Cell Biol. 2011, 21, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Shamas-Din, A.; Brahmbhatt, H.; Leber, B.; Andrews, D.W. BH3-Only Proteins: Orchestrators of Apoptosis. Biochim. Biophys. Acta 2011, 1813, 508–520. [Google Scholar] [CrossRef]
- Shamas-Din, A.; Kale, J.; Leber, B.; Andrews, D.W. Mechanisms of Action of Bcl-2 Family Proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a008714. [Google Scholar] [CrossRef]
- Duan, L.; Dong, S.; Huang, K.; Cong, Y.; Luo, S.; Zhang, J.Z.H. Computational Analysis of Binding Free Energies, Hotspots and the Binding Mechanism of Bcl-xL/Bcl-2 Binding to Bad/Bax. Phys. Chem. Chem. Phys. PCCP 2021, 23, 2025–2037. [Google Scholar] [CrossRef] [PubMed]
- Sora, V.; Papaleo, E. Structural Details of BH3 Motifs and BH3-Mediated Interactions: An Updated Perspective. Front. Mol. Biosci. 2022, 9, 864874. [Google Scholar] [CrossRef]
- Beigl, T.B.; Paul, A.; Fellmeth, T.P.; Nguyen, D.; Barber, L.; Weller, S.; Schäfer, B.; Gillissen, B.F.; Aulitzky, W.E.; Kopp, H.-G.; et al. BCL-2 and BOK Regulate Apoptosis by Interaction of Their C-Terminal Transmembrane Domains. EMBO Rep. 2024, 25, 3896–3924. [Google Scholar] [CrossRef]
- Wu, G.; Yang, F.; Cheng, X.; Mai, Z.; Wang, X.; Chen, T. Live-Cell Imaging Analysis on the Anti-Apoptotic Function of the Bcl-xL Transmembrane Carboxyl Terminal Domain. Biochem. Biophys. Res. Commun. 2023, 639, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Deng, X.; Carr, B.; May, W.S. Bcl-2 Phosphorylation Required for Anti-Apoptosis Function. J. Biol. Chem. 1997, 272, 11671–11673. [Google Scholar] [CrossRef]
- Wei, Y.; An, Z.; Zou, Z.; Sumpter, R., Jr.; Su, M.; Zang, X.; Sinha, S.; Gaestel, M.; Levine, B. The Stress-Responsive Kinases MAPKAPK2/MAPKAPK3 Activate Starvation-Induced Autophagy through Beclin 1 Phosphorylation. eLife 2015, 4, e05289. [Google Scholar] [CrossRef]
- Nakamura, M.; Keller, M.A.; Fefelova, N.; Zhai, P.; Liu, T.; Tian, Y.; Ikeda, S.; Del Re, D.P.; Li, H.; Xie, L.-H.; et al. Ser14 Phosphorylation of Bcl-xL Mediates Compensatory Cardiac Hypertrophy in Male Mice. Nat. Commun. 2023, 14, 5805. [Google Scholar] [CrossRef] [PubMed]
- Upreti, M.; Galitovskaya, E.N.; Chu, R.; Tackett, A.J.; Terrano, D.T.; Granell, S.; Chambers, T.C. Identification of the Major Phosphorylation Site in Bcl-xL Induced by Microtubule Inhibitors and Analysis of Its Functional Significance. J. Biol. Chem. 2008, 283, 35517–35525. [Google Scholar] [CrossRef]
- Wang, J.; Beauchemin, M.; Bertrand, R. Phospho-Bcl-xL(Ser62) Plays a Key Role at DNA Damage-Induced G2 Checkpoint. Cell Cycle 2012, 11, 2159–2169. [Google Scholar] [CrossRef]
- Gabellini, C.; Trisciuoglio, D.; Del Bufalo, D. Non-Canonical Roles of Bcl-2 and Bcl-xL Proteins: Relevance of BH4 Domain. Carcinogenesis 2017, 38, 579–587. [Google Scholar] [CrossRef]
- Gross, A.; Katz, S.G. Non-Apoptotic Functions of BCL-2 Family Proteins. Cell Death Differ. 2017, 24, 1348–1358. [Google Scholar] [CrossRef]
- Callens, M.; Kraskovskaya, N.; Derevtsova, K.; Annaert, W.; Bultynck, G.; Bezprozvanny, I.; Vervliet, T. The Role of Bcl-2 Proteins in Modulating Neuronal Ca2+ Signaling in Health and in Alzheimer’s Disease. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118997. [Google Scholar] [CrossRef]
- Ivanova, H.; Vervliet, T.; Monaco, G.; Terry, L.E.; Rosa, N.; Baker, M.R.; Parys, J.B.; Serysheva, I.I.; Yule, D.I.; Bultynck, G. Bcl-2-Protein Family as Modulators of IP3 Receptors and Other Organellar Ca2+ Channels. Cold Spring Harb. Perspect. Biol. 2020, 12, a035089. [Google Scholar] [CrossRef] [PubMed]
- Vervliet, T.; Clerix, E.; Seitaj, B.; Ivanova, H.; Monaco, G.; Bultynck, G. Modulation of Ca2+ Signaling by Anti-Apoptotic B-Cell Lymphoma 2 Proteins at the Endoplasmic Reticulum–Mitochondrial Interface. Front. Oncol. 2017, 7, 75. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.; Hayashi, T.; Wolozny, D.; Yin, B.; Su, T.-C.; Betenbaugh, M.J.; Su, T.-P. The Non-Apoptotic Action of Bcl-xL: Regulating Ca2+ Signaling and Bioenergetics at the ER-Mitochondrion Interface. J. Bioenerg. Biomembr. 2016, 48, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Valencia, I.; Zhong, F.; McColl, K.S.; Roderick, H.L.; Bootman, M.D.; Berridge, M.J.; Conway, S.J.; Holmes, A.B.; Mignery, G.A.; et al. Bcl-2 Functionally Interacts with Inositol 1,4,5-Trisphosphate Receptors to Regulate Calcium Release from the ER in Response to Inositol 1,4,5-Trisphosphate. J. Cell Biol. 2004, 166, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Monaco, G.; Decrock, E.; Akl, H.; Ponsaerts, R.; Vervliet, T.; Luyten, T.; De Maeyer, M.; Missiaen, L.; Distelhorst, C.W.; De Smedt, H.; et al. Selective Regulation of IP3-Receptor-Mediated Ca2+ Signaling and Apoptosis by the BH4 Domain of Bcl-2 versus Bcl-Xl. Cell Death Differ. 2012, 19, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Distelhorst, C.W. Bcl-2 Protein Family Members: Versatile Regulators of Calcium Signaling in Cell Survival and Apoptosis. Annu. Rev. Physiol. 2008, 70, 73–91. [Google Scholar] [CrossRef]
- Ivanova, H.; Ritaine, A.; Wagner, L.; Luyten, T.; Shapovalov, G.; Welkenhuyzen, K.; Seitaj, B.; Monaco, G.; De Smedt, H.; Prevarskaya, N.; et al. The Trans-Membrane Domain of Bcl-2α, but Not Its Hydrophobic Cleft, Is a Critical Determinant for Efficient IP3 Receptor Inhibition. Oncotarget 2016, 7, 55704–55720. [Google Scholar] [CrossRef] [PubMed]
- Foyouzi-Youssefi, R.; Arnaudeau, S.; Borner, C.; Kelley, W.L.; Tschopp, J.; Lew, D.P.; Demaurex, N.; Krause, K.H. Bcl-2 Decreases the Free Ca2+ Concentration within the Endoplasmic Reticulum. Proc. Natl. Acad. Sci. USA 2000, 97, 5723–5728. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Ferrari, D.; Magalhães, P.; Schulze-Osthoff, K.; Di Virgilio, F.; Pozzan, T.; Rizzuto, R. Reduced Loading of Intracellular Ca2+ Stores and Downregulation of Capacitative Ca2+ Influx in Bcl-2–Overexpressing Cells. J. Cell Biol. 2000, 148, 857–862. [Google Scholar] [CrossRef]
- Pinton, P.; Ferrari, D.; Rapizzi, E.; Virgilio, F.D.; Pozzan, T.; Rizzuto, R. The Ca2+ Concentration of the Endoplasmic Reticulum Is a Key Determinant of Ceramide-induced Apoptosis: Significance for the Molecular Mechanism of Bcl-2 Action. EMBO J. 2001, 20, 2690–2701. [Google Scholar] [CrossRef]
- He, H.; Lam, M.; McCormick, T.S.; Distelhorst, C.W. Maintenance of Calcium Homeostasis in the Endoplasmic Reticulum by Bcl-2. J. Cell Biol. 1997, 138, 1219–1228. [Google Scholar] [CrossRef] [PubMed]
- Monaco, G.; Vervliet, T.; Akl, H.; Bultynck, G. The Selective BH4-Domain Biology of Bcl-2-Family Members: IP3Rs and Beyond. Cell. Mol. Life Sci. CMLS 2013, 70, 1171–1183. [Google Scholar] [CrossRef] [PubMed]
- White, C.; Li, C.; Yang, J.; Petrenko, N.B.; Madesh, M.; Thompson, C.B.; Foskett, J.K. The Endoplasmic Reticulum Gateway to Apoptosis by Bcl-XL Modulation of the InsP3R. Nat. Cell Biol. 2005, 7, 1021–1028. [Google Scholar] [CrossRef]
- Yang, J.; Vais, H.; Gu, W.; Foskett, J.K. Biphasic Regulation of InsP3 Receptor Gating by Dual Ca2+ Release Channel BH3-like Domains Mediates Bcl-xL Control of Cell Viability. Proc. Natl. Acad. Sci. USA 2016, 113, E1953–E1962. [Google Scholar] [CrossRef]
- Rosa, N.; Ivanova, H.; Wagner, L.E.; Kale, J.; La Rovere, R.; Welkenhuyzen, K.; Louros, N.; Karamanou, S.; Shabardina, V.; Lemmens, I.; et al. Bcl-xL Acts as an Inhibitor of IP3R Channels, Thereby Antagonizing Ca2+-Driven Apoptosis. Cell Death Differ. 2022, 29, 788–805. [Google Scholar] [CrossRef]
- Li, C.; Fox, C.J.; Master, S.R.; Bindokas, V.P.; Chodosh, L.A.; Thompson, C.B. Bcl-XL Affects Ca2+ Homeostasis by Altering Expression of Inositol 1,4,5-Trisphosphate Receptors. Proc. Natl. Acad. Sci. USA 2002, 99, 9830–9835. [Google Scholar] [CrossRef]
- Li, C.; Wang, X.; Vais, H.; Thompson, C.B.; Foskett, J.K.; White, C. Apoptosis Regulation by Bcl-xL Modulation of Mammalian Inositol 1,4,5-Trisphosphate Receptor Channel Isoform Gating. Proc. Natl. Acad. Sci. USA 2007, 104, 12565–12570. [Google Scholar] [CrossRef]
- Woll, K.A.; Van Petegem, F. Calcium-Release Channels: Structure and Function of IP3 Receptors and Ryanodine Receptors. Physiol. Rev. 2022, 102, 209–268. [Google Scholar] [CrossRef] [PubMed]
- Vervliet, T.; Decrock, E.; Molgó, J.; Sorrentino, V.; Missiaen, L.; Leybaert, L.; De Smedt, H.; Kasri, N.N.; Parys, J.B.; Bultynck, G. Bcl-2 Binds to and Inhibits Ryanodine Receptors. J. Cell Sci. 2014, 127, 2782–2792. [Google Scholar] [CrossRef]
- Vervliet, T.; Lemmens, I.; Vandermarliere, E.; Decrock, E.; Ivanova, H.; Monaco, G.; Sorrentino, V.; Kasri, N.N.; Missiaen, L.; Martens, L.; et al. Ryanodine Receptors Are Targeted by Anti-Apoptotic Bcl-XL Involving Its BH4 Domain and Lys87 from Its BH3 Domain. Sci. Rep. 2015, 5, 9641. [Google Scholar] [CrossRef]
- Chiu, W.-T.; Chang, H.-A.; Lin, Y.-H.; Lin, Y.-S.; Chang, H.-T.; Lin, H.-H.; Huang, S.-C.; Tang, M.-J.; Shen, M.-R. Bcl-2 Regulates Store-Operated Ca2+ Entry to Modulate ER Stress-Induced Apoptosis. Cell Death Discov. 2018, 4, 37. [Google Scholar] [CrossRef]
- Bassik, M.C.; Scorrano, L.; Oakes, S.A.; Pozzan, T.; Korsmeyer, S.J. Phosphorylation of BCL-2 Regulates ER Ca2+ Homeostasis and Apoptosis. EMBO J. 2004, 23, 1207–1216. [Google Scholar] [CrossRef]
- Kuo, T.H.; Kim, H.R.; Zhu, L.; Yu, Y.; Lin, H.M.; Tsang, W. Modulation of Endoplasmic Reticulum Calcium Pump by Bcl-2. Oncogene 1998, 17, 1903–1910. [Google Scholar] [CrossRef]
- Dremina, E.S.; Sharov, V.S.; Kumar, K.; Zaidi, A.; Michaelis, E.K.; Schöneich, C. Anti-Apoptotic Protein Bcl-2 Interacts with and Destabilizes the Sarcoplasmic/Endoplasmic Reticulum Ca2+-ATPase (SERCA). Biochem. J. 2004, 383, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Garbincius, J.F.; Elrod, J.W. Mitochondrial Calcium Exchange in Physiology and Disease. Physiol. Rev. 2022, 102, 893–992. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, F.; Li, P.; Gao, Y. Mitochondrial Ca2+ Homeostasis: Emerging Roles and Clinical Significance in Cardiac Remodeling. Int. J. Mol. Sci. 2022, 23, 3025. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.L.; Gillet, G.; Prudent, J.; Popgeorgiev, N. Bcl-2 Family of Proteins in the Control of Mitochondrial Calcium Signalling: An Old Chap with New Roles. Int. J. Mol. Sci. 2021, 22, 3730. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Li, X.X.; Gottleib, E.; Hill, R.B.; Thompson, C.B.; Colombini, M. Bcl-xL Promotes the Open Configuration of the Voltage-Dependent Anion Channel and Metabolite Passage through the Outer Mitochondrial Membrane. J. Biol. Chem. 2001, 276, 19414–19419. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Chandel, N.S.; Schumacker, P.T.; Thompson, C.B. Bcl-xL Prevents Cell Death Following Growth Factor Withdrawal by Facilitating Mitochondrial ATP/ADP Exchange. Mol. Cell 1999, 3, 159–167. [Google Scholar] [CrossRef]
- Roy, S.S.; Madesh, M.; Davies, E.; Antonsson, B.; Danial, N.; Hajnóczky, G. Bad Targets the Permeability Transition Pore Independent of Bax or Bak to Switch between Ca2+-Dependent Cell Survival and Death. Mol. Cell 2009, 33, 377–388. [Google Scholar] [CrossRef]
- Szabó, I.; Zoratti, M. The Mitochondrial Permeability Transition Pore May Comprise VDAC Molecules. I. Binary Structure and Voltage Dependence of the Pore. FEBS Lett. 1993, 330, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Hu, X.; Eno, C.O.; Zhao, G.; Li, C.; White, C. An Interaction between Bcl-xL and the Voltage-Dependent Anion Channel (VDAC) Promotes Mitochondrial Ca2+ Uptake. J. Biol. Chem. 2013, 288, 19870–19881. [Google Scholar] [CrossRef]
- Monaco, G.; Decrock, E.; Arbel, N.; van Vliet, A.R.; La Rovere, R.M.; De Smedt, H.; Parys, J.B.; Agostinis, P.; Leybaert, L.; Shoshan-Barmatz, V.; et al. The BH4 Domain of Anti-Apoptotic Bcl-XL, but Not That of the Related Bcl-2, Limits the Voltage-Dependent Anion Channel 1 (VDAC1)-Mediated Transfer of pro-Apoptotic Ca2+ Signals to Mitochondria. J. Biol. Chem. 2015, 290, 9150–9161. [Google Scholar] [CrossRef]
- Shimizu, S.; Konishi, A.; Kodama, T.; Tsujimoto, Y. BH4 Domain of Antiapoptotic Bcl-2 Family Members Closes Voltage-Dependent Anion Channel and Inhibits Apoptotic Mitochondrial Changes and Cell Death. Proc. Natl. Acad. Sci. USA 2000, 97, 3100–3105. [Google Scholar] [CrossRef]
- Abu-Hamad, S.; Arbel, N.; Calo, D.; Arzoine, L.; Israelson, A.; Keinan, N.; Ben-Romano, R.; Friedman, O.; Shoshan-Barmatz, V. The VDAC1 N-Terminus Is Essential Both for Apoptosis and the Protective Effect of Anti-Apoptotic Proteins. J. Cell Sci. 2009, 122, 1906–1916. [Google Scholar] [CrossRef]
- Zhu, L.; Yu, Y.; Chua, B.H.; Ho, Y.S.; Kuo, T.H. Regulation of Sodium-Calcium Exchange and Mitochondrial Energetics by Bcl-2 in the Heart of Transgenic Mice. J. Mol. Cell. Cardiol. 2001, 33, 2135–2144. [Google Scholar] [CrossRef] [PubMed]
- Bas, J.; Nguyen, T.; Gillet, G. Involvement of Bcl-xL in Neuronal Function and Development. Int. J. Mol. Sci. 2021, 22, 3202. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, Y.-B.; Jones, A.; Sanger, R.H.; Collis, L.P.; Flannery, R.J.; McNay, E.C.; Yu, T.; Schwarzenbacher, R.; Bossy, B.; et al. Bcl-XL Induces Drp1-Dependent Synapse Formation in Cultured Hippocampal Neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 2169–2174. [Google Scholar] [CrossRef]
- Bleicken, S.; Hofhaus, G.; Ugarte-Uribe, B.; Schröder, R.; García-Sáez, A.J. cBid, Bax and Bcl-xL Exhibit Opposite Membrane Remodeling Activities. Cell Death Dis. 2016, 7, e2121. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, C.; Delivani, P.; Cullen, S.P.; Martin, S.J. Bax- or Bak-Induced Mitochondrial Fission Can Be Uncoupled from Cytochrome C Release. Mol. Cell 2008, 31, 570–585. [Google Scholar] [CrossRef]
- Bessou, M.; Lopez, J.; Gadet, R.; Deygas, M.; Popgeorgiev, N.; Poncet, D.; Nougarède, A.; Billard, P.; Mikaelian, I.; Gonzalo, P.; et al. The Apoptosis Inhibitor Bcl-xL Controls Breast Cancer Cell Migration through Mitochondria-Dependent Reactive Oxygen Species Production. Oncogene 2020, 39, 3056–3074. [Google Scholar] [CrossRef]
- Jonas, E. Contributions of Bcl-xL to Acute and Long Term Changes in Bioenergetics during Neuronal Plasticity. Biochim. Biophys. Acta 2014, 1842, 1168–1178. [Google Scholar] [CrossRef]
- Alavian, K.N.; Li, H.; Collis, L.P.; Bonanni, L.; Zeng, L.; Sacchetti, S.; Lazrove, E.; Nabili, P.; Flaherty, B.J.; Graham, M.; et al. Bcl-xL Regulates Metabolic Efficiency of Neurons through Interaction with the Mitochondrial F1FO ATP Synthase. Nat. Cell Biol. 2011, 13, 1224–1233. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-B.; Aon, M.A.; Hsu, Y.-T.; Soane, L.; Teng, X.; McCaffery, J.M.; Cheng, W.C.; Qi, B.; Li, H.; Alavian, K.N.; et al. Bcl-xL Regulates Mitochondrial Energetics by Stabilizing the Inner Membrane Potential. J. Cell Biol. 2011, 195, 263–276. [Google Scholar] [CrossRef]
- Jansen, J.; Scott, M.; Amjad, E.; Stumpf, A.; Lackey, K.H.; Caldwell, K.A.; Park, H.-A. Bcl-xL Is Required by Primary Hippocampal Neurons during Development to Support Local Energy Metabolism at Neurites. Biology 2021, 10, 772. [Google Scholar] [CrossRef] [PubMed]
- Fouqué, A.; Lepvrier, E.; Debure, L.; Gouriou, Y.; Malleter, M.; Delcroix, V.; Ovize, M.; Ducret, T.; Li, C.; Hammadi, M.; et al. The Apoptotic Members CD95, BclxL, and Bcl-2 Cooperate to Promote Cell Migration by Inducing Ca2+ Flux from the Endoplasmic Reticulum to Mitochondria. Cell Death Differ. 2016, 23, 1702–1716. [Google Scholar] [CrossRef]
- Juhaszova, M.; Kobrinsky, E.; Zorov, D.B.; Nuss, H.B.; Yaniv, Y.; Fishbein, K.W.; de Cabo, R.; Montoliu, L.; Gabelli, S.B.; Aon, M.A.; et al. ATP Synthase K+- and H+-Fluxes Drive ATP Synthesis and Enable Mitochondrial K+-“Uniporter” Function: II. Ion and ATP Synthase Flux Regulation. Function 2022, 3, zqac001. [Google Scholar] [CrossRef]
- Du, X.; Fu, X.; Yao, K.; Lan, Z.; Xu, H.; Cui, Q.; Yang, E. Bcl-2 Delays Cell Cycle through Mitochondrial ATP and ROS. Cell Cycle 2017, 16, 707–713. [Google Scholar] [CrossRef]
- Du, X.; Xiao, J.; Fu, X.; Xu, B.; Han, H.; Wang, Y.; Pei, X. A Proteomic Analysis of Bcl-2 Regulation of Cell Cycle Arrest: Insight into the Mechanisms. J. Zhejiang Univ. Sci. B 2021, 22, 839–855. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Ueta, E.; Kimura, T.; Yamamoto, T.; Osaki, T. Reactive Oxygen Species (ROS) Control the Expression of Bcl-2 Family Proteins by Regulating Their Phosphorylation and Ubiquitination. Cancer Sci. 2004, 95, 644–650. [Google Scholar] [CrossRef]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 Antiapoptotic Proteins Inhibit Beclin 1-Dependent Autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.G.; Rapp, U.R.; Reed, J.C. Bcl-2 Targets the Protein Kinase Raf-1 to Mitochondria. Cell 1996, 87, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Haughn, L.; Hawley, R.G.; Morrison, D.K.; von Boehmer, H.; Hockenbery, D.M. BCL-2 and BCL-XL Restrict Lineage Choice during Hematopoietic Differentiation. J. Biol. Chem. 2003, 278, 25158–25165. [Google Scholar] [CrossRef]
- Yang, C.-Y.; Liu, S.-H.; Su, C.-C.; Fang, K.-M.; Yang, T.-Y.; Liu, J.-M.; Chen, Y.-W.; Chang, K.-C.; Chuang, H.-L.; Wu, C.-T.; et al. Methylmercury Induces Mitochondria- and Endoplasmic Reticulum Stress-Dependent Pancreatic β-Cell Apoptosis via an Oxidative Stress-Mediated JNK Signaling Pathway. Int. J. Mol. Sci. 2022, 23, 2858. [Google Scholar] [CrossRef]
- Cunha, D.A.; Igoillo-Esteve, M.; Gurzov, E.N.; Germano, C.M.; Naamane, N.; Marhfour, I.; Fukaya, M.; Vanderwinden, J.-M.; Gysemans, C.; Mathieu, C.; et al. Death Protein 5 and P53-Upregulated Modulator of Apoptosis Mediate the Endoplasmic Reticulum Stress–Mitochondrial Dialog Triggering Lipotoxic Rodent and Human β-Cell Apoptosis. Diabetes 2012, 61, 2763–2775. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Kong, X.; Lv, S.; Shu, X.; Ma, X.; Ai, X.; Yan, D.; Ying, Y. FXR-Regulated COX6A2 Triggers Mitochondrial Apoptosis of Pancreatic β-Cell in Type 2 Diabetes. Cell Death Dis. 2024, 15, 920. [Google Scholar] [CrossRef]
- Park, J.H.; Oh, J.E.; Kim, N.; Kwak, Y.-L. Dexmedetomidine Alleviates CoCl2-Induced Hypoxic Cellular Damage in INS-1 Cells by Regulating Autophagy. Korean J. Anesthesiol. 2024, 77, 623–634. [Google Scholar] [CrossRef]
- Lu, H.; Hao, L.; Li, S.; Lin, S.; Lv, L.; Chen, Y.; Cui, H.; Zi, T.; Chu, X.; Na, L.; et al. Elevated Circulating Stearic Acid Leads to a Major Lipotoxic Effect on Mouse Pancreatic Beta Cells in Hyperlipidaemia via a miR-34a-5p-Mediated PERK/P53-Dependent Pathway. Diabetologia 2016, 59, 1247–1257. [Google Scholar] [CrossRef]
- Lin, N.; Niu, Y.; Zhang, W.; Li, X.; Yang, Z.; Su, Q. microRNA-802 Is Involved in Palmitate-Induced Damage to Pancreatic β Cells through Repression of Sirtuin 6. Int. J. Clin. Exp. Pathol. 2017, 10, 11300–11307. [Google Scholar]
- Lee, Y.-S.; Li, N.; Shin, S.; Jun, H.-S. Role of Nitric Oxide in the Pathogenesis of Encephalomyocarditis Virus-Induced Diabetes in Mice. J. Virol. 2009, 83, 8004–8011. [Google Scholar] [CrossRef]
- Dawson, S.-J.; Makretsov, N.; Blows, F.M.; Driver, K.E.; Provenzano, E.; Le Quesne, J.; Baglietto, L.; Severi, G.; Giles, G.G.; McLean, C.A.; et al. BCL2 in Breast Cancer: A Favourable Prognostic Marker across Molecular Subtypes and Independent of Adjuvant Therapy Received. Br. J. Cancer 2010, 103, 668–675. [Google Scholar] [CrossRef]
- Ikezawa, K.; Hikita, H.; Shigekawa, M.; Iwahashi, K.; Eguchi, H.; Sakamori, R.; Tatsumi, T.; Takehara, T. Increased Bcl-xL Expression in Pancreatic Neoplasia Promotes Carcinogenesis by Inhibiting Senescence and Apoptosis. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 185–200.e1. [Google Scholar] [CrossRef] [PubMed]
- Moriishi, T.; Maruyama, Z.; Fukuyama, R.; Ito, M.; Miyazaki, T.; Kitaura, H.; Ohnishi, H.; Furuichi, T.; Kawai, Y.; Masuyama, R.; et al. Overexpression of Bcl2 in Osteoblasts Inhibits Osteoblast Differentiation and Induces Osteocyte Apoptosis. PLoS ONE 2011, 6, e27487. [Google Scholar] [CrossRef]
- Shimora, H.; Matsuda, M.; Nakayama, Y.; Maeyama, H.; Tanioka, R.; Tanaka, Y.; Kitatani, K.; Nabe, T. Involvement of Janus Kinase-Dependent Bcl-xL Overexpression in Steroid Resistance of Group 2 Innate Lymphoid Cells in Asthma. Immunology 2024, 172, 653–668. [Google Scholar] [CrossRef]
- Kodama, T.; Takehara, T.; Hikita, H.; Shimizu, S.; Shigekawa, M.; Li, W.; Miyagi, T.; Hosui, A.; Tatsumi, T.; Ishida, H.; et al. BH3-Only Activator Proteins Bid and Bim Are Dispensable for Bak/Bax-Dependent Thrombocyte Apoptosis Induced by Bcl-xL Deficiency: Molecular Requisites for the Mitochondrial Pathway to Apoptosis in Platelets. J. Biol. Chem. 2011, 286, 13905–13913. [Google Scholar] [CrossRef] [PubMed]
- Afreen, S.; Bohler, S.; Müller, A.; Demmerath, E.-M.; Weiss, J.M.; Jutzi, J.S.; Schachtrup, K.; Kunze, M.; Erlacher, M. BCL-XL Expression Is Essential for Human Erythropoiesis and Engraftment of Hematopoietic Stem Cells. Cell Death Dis. 2020, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, K.; Waring, P.; Glaser, S.P.; Wimmer, V.; Cottle, D.L.; Tham, M.S.; Nhu, D.; Whitehead, L.; Delbridge, A.R.; Lessene, G.; et al. BCL-XL Exerts a Protective Role against Anemia Caused by Radiation-Induced Kidney Damage. EMBO J. 2020, 39, e105561. [Google Scholar] [CrossRef]
- Nakamura, A.; Swahari, V.; Plestant, C.; Smith, I.; McCoy, E.; Smith, S.; Moy, S.S.; Anton, E.S.; Deshmukh, M. Bcl-xL Is Essential for the Survival and Function of Differentiated Neurons in the Cortex That Control Complex Behaviors. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 5448–5461. [Google Scholar] [CrossRef] [PubMed]
- Ceizar, M.; Dhaliwal, J.; Xi, Y.; Smallwood, M.; Kumar, K.L.; Lagace, D.C. Bcl-2 Is Required for the Survival of Doublecortin-Expressing Immature Neurons. Hippocampus 2016, 26, 211–219. [Google Scholar] [CrossRef]
- Zhou, Y.-P.; Pena, J.C.; Roe, M.W.; Mittal, A.A.; Levisetti, M.G.; Baldwin, A.C.; Pugh, W.; Ostrega, D.; Ahmed, N.; Bindokas, V.P.; et al. Overexpression of Bcl-xL in β-Cells Prevents Cell Death but Impairs Mitochondrial Signal for Insulin Secretion. Am. J. Physiol.-Endocrinol. Metab. 2000, 278, E340–E351. [Google Scholar] [CrossRef] [PubMed]
- Klein, D.; Ribeiro, M.M.; Mendoza, V.; Jayaraman, S.; Kenyon, N.S.; Pileggi, A.; Molano, R.D.; Inverardi, L.; Ricordi, C.; Pastori, R.L. Delivery of Bcl-XL or Its BH4 Domain by Protein Transduction Inhibits Apoptosis in Human Islets. Biochem. Biophys. Res. Commun. 2004, 323, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Holohan, C.; Szegezdi, E.; Ritter, T.; O’Brien, T.; Samali, A. Cytokine-Induced β-Cell Apoptosis Is NO-Dependent, Mitochondria-Mediated and Inhibited by BCL-XL. J. Cell. Mol. Med. 2008, 12, 591–606. [Google Scholar] [CrossRef] [PubMed]
- Allison, J.; Thomas, H.; Beck, D.; Brady, J.L.; Lew, A.M.; Elefanty, A.; Kosaka, H.; Kay, T.W.; Huang, D.C.S.; Strasser, A. Transgenic Overexpression of Human Bcl-2 in Islet β Cells Inhibits Apoptosis but Does Not Prevent Autoimmune Destruction. Int. Immunol. 2000, 12, 9–17. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Barthson, J.; Germano, C.M.; Moore, F.; Maida, A.; Drucker, D.J.; Marchetti, P.; Gysemans, C.; Mathieu, C.; Nuñez, G.; Jurisicova, A.; et al. Cytokines Tumor Necrosis Factor-α and Interferon-γ Induce Pancreatic β-Cell Apoptosis through STAT1-Mediated Bim Protein Activation. J. Biol. Chem. 2011, 286, 39632–39643. [Google Scholar] [CrossRef] [PubMed]
- Carrington, E.M.; McKenzie, M.D.; Jansen, E.S.; Myers, M.; Fynch, S.; Kos, C.; Strasser, A.; Kay, T.W.H.; Scott, C.L.; Allison, J. Islet β-Cells Deficient in Bcl-xL Develop but Are Abnormally Sensitive to Apoptotic Stimuli. Diabetes 2009, 58, 2316–2323. [Google Scholar] [CrossRef]
- Gurzov, E.N.; Germano, C.M.; Cunha, D.A.; Ortis, F.; Vanderwinden, J.-M.; Marchetti, P.; Zhang, L.; Eizirik, D.L. P53 Up-Regulated Modulator of Apoptosis (PUMA) Activation Contributes to Pancreatic β-Cell Apoptosis Induced by Proinflammatory Cytokines and Endoplasmic Reticulum Stress*. J. Biol. Chem. 2010, 285, 19910–19920. [Google Scholar] [CrossRef] [PubMed]
- Lupi, R.; Dotta, F.; Marselli, L.; Del Guerra, S.; Masini, M.; Santangelo, C.; Patanè, G.; Boggi, U.; Piro, S.; Anello, M.; et al. Prolonged Exposure to Free Fatty Acids Has Cytostatic and Pro-Apoptotic Effects on Human Pancreatic Islets: Evidence That β-Cell Death Is Caspase Mediated, Partially Dependent on Ceramide Pathway, and Bcl-2 Regulated. Diabetes 2002, 51, 1437–1442. [Google Scholar] [CrossRef] [PubMed]
- Iwahashi, H.; Hanafusa, T.; Eguchi, Y.; Nakajima, H.; Miyagawa, J.; Itoh, N.; Tomita, K.; Namba, M.; Kuwajima, M.; Noguchi, T.; et al. Cytokine-Induced Apoptotic Cell Death in a Mouse Pancreatic Beta-Cell Line: Inhibition by Bcl-2. Diabetologia 1996, 39, 530–536. [Google Scholar] [CrossRef]
- Litwak, S.A.; Wali, J.A.; Pappas, E.G.; Saadi, H.; Stanley, W.J.; Varanasi, L.C.; Kay, T.W.H.; Thomas, H.E.; Gurzov, E.N. Lipotoxic Stress Induces Pancreatic β-Cell Apoptosis through Modulation of Bcl-2 Proteins by the Ubiquitin-Proteasome System. J. Diabetes Res. 2015, 2015, 280615. [Google Scholar] [CrossRef] [PubMed]
- Miani, M.; Barthson, J.; Colli, M.L.; Brozzi, F.; Cnop, M.; Eizirik, D.L. Endoplasmic Reticulum Stress Sensitizes Pancreatic Beta Cells to Interleukin-1β-Induced Apoptosis via Bim/A1 Imbalance. Cell Death Dis. 2013, 4, e701. [Google Scholar] [CrossRef]
- Loo, L.S.W.; Soetedjo, A.A.P.; Lau, H.H.; Ng, N.H.J.; Ghosh, S.; Nguyen, L.; Krishnan, V.G.; Choi, H.; Roca, X.; Hoon, S.; et al. BCL-xL/BCL2L1 Is a Critical Anti-Apoptotic Protein That Promotes the Survival of Differentiating Pancreatic Cells from Human Pluripotent Stem Cells. Cell Death Dis. 2020, 11, 378. [Google Scholar] [CrossRef] [PubMed]
- Garchon, H.J.; Luan, J.J.; Eloy, L.; Bédossa, P.; Bach, J.F. Genetic Analysis of Immune Dysfunction in Non-Obese Diabetic (NOD) Mice: Mapping of a Susceptibility Locus Close to the Bcl-2 Gene Correlates with Increased Resistance of NOD T Cells to Apoptosis Induction. Eur. J. Immunol. 1994, 24, 380–384. [Google Scholar] [CrossRef]
- Brozzi, F.; Jacovetti, C.; Cosentino, C.; Menoud, V.; Wu, K.; Bayazit, M.B.; Abdulkarim, B.; Iseli, C.; Guex, N.; Guay, C.; et al. tRNA-Derived Fragments in T Lymphocyte–Beta Cell Crosstalk and in Type 1 Diabetes Pathogenesis in NOD Mice. Diabetologia 2024, 67, 2260–2274. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, P.I.; Juárez-Vicente, F.; Cobo-Vuilleumier, N.; García-Domínguez, M.; Gauthier, B.R. The Diabetes-Linked Transcription Factor PAX4: From Gene to Functional Consequences. Genes 2017, 8, 101. [Google Scholar] [CrossRef]
- Ko, J.; Fonseca, V.A.; Wu, H. Pax4 in Health and Diabetes. Int. J. Mol. Sci. 2023, 24, 8283. [Google Scholar] [CrossRef] [PubMed]
- Sosa-Pineda, B.; Chowdhury, K.; Torres, M.; Oliver, G.; Gruss, P. The Pax4 Gene Is Essential for Differentiation of Insulin-Producing Beta Cells in the Mammalian Pancreas. Nature 1997, 386, 399–402. [Google Scholar] [CrossRef]
- Brun, T.; Franklin, I.; St-Onge, L.; Biason-Lauber, A.; Schoenle, E.J.; Wollheim, C.B.; Gauthier, B.R. The Diabetes-Linked Transcription Factor PAX4 Promotes β-Cell Proliferation and Survival in Rat and Human Islets. J. Cell Biol. 2004, 167, 1123–1135. [Google Scholar] [CrossRef]
- Mellado-Gil, J.M.; Jiménez-Moreno, C.M.; Martin-Montalvo, A.; Alvarez-Mercado, A.I.; Fuente-Martin, E.; Cobo-Vuilleumier, N.; Lorenzo, P.I.; Bru-Tari, E.; de Gracia Herrera-Gómez, I.; López-Noriega, L.; et al. PAX4 Preserves Endoplasmic Reticulum Integrity Preventing Beta Cell Degeneration in a Mouse Model of Type 1 Diabetes Mellitus. Diabetologia 2016, 59, 755–765. [Google Scholar] [CrossRef] [PubMed]
- He, K.H.; Lorenzo, P.I.; Brun, T.; Jimenez Moreno, C.M.; Aeberhard, D.; Vallejo Ortega, J.; Cornu, M.; Thorel, F.; Gjinovci, A.; Thorens, B.; et al. In Vivo Conditional Pax4 Overexpression in Mature Islet β-Cells Prevents Stress-Induced Hyperglycemia in Mice. Diabetes 2011, 60, 1705–1715. [Google Scholar] [CrossRef]
- Brun, T.; Duhamel, D.L.; He, K.H.; Wollheim, C.B.; Gauthier, B.R. The Transcription Factor PAX4 Acts as a Survival Gene in INS-1E Insulinoma Cells. Oncogene 2007, 26, 4261–4271. [Google Scholar] [CrossRef] [PubMed]
- Brun, T.; He, K.H.H.; Lupi, R.; Boehm, B.; Wojtusciszyn, A.; Sauter, N.; Donath, M.; Marchetti, P.; Maedler, K.; Gauthier, B.R. The Diabetes-Linked Transcription Factor Pax4 Is Expressed in Human Pancreatic Islets and Is Activated by Mitogens and GLP-1. Hum. Mol. Genet. 2008, 17, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Welsh, N.; Jonas, J.-C.; Jörns, A.; Lenzen, S.; Eizirik, D.L. Mechanisms of Pancreatic Beta-Cell Death in Type 1 and Type 2 Diabetes: Many Differences, Few Similarities. Diabetes 2005, 54, S97–S107. [Google Scholar] [CrossRef]
- Gurzov, E.N.; Ortis, F.; Cunha, D.A.; Gosset, G.; Li, M.; Cardozo, A.K.; Eizirik, D.L. Signaling by IL-1beta+IFN-Gamma and ER Stress Converge on DP5/Hrk Activation: A Novel Mechanism for Pancreatic Beta-Cell Apoptosis. Cell Death Differ. 2009, 16, 1539–1550. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.A.; Kutlu, B.; Velmurugan, K.; Kizaka-Kondoh, S.; Lee, C.E.; Wong, R.; Valentine, A.; Davidson, H.W.; Hutton, J.C.; Pugazhenthi, S. Cytokine-Mediated Induction of Anti-Apoptotic Genes That Are Linked to Nuclear Factor Kappa-B (NF-kappaB) Signalling in Human Islets and in a Mouse Beta Cell Line. Diabetologia 2009, 52, 1092–1101. [Google Scholar] [CrossRef][Green Version]
- Gurzov, E.N.; Ortis, F.; Bakiri, L.; Wagner, E.F.; Eizirik, D.L. JunB Inhibits ER Stress and Apoptosis in Pancreatic Beta Cells. PLoS ONE 2008, 3, e3030. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Labuda, T.; Xia, Y.; Gallagher, E.; Fang, D.; Liu, Y.-C.; Karin, M. Jun Turnover Is Controlled through JNK-Dependent Phosphorylation of the E3 Ligase Itch. Science 2004, 306, 271–275. [Google Scholar] [CrossRef]
- Inoshita, S.; Takeda, K.; Hatai, T.; Terada, Y.; Sano, M.; Hata, J.; Umezawa, A.; Ichijo, H. Phosphorylation and Inactivation of Myeloid Cell Leukemia 1 by JNK in Response to Oxidative Stress. J. Biol. Chem. 2002, 277, 43730–43734. [Google Scholar] [CrossRef]
- Allagnat, F.; Cunha, D.; Moore, F.; Vanderwinden, J.M.; Eizirik, D.L.; Cardozo, A.K. Mcl-1 Downregulation by pro-Inflammatory Cytokines and Palmitate Is an Early Event Contributing to β-Cell Apoptosis. Cell Death Differ. 2011, 18, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, R.S.; Marroqui, L.; Grieco, F.A.; Marselli, L.; Suleiman, M.; Henz, S.R.; Marchetti, P.; Wernersson, R.; Eizirik, D.L. Protective Role of Complement C3 Against Cytokine-Mediated β-Cell Apoptosis. Endocrinology 2017, 158, 2503–2521. [Google Scholar] [CrossRef] [PubMed]
- Moore, F.; Naamane, N.; Colli, M.L.; Bouckenooghe, T.; Ortis, F.; Gurzov, E.N.; Igoillo-Esteve, M.; Mathieu, C.; Bontempi, G.; Thykjaer, T.; et al. STAT1 Is a Master Regulator of Pancreatic {beta}-Cell Apoptosis and Islet Inflammation. J. Biol. Chem. 2011, 286, 929–941. [Google Scholar] [CrossRef]
- Kim, H.; Rafiuddin-Shah, M.; Tu, H.-C.; Jeffers, J.R.; Zambetti, G.P.; Hsieh, J.J.-D.; Cheng, E.H.-Y. Hierarchical Regulation of Mitochondrion-Dependent Apoptosis by BCL-2 Subfamilies. Nat. Cell Biol. 2006, 8, 1348–1358. [Google Scholar] [CrossRef]
- Stanley, W.J.; Trivedi, P.M.; Sutherland, A.P.; Thomas, H.E.; Gurzov, E.N. Differential Regulation of Pro-Inflammatory Cytokine Signalling by Protein Tyrosine Phosphatases in Pancreatic β-Cells. J. Mol. Endocrinol. 2017, 59, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Mehmeti, I.; Lenzen, S.; Lortz, S. Modulation of Bcl-2-Related Protein Expression in Pancreatic Beta Cells by pro-Inflammatory Cytokines and Its Dependence on the Antioxidative Defense Status. Mol. Cell. Endocrinol. 2011, 332, 88–96. [Google Scholar] [CrossRef]
- Sims, E.K.; Lakhter, A.J.; Anderson-Baucum, E.; Kono, T.; Tong, X.; Evans-Molina, C. MicroRNA 21 Targets BCL2 mRNA to Increase Apoptosis in Rat and Human Beta Cells. Diabetologia 2017, 60, 1057–1065. [Google Scholar] [CrossRef]
- Kharbanda, S.; Saxena, S.; Yoshida, K.; Pandey, P.; Kaneki, M.; Wang, Q.; Cheng, K.; Chen, Y.-N.; Campbell, A.; Sudha, T.; et al. Translocation of SAPK/JNK to Mitochondria and Interaction with Bcl-xL in Response to DNA Damage*. J. Biol. Chem. 2000, 275, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Sinha, S.C.; Levine, B. Dual Role of JNK1-Mediated Phosphorylation of Bcl-2 in Autophagy and Apoptosis Regulation. Autophagy 2008, 4, 949–951. [Google Scholar] [CrossRef]
- Edlich, F.; Banerjee, S.; Suzuki, M.; Cleland, M.M.; Arnoult, D.; Wang, C.; Neutzner, A.; Tjandra, N.; Youle, R.J. Bcl-xL Retrotranslocates Bax from the Mitochondria into the Cytosol. Cell 2011, 145, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Federici, M.; Hribal, M.L.; Perego, L.; Ranalli, M.; Caradonna, Z.; Perego, C.; Usellini, L.; Nano, R.; Bonini, P.; Bertuzzi, F.; et al. High Glucose Causes Apoptosis in Cultured Human Pancreatic Islets of Langerhans: A Potential Role for Regulation of Specific Bcl Family Genes Toward an Apoptotic Cell Death Program. Diabetes 2001, 50, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Koh, P.-O. Streptozotocin-Induced Diabetes Increases the Interaction of Bad/Bcl-XL and Decreases the Binding of pBad/14-3-3 in Rat Testis. Life Sci. 2007, 81, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Szymczak, F.; Alvelos, M.I.; Marín-Cañas, S.; Castela, Â.; Demine, S.; Colli, M.L.; de Beeck, A.O.; Thomaidou, S.; Marselli, L.; Zaldumbide, A.; et al. Transcription and Splicing Regulation by NLRC5 Shape the Interferon Response in Human Pancreatic β Cells. Sci. Adv. 2022, 8, eabn5732. [Google Scholar] [CrossRef] [PubMed]
- Marroquí, L.; Masini, M.; Merino, B.; Grieco, F.A.; Millard, I.; Dubois, C.; Quesada, I.; Marchetti, P.; Cnop, M.; Eizirik, D.L. Pancreatic α Cells Are Resistant to Metabolic Stress-Induced Apoptosis in Type 2 Diabetes. eBioMedicine 2015, 2, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.A.; Redick, S.D.; Blodgett, D.M.; Richardson, S.J.; Leete, P.; Krogvold, L.; Dahl-Jørgensen, K.; Bottino, R.; Brissova, M.; Spaeth, J.M.; et al. HLA Class II Antigen Processing and Presentation Pathway Components Demonstrated by Transcriptome and Protein Analyses of Islet β-Cells From Donors with Type 1 Diabetes. Diabetes 2019, 68, 988–1001. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Yang, H.; Boffa, D.J.; Ding, R.; Sharma, V.K.; Lagman, M.; Li, B.; Hering, B.; Mohanakumar, T.; Lakey, J.; et al. Proapoptotic Bax Is Hyperexpressed in Isolated Human Islets Compared with Antiapoptotic Bcl-21. Transplantation 2002, 74, 1489–1496. [Google Scholar] [CrossRef] [PubMed]
- Brondani, L.A.; Rech, T.H.; Boelter, G.; Bauer, A.C.; Leitão, C.B.; Eizirik, D.L.; Crispim, D. UCP2 Expression Is Increased in Pancreas From Brain-Dead Donors and Involved in Cytokine-Induced β Cells Apoptosis. Transplantation 2017, 101, e59–e67. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Ythier, D.; Brozzi, F.; Eizirik, D.L.; Thorens, B. Clic4, a Novel Protein That Sensitizes β-Cells to Apoptosis. Mol. Metab. 2015, 4, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Cunha, D.A.; Ladrière, L.; Ortis, F.; Igoillo-Esteve, M.; Gurzov, E.N.; Lupi, R.; Marchetti, P.; Eizirik, D.L.; Cnop, M. Glucagon-like Peptide-1 Agonists Protect Pancreatic Beta-Cells from Lipotoxic Endoplasmic Reticulum Stress through Upregulation of BiP and JunB. Diabetes 2009, 58, 2851–2862. [Google Scholar] [CrossRef] [PubMed]
- Natalicchio, A.; De Stefano, F.; Orlando, M.R.; Melchiorre, M.; Leonardini, A.; Cignarelli, A.; Labarbuta, R.; Marchetti, P.; Perrini, S.; Laviola, L.; et al. Exendin-4 Prevents c-Jun N-Terminal Protein Kinase Activation by Tumor Necrosis Factor-α (TNFα) and Inhibits TNFα-Induced Apoptosis in Insulin-Secreting Cells. Endocrinology 2010, 151, 2019–2029. [Google Scholar] [CrossRef] [PubMed]
- Takayama, S.; Sato, T.; Krajewski, S.; Kochel, K.; Irie, S.; Millan, J.A.; Reed, J.C. Cloning and Functional Analysis of BAG-1: A Novel Bcl-2-Binding Protein with Anti-Cell Death Activity. Cell 1995, 80, 279–284. [Google Scholar] [CrossRef]
- Ferland, C.L.; Harris, E.P.; Lam, M.; Schrader, L.A. Facilitation of the HPA Axis to a Novel Acute Stress Following Chronic Stress Exposure Modulates Histone Acetylation and the ERK/MAPK Pathway in the Dentate Gyrus of Male Rats. Endocrinology 2014, 155, 2942–2952. [Google Scholar] [CrossRef]
- Kurek, A.; Głombik, K.; Detka, J.; Basta-Kaim, A.; Kubera, M.; Lasoń, W.; Budziszewska, B. Regulators of Glucocorticoid Receptor Function in an Animal Model of Depression and Obesity. J. Neuroendocrinol. 2018, 30, e12591. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Gao, Z.; Robert, C.E.; Greene, S.; Xu, G.; Xu, W.; Bell, E.; Campbell, D.; Zhu, Y.; Young, R.; et al. Pancreatic-Derived Factor (FAM3B), a Novel Islet Cytokine, Induces Apoptosis of Insulin-Secreting β-Cells. Diabetes 2003, 52, 2296–2303. [Google Scholar] [CrossRef]
- Yang, J.; Robert, C.E.; Burkhardt, B.R.; Young, R.A.; Wu, J.; Gao, Z.; Wolf, B.A. Mechanisms of Glucose-Induced Secretion of Pancreatic-Derived Factor (PANDER or FAM3B) in Pancreatic Beta-Cells. Diabetes 2005, 54, 3217–3228. [Google Scholar] [CrossRef]
- Zhu, Y.; Xu, G.; Patel, A.; McLaughlin, M.M.; Silverman, C.; Knecht, K.; Sweitzer, S.; Li, X.; McDonnell, P.; Mirabile, R.; et al. Cloning, Expression, and Initial Characterization of a Novel Cytokine-like Gene Family. Genomics 2002, 80, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Mou, H.; Li, Z.; Yao, P.; Zhuo, S.; Luan, W.; Deng, B.; Qian, L.; Yang, M.; Mei, H.; Le, Y. Knockdown of FAM3B Triggers Cell Apoptosis through P53-Dependent Pathway. Int. J. Biochem. Cell Biol. 2013, 45, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Yang, J.; Burkhardt, B.R.; Gao, Z.; Wong, R.K.; Greene, S.R.; Wu, J.; Wolf, B.A. Effects of Overexpression of Pancreatic Derived Factor (FAM3B) in Isolated Mouse Islets and Insulin-Secreting βTC3 Cells. Am. J. Physiol.-Endocrinol. Metab. 2005, 289, E543–E550. [Google Scholar] [CrossRef] [PubMed]
- Arda, H.E.; Li, L.; Tsai, J.; Torre, E.A.; Rosli, Y.; Peiris, H.; Spitale, R.C.; Dai, C.; Gu, X.; Qu, K.; et al. Age-Dependent Pancreatic Gene Regulation Reveals Mechanisms Governing Human β Cell Function. Cell Metab. 2016, 23, 909–920. [Google Scholar] [CrossRef]
- Helman, A.; Klochendler, A.; Azazmeh, N.; Gabai, Y.; Horwitz, E.; Anzi, S.; Swisa, A.; Condiotti, R.; Granit, R.Z.; Nevo, Y.; et al. P16Ink4a-Induced Senescence of Pancreatic Beta Cells Enhances Insulin Secretion. Nat. Med. 2016, 22, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, E.; Krogvold, L.; Zhitomirsky, S.; Swisa, A.; Fischman, M.; Lax, T.; Dahan, T.; Hurvitz, N.; Weinberg-Corem, N.; Klochendler, A.; et al. β-Cell DNA Damage Response Promotes Islet Inflammation in Type 1 Diabetes. Diabetes 2018, 67, 2305–2318. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.J.; Shah, A.; Ntranos, V.; Gool, F.V.; Atkinson, M.; Bhushan, A. Targeted Elimination of Senescent Beta Cells Prevents Type 1 Diabetes. Cell Metab. 2019, 29, 1045–1060.e10. [Google Scholar] [CrossRef]
- Rampazzo Morelli, N.; Préfontaine, C.; Pipella, J.; Thompson, P.J. Secreted GDF15 Maintains Transcriptional Responses during DNA Damage-Mediated Senescence in Human β-Cells. Am. J. Physiol.-Endocrinol. Metab. 2024, 327, E552–E562. [Google Scholar] [CrossRef] [PubMed]
- Pasula, D.J.; Shi, R.; Vanderkruk, B.; Shih, A.Z.L.; Zou, Y.; Chaudhry, A.; Hoffman, B.G.; Luciani, D.S. Bcl-xL Restricts Transcriptional, Morphological and Functional Decompensation of β-Cell Mitochondria under Chronic Glucose Excess. bioRxiv 2021, 10.25.465491. [Google Scholar] [CrossRef]
- Soria, B.; Gauthier, B.R. Dual Trade of Bcl-2 and Bcl-xL in Islet Physiology: Balancing Life and Death with Metabolism Secretion Coupling. Diabetes 2012, 62, 18–21. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Singh, K.; Bricard, O.; Haughton, J.; Björkqvist, M.; Thorstensson, M.; Luo, Z.; Mascali, L.; Pasciuto, E.; Mathieu, C.; Dooley, J.; et al. Gene Delivery of Manf to Beta-Cells of the Pancreatic Islets Protects NOD Mice from Type 1 Diabetes Development. Biomolecules 2022, 12, 1493. [Google Scholar] [CrossRef] [PubMed]
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.L.; Gao, G. Viral Vector Platforms within the Gene Therapy Landscape. Signal Transduct. Target. Ther. 2021, 6, 53. [Google Scholar] [CrossRef]
- Sabatino, D.E.; Bushman, F.D.; Chandler, R.J.; Crystal, R.G.; Davidson, B.L.; Dolmetsch, R.; Eggan, K.C.; Gao, G.; Gil-Farina, I.; Kay, M.A.; et al. Evaluating the State of the Science for Adeno-Associated Virus Integration: An Integrated Perspective. Mol. Ther. 2022, 30, 2646–2663. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-H.; Gessler, D.J.; Zhan, W.; Gallagher, T.L.; Gao, G. Adeno-Associated Virus as a Delivery Vector for Gene Therapy of Human Diseases. Signal Transduct. Target. Ther. 2024, 9, 78. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, Y.; Xu, C.; Liu, J.; Chen, J.; Li, G.; Huang, B.; Pan, Y.; Zhang, Y.; Wei, Q.; et al. Circular RNA circGlis3 Protects against Islet β-Cell Dysfunction and Apoptosis in Obesity. Nat. Commun. 2023, 14, 351. [Google Scholar] [CrossRef]
- Zhong, L.; Granelli-Piperno, A.; Choi, Y.; Steinman, R.M. Recombinant Adenovirus Is an Efficient and Non-Perturbing Genetic Vector for Human Dendritic Cells. Eur. J. Immunol. 1999, 29, 964–972. [Google Scholar] [CrossRef]
- Naik, P.; Karrim, J.; Hanahan, D. The Rise and Fall of Apoptosis during Multistage Tumorigenesis: Down-Modulation Contributes to Tumor Progression from Angiogenic Progenitors. Genes Dev. 1996, 10, 2105–2116. [Google Scholar] [CrossRef] [PubMed]
- Kapturczak, M.H.; Flotte, T.; Atkinson, M.A. Adeno-Associated Virus (AAV) as a Vehicle for Therapeutic Gene Delivery Improvements in Vector Design and Viral Production Enhance Potential to Prolong Graft Survival in Pancreatic Islet Cell Transplantation for the Reversal of Type 1 Diabetes. Curr. Mol. Med. 2001, 1, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Bevacqua, R.J.; Dai, X.; Lam, J.Y.; Gu, X.; Friedlander, M.S.H.; Tellez, K.; Miguel-Escalada, I.; Bonàs-Guarch, S.; Atla, G.; Zhao, W.; et al. CRISPR-Based Genome Editing in Primary Human Pancreatic Islet Cells. Nat. Commun. 2021, 12, 2397. [Google Scholar] [CrossRef] [PubMed]
- Aguayo-Mazzucato, C.; Andle, J.; Lee, T.B.; Midha, A.; Talemal, L.; Chipashvili, V.; Hollister-Lock, J.; van Deursen, J.; Weir, G.; Bonner-Weir, S. Acceleration of β Cell Aging Determines Diabetes and Senolysis Improves Disease Outcomes. Cell Metab. 2019, 30, 129–142.e4. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, K.; Abarca, C.; Aguayo-Mazzucato, C. Regulation of Cellular Senescence in Type 2 Diabetes Mellitus: From Mechanisms to Clinical Applications. Diabetes Metab. J. 2023, 47, 441–453. [Google Scholar] [CrossRef]
- Rorsman, P.; Ashcroft, F.M. Pancreatic β-Cell Electrical Activity and Insulin Secretion: Of Mice and Men. Physiol. Rev. 2018, 98, 117–214. [Google Scholar] [CrossRef] [PubMed]
- Brozzi, F.; Nardelli, T.R.; Lopes, M.; Millard, I.; Barthson, J.; Igoillo-Esteve, M.; Grieco, F.A.; Villate, O.; Oliveira, J.M.; Casimir, M.; et al. Cytokines Induce Endoplasmic Reticulum Stress in Human, Rat and Mouse Beta Cells via Different Mechanisms. Diabetologia 2015, 58, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Balboa, D.; Saarimäki-Vire, J.; Otonkoski, T. Concise Review: Human Pluripotent Stem Cells for the Modeling of Pancreatic β-Cell Pathology. Stem Cells 2019, 37, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Blanchi, B.; Taurand, M.; Colace, C.; Thomaidou, S.; Audeoud, C.; Fantuzzi, F.; Sawatani, T.; Gheibi, S.; Sabadell-Basallote, J.; Boot, F.W.J.; et al. EndoC-βH5 Cells Are Storable and Ready-to-Use Human Pancreatic Beta Cells with Physiological Insulin Secretion. Mol. Metab. 2023, 76, 101772. [Google Scholar] [CrossRef]
- Marasco, M.R.; Linnemann, A.K. β-Cell Autophagy in Diabetes Pathogenesis. Endocrinology 2018, 159, 2127–2141. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).