Abstract
Malignant melanoma (MM) is a malignant tumor, resulting from mutations in melanocytes of the skin and mucous membranes. Its mortality rate accounts for 90% of all dermatologic tumor mortality. Traditional treatments such as surgery, chemotherapy, and radiotherapy are unable to achieve the expected results due to MM’s low sensitivity, high drug resistance, and toxic side effects. As treatment advances, immunotherapy and targeted therapy have made significant breakthroughs in the treatment of MM and have demonstrated promising application prospects. However, the heterogeneity of tumor immune response causes more than half of patients to not benefit from clinical immunotherapy and targeted therapy, which delays the patient’s condition and causes them to suffer adverse immune events’ side effects. The combination of immunotherapy and targeted therapy can help improve therapeutic effects, delay drug resistance, and mitigate adverse effects. This review provides a comprehensive overview of the current development status and research progress of immune checkpoints, targeted genes, and their inhibitors, with a view to providing a reference for the clinical treatment of MM.
1. Introduction
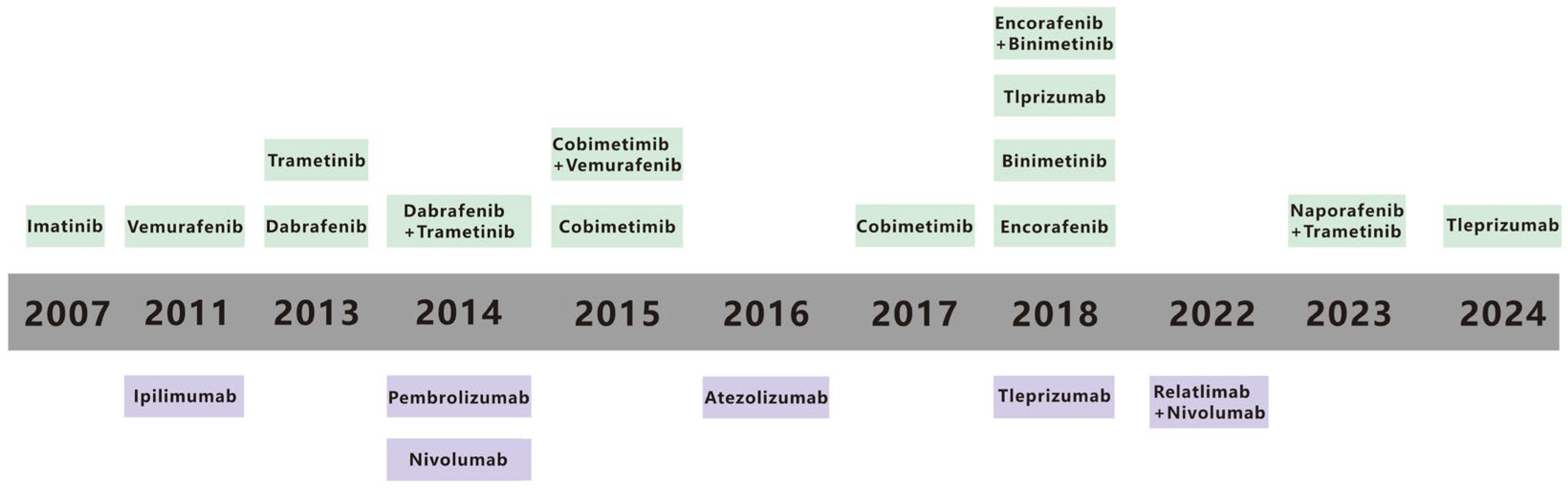
Malignant melanoma (MM) accounts for 90% of mortality from all cutaneous tumors [1]. MM is one of the most aggressive metastatic cancers and can spread from a relatively small primary tumor and metastasize to multiple sites, including the lungs [2], liver, brain [3], lymph node [4], and bones [5], and the 5-year survival rate for metastatic MM is only 10% [6]. The spread of MM cells to these organs often leads to the development of multiple organ failure and ultimately contributes to the high mortality rate [7]. An estimated 331,647 new cases and 58,645 deaths of MM occurred in 2022 [8]. Traditional treatments for MM, including radiation, chemotherapy, and surgery, are poorly targeted and result in poor patient prognosis [9,10]. Early primary MM can be treated with surgical resection, but metastatic MM has metastasized to other organs and cannot be surgically removed [7]. In contrast, recent advances in immunotherapy and targeted therapy offer new hope for enhancing treatment efficacy [11]. Immunotherapy and targeted therapy have emerged as important treatment modalities in recent years [12]. Immunotherapy, which aims to enhance the body’s immune response against cancer cells, and targeted therapy, which focuses on specific molecular targets within cancer cells, have shown certain positive effects [13,14]. Immunotherapy has been a major driving force in MM treatment for decades, despite historically poor clinical outcomes [9,10]. Targeted therapies, which attack specific gene mutations, pathways, or proteins associated with the development of MM, have also been pivotal [15]. Metastatic melanoma survival has also gotten better in recent years because of progress in targeted therapies and immunotherapies [16,17]. Still, long term survival is restricted. Recent data show that the 5-year survival rate for metastatic melanoma is about 22.5%, but this number changes based on the metastases’ location and the tumor burden [18,19]. Over the past few decades, a range of immunotherapy and targeted therapy drugs, along with combination approaches, have been approved by the US Food and Drug Administration (FDA) (Figure 1) [15,20]. Although these current therapies have improved the prognosis of patients with MM, challenges such as low efficacy rates and inevitable treatment resistance remain. This review paper summarizes the relevant literature to provide a reference for achieving precise treatment of MM.
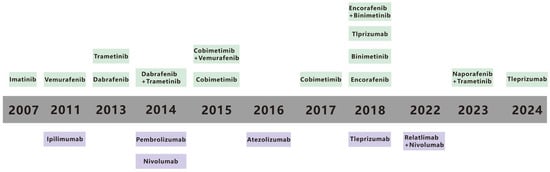
Figure 1.
Timeline for FDA-approved therapies for metastatic melanoma. The timeline highlights key advancements in the fields of immunotherapy and targeted therapy from 1990 to 2021. Therapies are categorized into immunotherapies (in purple) and targeted therapies (in green), reflecting the evolution of treatment strategies over time. Reproduced from ref [15].
2. Immunotherapy and Inhibitors
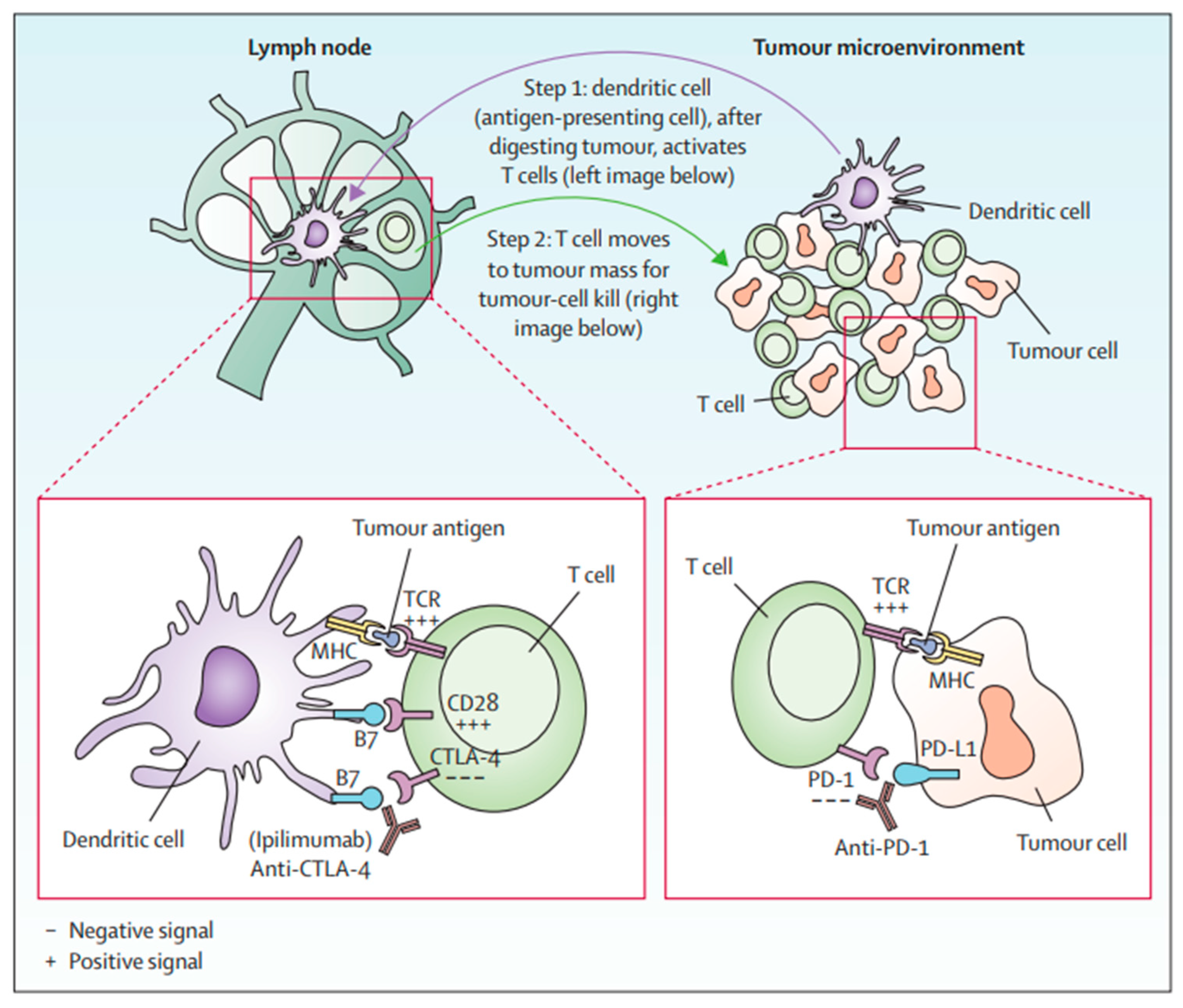
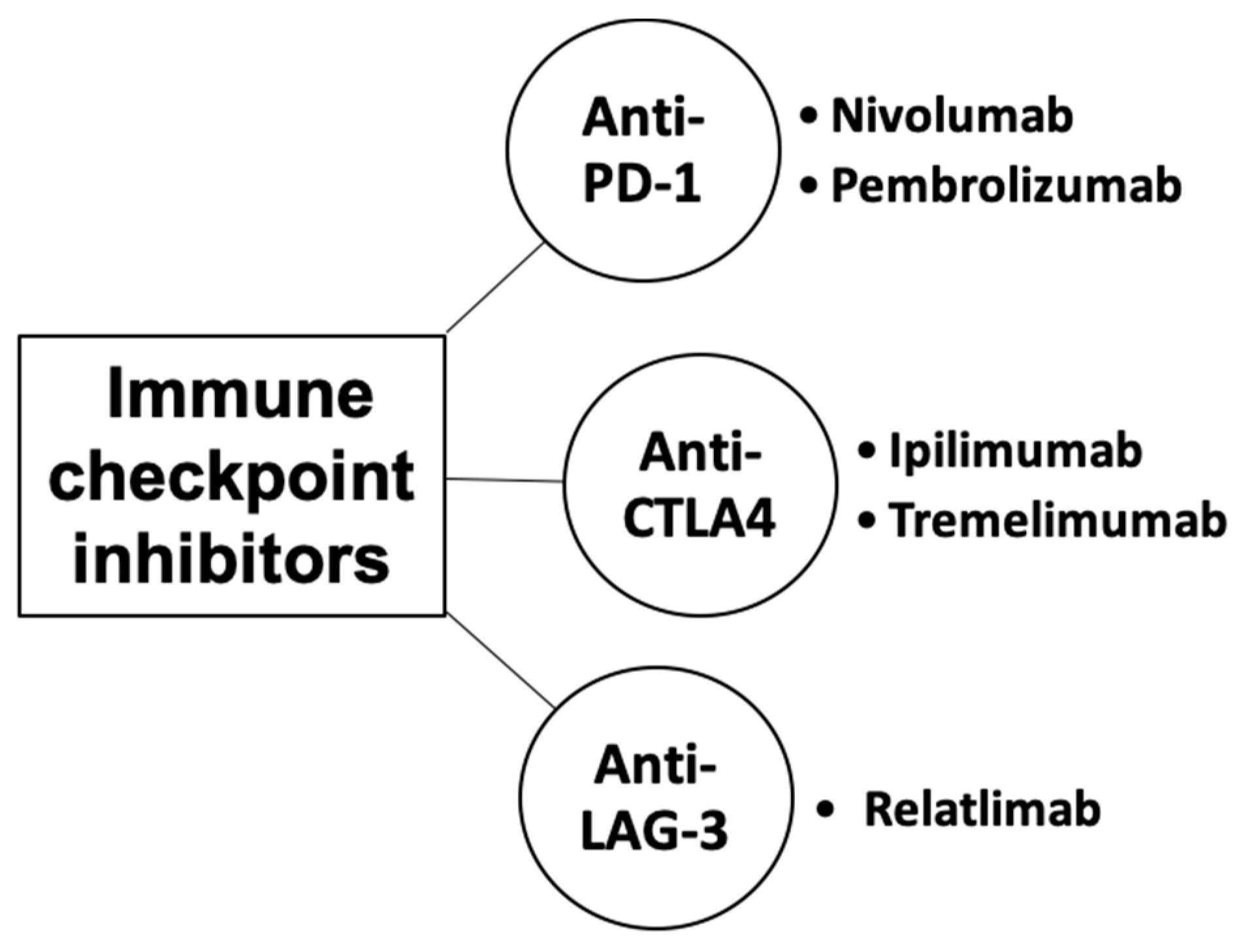
Immune checkpoints are cell-surface proteins expressed by immune cells, the function of which is to control the initiation, duration, and magnitude of immune responses, and particularly relevant to T-cell function (Figure 2) [12]. Immune checkpoint inhibitors (ICIs) are a type of immunotherapy that work by blocking proteins on immune cells or tumor cells that prevent the immune system from attacking cancer cells. These proteins are normally involved in regulating the immune response to prevent it from attacking healthy cells [21]. However, tumor cells can use these checkpoints to evade the immune system. MM cells exhibit substantial immunogenicity. The discovery of immune checkpoint proteins such as PD-1/PDL-1 and CTLA-4 was a major breakthrough in the field of cancer immunotherapy. Humanized monoclonal antibodies against these immune checkpoint proteins have been successfully used in patients with MM. ICIs improve therapeutic response by increasing the sensitivity of the immune system to tumor cells [22]. The efficacy of ICIs varies across melanoma subtypes [23,24]. Currently, ICIs, particularly those targeting PD-1, CTLA-4, and LAG-3, have demonstrated superior efficacy compared to conventional therapies in treating MM (Figure 3) [25,26,27].
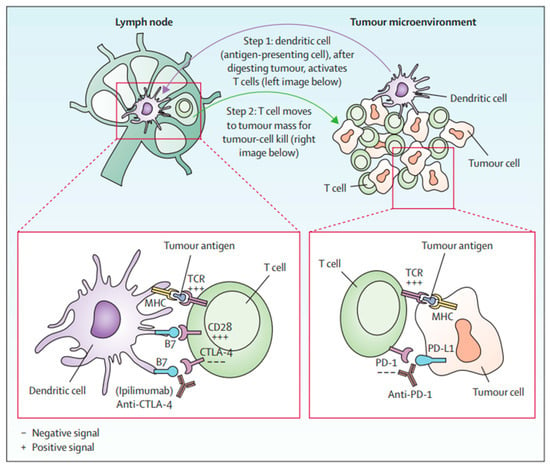
Figure 2.
T-cell activation by anti-CTLA-4 and anti-PD1 [12].
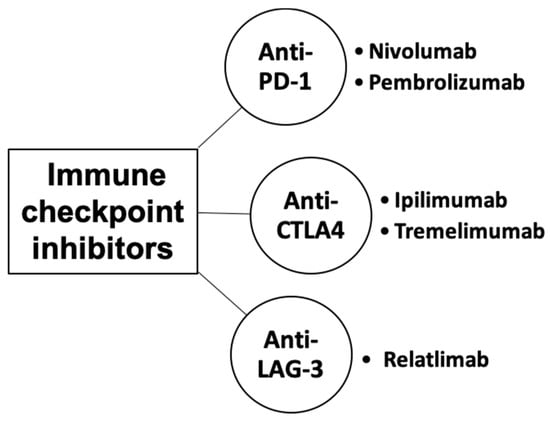
Figure 3.
Classes of immune checkpoint inhibitors for MM.
2.1. Programmed Cell Death Protein-1 (PD-1) Inhibitors
PD-1 is a member of the immunoglobulin superfamily and is primarily expressed on immune cells such as macrophages, dendritic cells, natural killer cells, T cells, and B cells [28]. PD-1 plays a critical role in programmed death signaling and regulates T cell-mediated responses. When PD-1 bind to its ligands, it disrupts the downstream signaling pathways essential for T-cell activation and inhibits transcription, thereby suppressing T-cell immune responses [29,30]. Currently, the primary anti-PD-1 monoclonal antibodies available are nivolumab and pembrolizumab. These antibodies specifically target the interactions between PD-1 and its ligands PD-L1 and PD-L2, setting the stage for a detailed discussion of their roles in the treatment of malignant melanoma [31,32].
Nivolumab, a humanized immunoglobulin monoclonal antibody, has been approved by the FDA as a single agent for patients with BRAF V600 wild-type unresectable or metastatic MM [33]. Clinical trials have demonstrated that in patients with stage IIIB/C or stage IV melanoma, those who received adjuvant treatment with nivolumab and the anti-CTLA-4 antibody Ipilimumab, after surgical resection of tumors, experienced significantly higher 12-month progression-free survival (RFS) compared to those treated only with ipilimumab. Additionally, the incidence of grade 3–4 side effects was significantly lower in the Nivolumab-treated group [34]. Because Nivolumab has shown significant effects in the treatment of patients with MM, the FDA has approved it for use in the adjuvant treatment of patients with advanced lymph node dissection or metastatic MM.
Pembrolizumab, an IgG4-kappa monoclonal antibody, is the first anti-programmed-death-1 (PD-1) drug licensed by the FDA for the treatment of advanced MM [35]. Pembrolizumab has demonstrated durable antitumor activity and tolerability in the treatment of patients with advanced MM [36]. Pembrolizumab also prolongs PFS and overall survival (OS) in patients with advanced MM, with lower toxicity than Ipilimumab [37]. In addition, clinical trial results showed that Pembrolizumab significantly reduced the risk of disease recurrence or death as an adjuvant treatment for stage IIB or IIC MM [38].
2.2. Cytotoxic T-Lym-Phocyte Antigen 4 (CTLA-4) Inhibitors
CTLA-4, a member of the immunoglobulin-associated receptor family, is expressed solely on T cells and governs the amplitude of T-cell activation throughout the early phases. CTLA-4 primarily inhibits the function of CD28 [39,40]. CTLA-4 inhibits T-cell activation and enhances the immunosuppressive activity of T regulatory cells, acting as a negative regulator in the immune process [41]. Currently, anti-CTLA-4 antibodies mainly include Ipilimumab and Tremelimumab.
Ipilimumab is the first FDA-approved specific antibody that can inhibit the function of CTLA-4, and its emergence provides new possibilities for immune checkpoint blockade (ICB) therapy [42,43]. Compared to interferon, treatment with Ipilimumab significantly prolongs OS in postoperative stage III MM patients with a high risk of cutaneous recurrence [44]. Ipilimumab promotes T-cell activation and proliferation, and it can kill MM cells by activating the immune system. However, it also can cause other immune-related side effects. It has been noted that the proportion of adverse events resulting in treatment discontinuation after treatment with high doses of Ipilimumab is about 53%, and most of them are grade 3–4 adverse events [45]. Since CTLA-4 is involved in autoimmune prevention as a negative regulator of T-cell immune responses, blockade of it by ibritumomab may lead to immune-related adverse effects (irAEs), such as colitis and enterocolitis [46]. Further studies are needed to investigate the therapeutic limitations and resistance mechanisms of Ipilimumab-mediated immunotherapy.
Tremelimumab was the first anti-CTLA-4 antibody to be investigated. In the results of a clinical trial, it was noted that treatment with Tremelimumab significantly improved the duration of response to the drug in patients with inoperable stage III/IV MM compared to the use of standard chemotherapeutic agents (Temozolomide or Dacarbazine). Due to the similar objective response rate of patients in the Tremelimumab-treatment group and the final death of 340 patients, the trial was stopped, and the effectiveness of Tremelimumab in the treatment of MM was not successfully confirmed. However, the duration of response was longer than that in the chemotherapy group, which also had certain clinical significance [47].
2.3. Lymphocyte Activation Gene-3 (LAG-3) Inhibitors
LAG-3 is a type I transmembrane protein, highly structurally homologous to CD4, and is mainly expressed on CD4+ T cells, CD8+ T cells, natural killer cells, and Treg cells [48]. Previous studies have shown that blocking LAG3 increased the proliferation of CD4+ and CD8+ T cells [46]. Anti-PD-1 antibodies could only activate T cells but not inhibit regulatory T-cell activity, whereas anti-LAG-3 antibodies not only restored T-cell function but also inhibited regulatory T-cell activity. Therefore, in clinical development, anti-LAG-3 antibodies are often used in combination with anti-PD-1/PD-L1 antibodies. The combination of the LAG-3 monoclonal antibody Relatlimab and the anti-PD-1 antibody Nivolumab is effective in prolonging PFS and progression-free survival rate in patients with unresectable or metastatic MM [49]. As a result, Relatlimab, a LAG3-blocking antibody, combined with Nivolumab for the treatment of unresectable or metastatic MM, was approved by the FDA in 2022 [50].
Although only one of the anti-LAG-3 antibodies has been approved, several anti-LAG-3 antibodies have entered clinical studies, such as [51], BI 754111 [52], and Ieramilimab [53], and the combination of IMP321 with Pembrolizumab (NCT02676869). Information on safety and efficacy of these antibodies is limited.
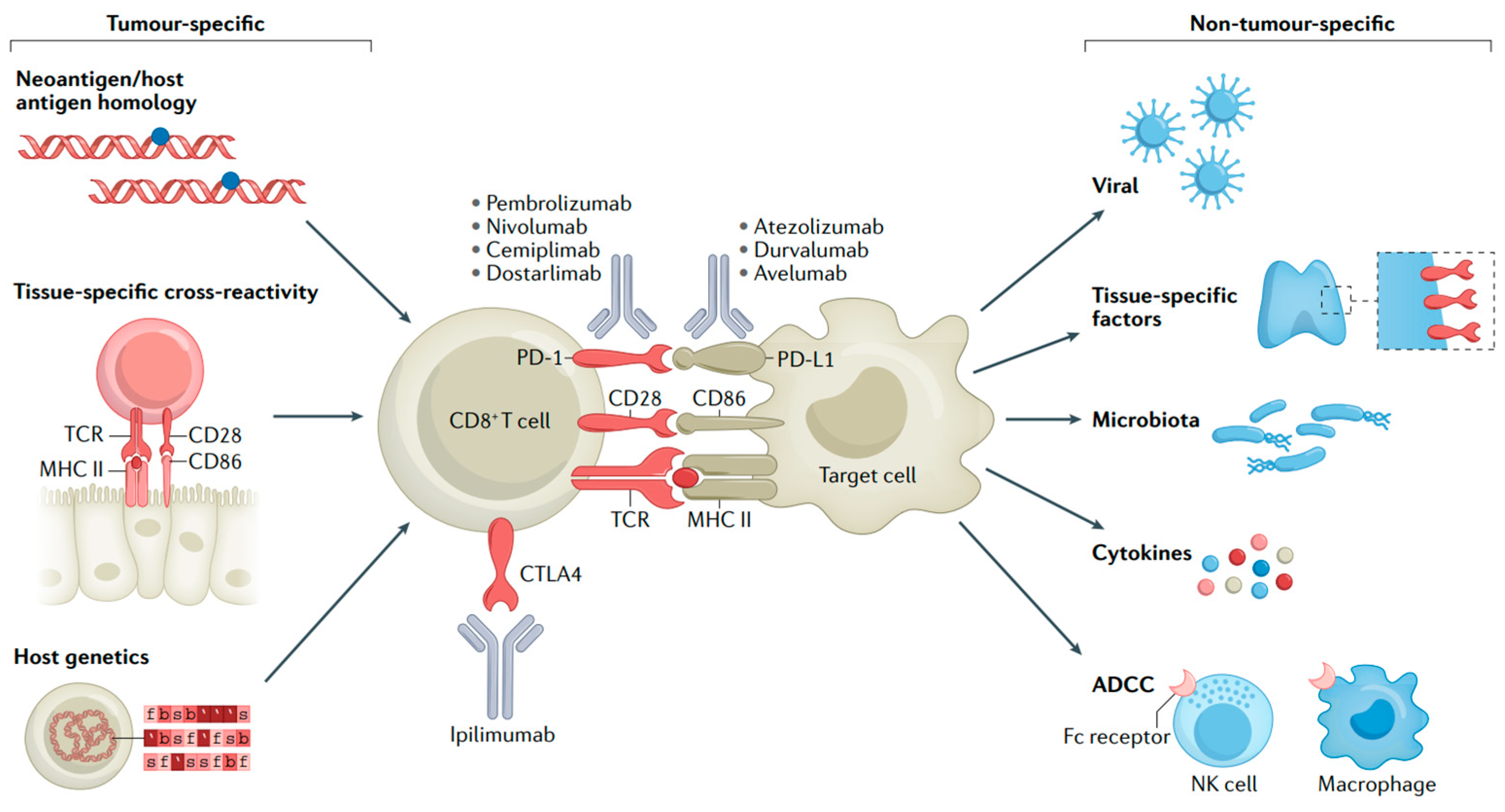
Despite immunotherapy having achieved remarkable success in MM treatment, it also has certain drawbacks. Firstly, ICIs may lead the immune system to attack other organs in the body, resulting in irAEs like pneumonia, hepatitis, and endocrine diseases [54,55]. The immune activation that underlies most irAEs might be coupled with the activity required for antitumor immune responses (Figure 4) [56]. Secondly, there is the issue of drug resistance. Some patients may become resistant to ICIs, which causes a decline in treatment effectiveness [57,58]. Furthermore, the high cost of these inhibitors may limit their use among some patients. Future studies need to further explore ways to enhance the efficacy of these drugs, reduce adverse effects, and cut down the treatment cost.
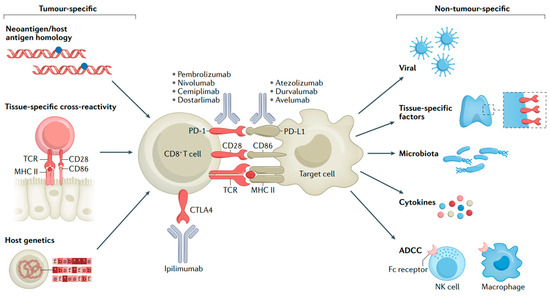
Figure 4.
Mechanisms of irAEs. T-cell interactions with malignant or non-malignant cells and molecular mechanisms of immune checkpoint blockade [56].
3. Targeted Therapy and Inhibitors
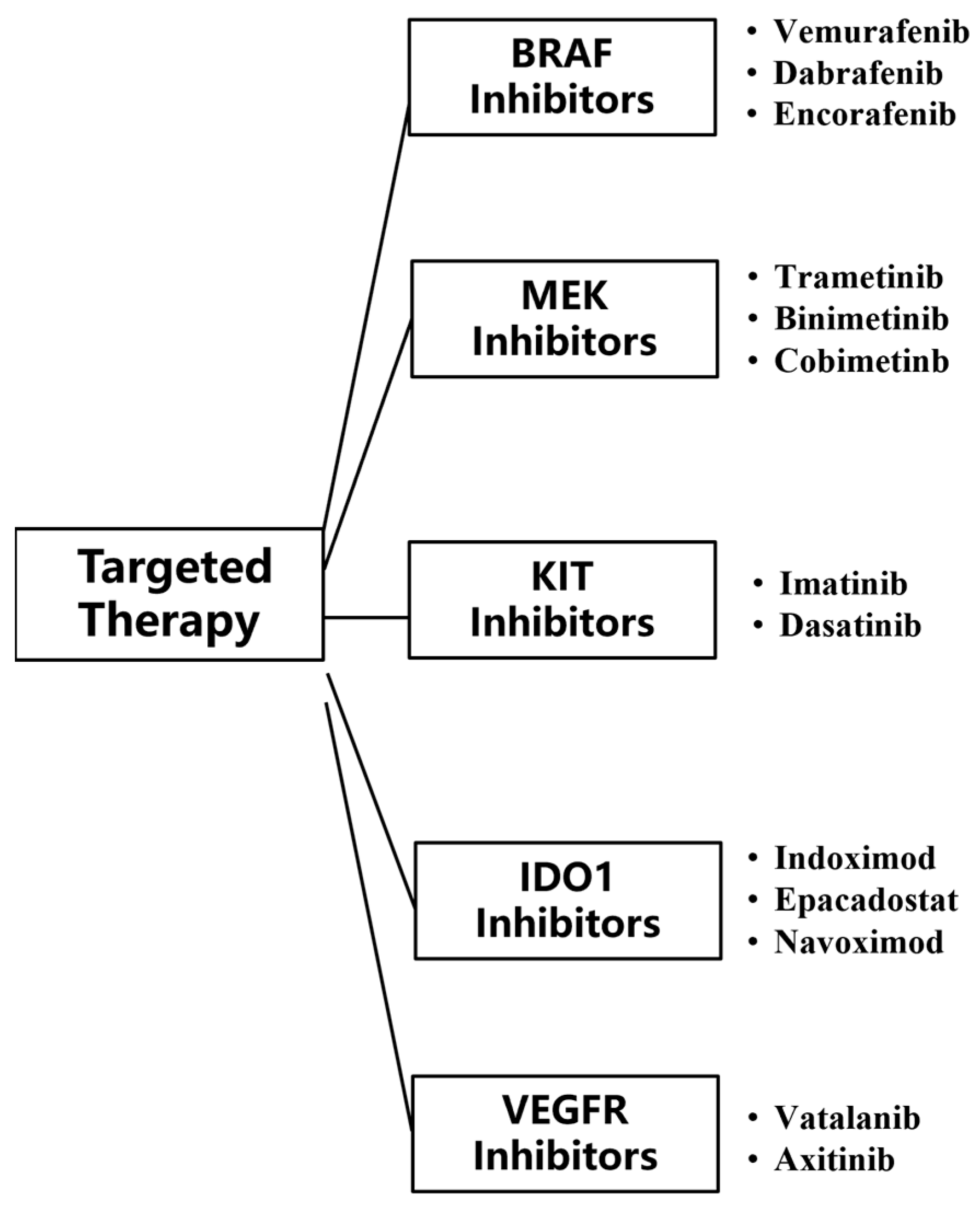
Targeted therapies are drugs that target specific genetic mutations, pathways, or proteins associated with cancer development. Compared to conventional chemotherapy, the inhibitors used in targeted therapy can pinpoint mutated tumor genes, and these inhibitors do not harm the normal tissue cells surrounding the tumor, making targeted therapy more accurate and effective compared with other treatment [59]. Currently, targeted therapy for MM mainly uses two types of monoclonal antibodies, BRAF and MEK. BRAF inhibitors can inhibit activated BRAF-mutated kinase, and MEK inhibitors can inhibit the activation of MEK (Figure 5).
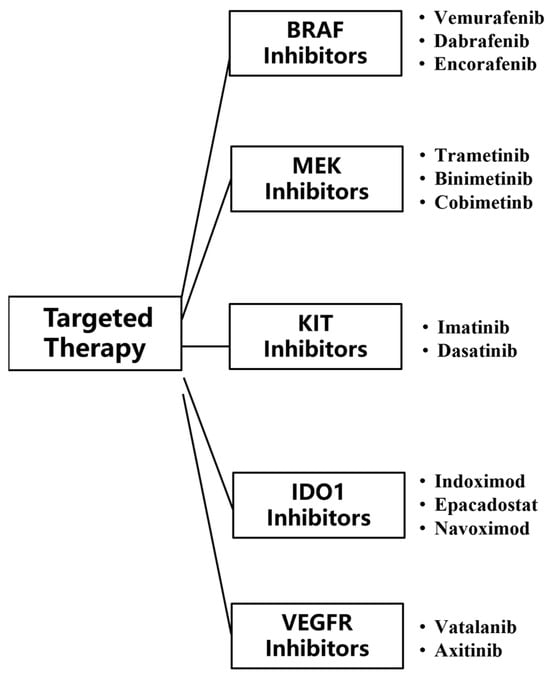
Figure 5.
Classes of targeted therapy checkpoint inhibitors for MM.
3.1. V-Raf Murine Sarcoma Viral Oncogene Homolog B1 (BRAF) Inhibitors
Activating mutations in BRAF occur in about 50% of skin melanomas [45]. The BRAF protein consists of three conserved structural domains. When the Valine at position 600 of the BRAF protein is replaced by a Glutamate, a BRAF V600E mutation occurs, leading to a significant increase in BRAF kinase activity, which activates the MAPK signaling pathway and thus transforms melanocytes into MM cells [60,61]. BRAF inhibitors are the first targeted therapeutic agents for patients with MM [48], and currently the most representative BRAF inhibitors are Vemurafenib [62], Dabrafenib [63], and Encorafenib [64].
Vemurafenib reduces the tumor size in more than half of advanced BRAF V600E mutation MM patients, while also keeping patients stable [63]. The results confirmed that the Vemurafenib-treatment group significantly prolonged patients’ PFS [65]. Moreover, compared to the chemotherapy group, BRAF V600E mutant metastatic MM patients with long-term survival in the Vemurafenib-treatment group did not frequently experience adverse skin events [66]. Although Vemurafenib has shown significant effects in the pre-treatment of patients with BRAF V600E mutation, patients are prone to develop resistance to it, such that 80% of MM patients treated for 3 years have shown resistance to Vemurafenib [67]. To better address this issue, recent findings suggest that the sensitivity of BRAF V600E mutant MM patients to Vemurafenib-treatment can be improved by targeting Glutathione Peroxidase 4 (GPX4) and ferroptosis through the plant sesquiterpene lactones DET and DETD-3 [68].
Compared with the Dacarbazine-chemotherapy group, the Dabrafenib-treatment group not only improved the PFS of patients with BRAF V600E mutant metastatic MM but also increased the partial emission rate (PR) and complete remission (CR) rates. Therefore, in 2013, the FDA approved Dabrafenib as a single agent in the treatment of MM patients with BRAF V600E mutation [69]. In addition, a trial validated that BRAF inhibitors combined with MEK inhibitors could improve the OS of metastatic MM patients with BRAF mutations [70]; so, a year later, the FDA approved Dabrafenib in combination with the MEK inhibitor Trametinib for MM treatment. A recent clinical trial again validated that Dabrafenib combined with Trametinib treatment significantly improved PFS and PR in advanced MM patients [71].
Encorafenib has also been approved by the FDA for the treatment of MM because of its ability to bind to mutant BRAF receptors and its long dissociation half-life [72]. The results of a clinical trial conducted in 2018 showed that PFS was increased approximately twofold in MM patients treated with the combination of Encorafenib and the MEK inhibitor Binimetinib [73]. The latest clinical trial demonstrated yet another improvement in OS in patients with BRAF V600E mutant MM treated with Encorafenib in combination with Binimetinib [74].
3.2. Mitogen-Activated Protein (MEK) Inhibitors
MEK is a mitogen-activated protease downstream of BRAF [75], and MEK inhibitors reduce the activity of the MAPK signaling pathway by blocking the expression of MEK genes located downstream of BRAF genes and RAS genes, thereby inhibiting tumor cell proliferation [76]. Trametinib, Binimetinib, and Cobimetinib are the main MEK inhibitors approved by the FDA for combination therapy or single therapy of metastatic MM.
Trametinib is orally effective, has a long half-life, and is well tolerated. The results of a phase III clinical trial showed that Trametinib improved PFS and OS in advanced MM patients compared to treatment with Dacarbazine or Paclitaxel alone [77]. However, MEK inhibitors have greater side effects when treating MM alone. In order to reduce the side effects and improve the therapeutic efficacy of MEK inhibitors during treatment, combining MEK inhibitors remains the focus of current research. For example, the BRAF inhibitor Vemurafenib combined with the MEK inhibitor Cobimetinib significantly improved PFS and PR in locally advanced or metastatic MM patients with BRAF V600E mutation [78].
3.3. V-Kit Hardy-Zuckerman 4 Feline Sarcoma Viral Oncogene Homolog (KIT) Inhibitors
KIT is recognized as one of the rational therapeutic targets. The KIT gene is an oncogene that encodes a transmembrane glycoprotein belonging to the tyrosine kinase (PTK) receptor family [79]. KIT mutations have been identified in more than 35% of acral and mucosal melanomas [80]. The KIT mutation lead to spontaneous ligand-independent receptor dimerization of KIT protein, stimulating excessive cell proliferation and anti-apoptotic signaling, thus playing an important role in MM growth [81,82]. In a genetic analysis of MM patients conducted in 2011, it was shown that 17% of MM patients in China develop the disease due to C-Kit mutation. Therefore, targeted therapies for C-Kit mutation play a key role in the treatment of Chinese MM patients [83]. The National Comprehensive Cancer Network (NCCN) treatment guidelines approved Imatinib as a guideline drug for the treatment of MM caused by KIT mutation in 2013 through a study of its therapeutic effects and side effects [84].
A phase II clinical trial conducted in China reported for the first time that Imatinib was effective in prolonging the PFS of advanced metastatic MM patients caused by KIT mutations and in improving the rate of disease control and treatment efficiency [51]. In addition, in some phase II clinical trials of mucosal MM caused by KIT mutations, the CR in the Dasatinib-treatment group, which is also a Kit inhibitor, reached only 18%, a significant decrease compared to Imatinib-treatment [52]. Therefore, even though KIT inhibitors are subject to more side effects in practice, Imatinib is currently still the first choice for targeted treatment of metastatic mucosal MM [66].
3.4. Indoleamine 2,3-Dioxygenase (IDO1) Inhibitors
IDO1 is the catalytic rate-limiting enzyme of the first step of the major metabolic pathway of L-tryptophan (L-Trp) [85], and IDO1 overexpression can lead to L-Trp depletion in the local microenvironment and subsequent impaired T-cell function [86,87]. In recent years, several new structural types of IDO1 inhibitors have been discovered, and a few have entered the clinical stage, such as Indoximod [88], Epacadostat [89] and Navoximod [90], but no successful drugs have been marketed yet.
Tryptophan-derived inhibitors were among the first reported IDO1 inhibitors [91], the most representative of which is Indoximod. The results of a clinical trial showed that the combination of Indoximod and the anti-PD-1 antibody Pembrolizumab in advanced MM patients had a PR of 61%, a median PFS of 12.9 months, and a 1-year survival rate of 56%, suggesting that Indoximod and Pembrolizumab have a significant synergistic anti-tumor effect [92].
Epacadostat is a competitive substrate for the L-Trp, which is an IDO1 substrate [93]. Epacadostat can interfere with the abnormal metabolism of Trp in tumor cells by competing for binding to the enzymatically active catalytic structural domain of IDO1 [94], thereby inhibiting tumor growth. Data from clinical trials in MM showed that Epacadostat in combination with the anti-CTLA-4 antibody Ipilimumab not only reduced tumor size but also improved disease control rates and PFS in patients [95]. Meanwhile, the combination of the anti-PD-1 antibody Pembrolizumab and Epacadostat was tried in a randomized phase III study. Despite having better clinical outcomes in early trials, Epacadostat failed to prevent IDO1-dependent MM immune escape and did not increase PFS and OS when treated with Pembrolizumab monotherapy [96].
Navoximod is a small molecule inhibitor of IDO1 under investigation [97]. In vivo studies have shown that oral administration of Navoximod reduces Kynurenine concentrations in plasma and tissues by approximately 50% and causes effector T cells to show dose-dependent activation and proliferation, thereby reducing the size of MM [98]. Phase I clinical trials of Navoximod alone and in combination with the anti-PD-L1 antibody Atezolizumab for the treatment of solid tumors have been completed, but no published data are available at this time [99].
BMS-986205 is a novel IDO1 inhibitor developed by Bristol-Myers Squibb [100]. Preclinical trials have demonstrated dose-dependent efficacy of BMS-986205 [101]. Though BMS-986205 successfully inhibited IDO1 and reduced human canine urinary quinolinic acid serum levels even at low concentrations, it had better efficacy and pharmacokinetics compared to Epacadostat [102].
3.5. VEGFR (Vascular Endothelial Growth Factor Receptor) Inhibitors
VEGF is involved in regulating processes such as angiogenesis development and embryopoiesis, and it has been found that the production of VEGFR-1 and laminin is required for tumor growth. VEGFR and its family members are often at abnormal levels in malignant tumors [103,104]. A growing number of preclinical and clinical studies have demonstrated the role of VEGF signaling in melanoma progression, treatment response, and OS [105,106]. The results of clinical trials showed that the novel VEGFR inhibitor Vatalanib was able to delay disease progression in a subset of patients, although it did not significantly improve OS of patients [107]. Another clinical trial showed that Axitinib, a second-generation inhibitor of VEGFR, was effective in inhibiting the progression of stage II metastatic MM and was well tolerated by patients [108]. Another trial demonstrated that Axitinib in combination with Paclitaxel and Carboplatin was effective in prolonging OS in MM patients [109].
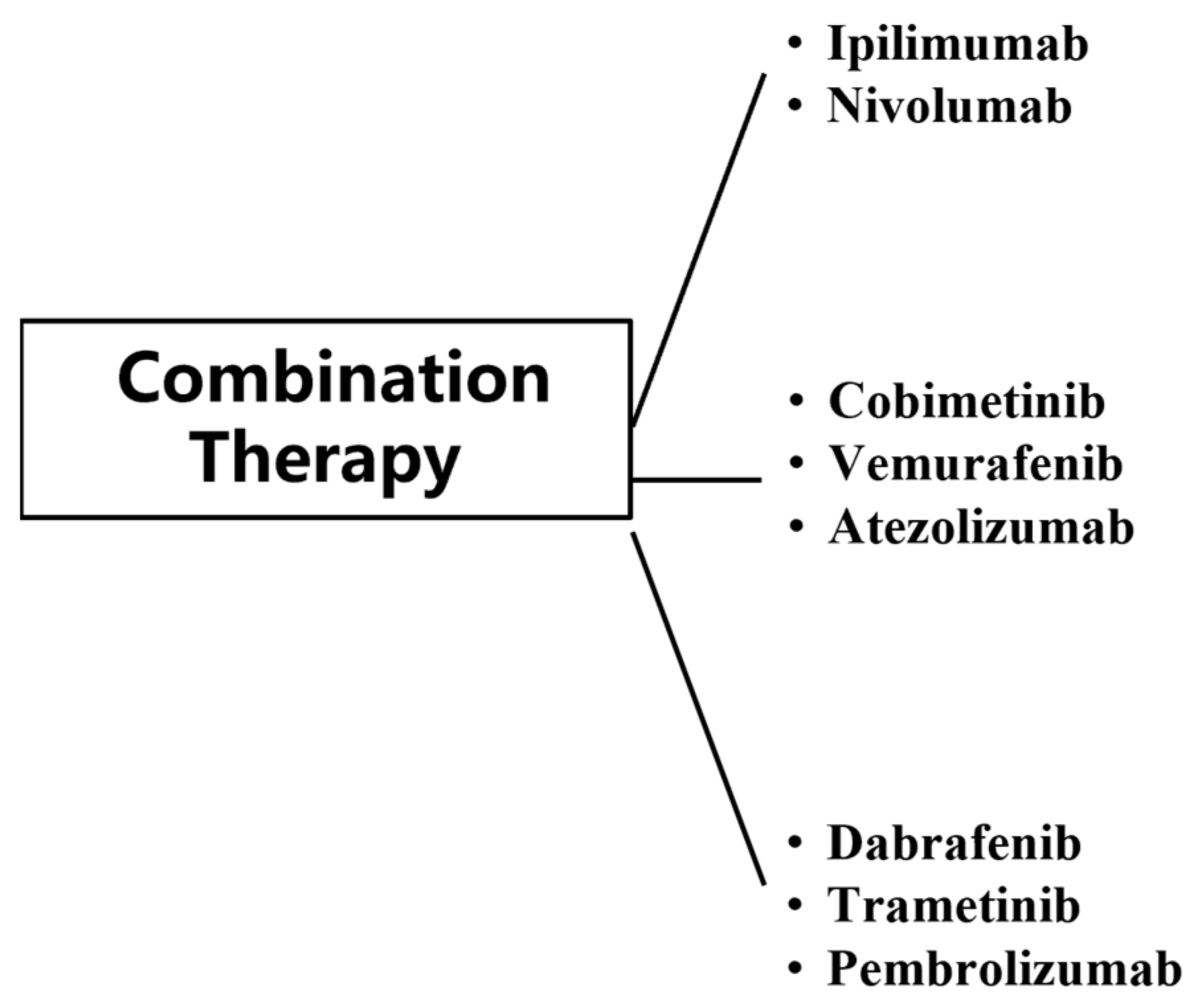
4. Combination Therapy
Combination therapy has substantially improved clinical outcomes in patients with metastatic MM, with approximately 50% of patients responding to treatment [110]. The use of immunotherapy and targeted therapy has broken the limitations of conventional MM treatment, but both immunotherapy and targeted therapy have shown resistance and toxicity in the treatment of MM. In order to better address this problem, trials on their combination regimens have been conducted at this stage (Figure 6).
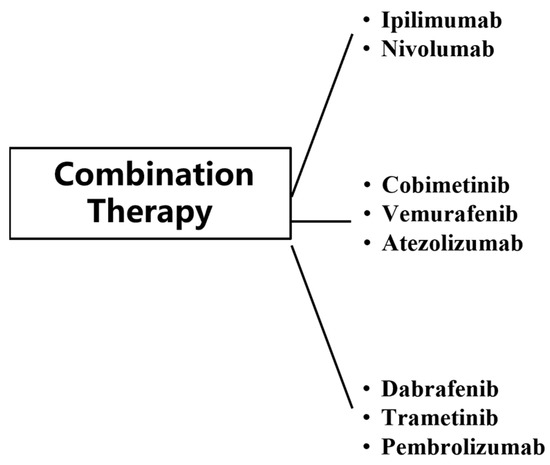
Figure 6.
Classes of combination therapy for MM.
The combination of Ipilimumab and Nivolumab is widely recognized as the most effective first-line treatment for patients with advanced MM [101]. The combination of the MEK inhibitor Cobimetinib and BRAF inhibitor Vemurafenib with anti-PD-1 antibody Atezolizumab showed better PFS in patients with metastatic MM than those treated with Cobimetinib and Vemurafenib [111]. The safety shown by the three-drug combination group during treatment is consistent with the known safety of each drug. As a result, Cobimetinib + Vemurafenib + Atezolizumab became the first three-drug combination group approved by the FDA for patients with BRAF mutation, unresectable or metastatic MM on 30 July 2020. In addition, clinical trial results for the three-drug combination of Dabrafenib + Trametinib + Pembrolizumab showed that the three-drug combination group improved PR and CR in MM patients with resectable stage III BRAF-mutant [112].
Only one three-drug combination group has been approved by the FDA, but several three-drug combination group regimens have entered clinical trials. For example, the stage II clinical trial for the anti-PD-1 antibody Camrelizumab in combination with the VEGFR inhibitor Apatinib and the chemotherapeutic agent Temozolomide in patients with advanced limb MM noted that the 66.7% objective response rate demonstrated by this combination regimen over the course of treatment exceeds that of any other drug combination explored in the literature. With a significant improvement in PFS, the regimen is expected to meet the urgent international need for treatment options for limb-end MM [113].
Trials for the tryptophan inhibitor ADI PEG 20 + PD-1 monoclonal antibody Nivolumab + CTLA-4 monoclonal antibody Ipilimumab in patients at high risk of metastatic uveal MM and for the PD-1 monoclonal antibody Spartalizumab + BRAF inhibitors Dabrafenib and Trametinib in patients with unresectable or metastatic BRAF V600 mutation-positive cutaneous MM have also concluded [114,115]. Although the study did not meet the primary endpoint of PFS, it did provide additional reference data for the development of combination regimens for the treatment of MM. The results of these trials suggest that three-drug combination therapy may be the newest approach for the treatment of metastatic MM, but longer follow-up is needed because the results are still immature [111].
5. Conclusions
MM is characterized by an insidious onset, aggressiveness, and poor prognosis. In recent years, immunotherapy and targeted therapy have emerged as important treatment modalities. They can indeed improve the prognosis of patients with advanced MM to some extent. In patients with primary MM, these therapies also contribute to improved survival. However, when it comes to some advanced patients with metastases, the situation is far from satisfactory. One of the main problems is that patients may suffer from varying degrees of drug resistance. In addition, irAEs are an issue. Combination therapy, which combines different treatments, such as immunotherapy and targeted therapy or other traditional therapies, has made great advances. Combination therapy has the potential to overcome some of the limitations of single-agent therapy. The effectiveness of the treatment, in terms of whether it can truly and consistently deliver better results in a larger patient population, still needs further research. Similarly, the safety of combination therapy needs to be more fully assessed through clinical validation. Future studies will focus on new immunotherapy targets and the combined use of multiple treatments, which may lead to more personalized and effective MM treatment, better clinical trial options, and improved overall survival and quality of life.
Author Contributions
Conceptualization, X.W.; writing—original draft preparation, X.W. and W.G.; writing—review and editing, S.M., S.Z., L.Z. and W.G. funding acquisition, W.G. and L.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by The National Natural Science Foundation of China (Grant No. 32070395 and 82103055) and The Key Science and Technology Research Project of Henan Province of China (Grant No. 242102311166).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Chinese guidelines for diagnosis and treatment of melanoma 2018 (English version). Chin. J. Cancer Res. 2019, 31, 578–585. [CrossRef]
- Sabit, H.; Kaliyadan, F.; Menezes, R.G. Malignant melanoma: Underlying epigenetic mechanisms. Indian J. Dermatol. Venereol. Leprol. 2020, 86, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Seervi, M.K.; Jain, S.; Meena, U.S.; Purohit, D.K. Metastatic Malignant Melanoma of Brain: A Rare Case Report. Asian J. Neurosurg. 2024, 19, 777–781. [Google Scholar] [CrossRef]
- Huang, J.; Gao, Z.; Xuan, J.; Gao, N.; Wei, C.; Gu, J. Metabolic insights into tumor lymph node metastasis in melanoma. Cell. Oncol. 2024, 47, 2099–2112. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.R.; de Groot, T.M.; Twining, P.K.; Kobes, T.; Ferrone, M.; Raskin, K.; Jutte, P.C.; Cohen, S.; Lozano-Calderon, S.; Groot, O.Q. Factors associated with skeletal-related events in patients with bone metastatic melanoma: A retrospective study of 481 patients. J. Surg. Oncol. 2024, 130, 310–321. [Google Scholar] [CrossRef]
- Orecchini, E.; Belladonna, M.L.; Pallotta, M.T.; Volpi, C.; Zizi, L.; Panfili, E.; Gargaro, M.; Fallarino, F.; Rossini, S.; Suvieri, C.; et al. The signaling function of IDO1 incites the malignant progression of mouse B16 melanoma. Oncoimmunology 2023, 12, 2170095. [Google Scholar] [CrossRef] [PubMed]
- Kudchadkar, R.R.; Lowe, M.C.; Khan, M.K.; McBrien, S.M. Metastatic melanoma. CA Cancer J. Clin. 2020, 70, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Franken, M.G.; Leeneman, B.; Gheorghe, M.; Uyl-de Groot, C.A.; Haanen, J.; van Baal, P.H.M. A systematic literature review and network meta-analysis of effectiveness and safety outcomes in advanced melanoma. Eur. J. Cancer 2019, 123, 58–71. [Google Scholar] [CrossRef]
- Tran, K.B.; Buchanan, C.M.; Shepherd, P.R. Evolution of Molecular Targets in Melanoma Treatment. Curr. Pharm. Des. 2020, 26, 396–414. [Google Scholar] [CrossRef] [PubMed]
- Reschke, R.; Enk, A.H.; Hassel, J.C. Prognostic Biomarkers in Evolving Melanoma Immunotherapy. Am. J. Clin. Dermatol. 2024, Online ahead of print. [Google Scholar] [CrossRef]
- Carlino, M.S.; Larkin, J.; Long, G.V. Immune checkpoint inhibitors in melanoma. Lancet 2021, 398, 1002–1014. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yuan, S.; Zhang, Z.; Fu, S.; Liu, S.; Liu, J.; Ma, Q.; Xia, Z.; Gu, P.; Gao, S.; et al. Regulating tumor cells to awaken T cell antitumor function and enhance melanoma immunotherapy. Biomaterials 2024, 316, 123034. [Google Scholar] [CrossRef] [PubMed]
- Danelli, L. Personalized neoantigen therapy for melanoma immunotherapy. Nat. Cancer 2024, 5, 1783. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Wang, H.; Li, C. Signal pathways of melanoma and targeted therapy. Signal. Transduct. Target. Ther. 2021, 6, 424. [Google Scholar] [CrossRef] [PubMed]
- Pekarek, L.; Sanchez Cedra, A.; Jaudenes, Y.D.Y.; Ospino, L.R.; Iglesias Pedrejon, B.; Bernier, L.; Roberts Cervantes, E.D.; Sanchez Cendra, C.; Cassinello, J.; Trasobares, L.; et al. Paradigm of biomarkers in metastatic melanoma (Review). Oncol. Lett. 2025, 29, 78. [Google Scholar] [CrossRef]
- Chen, J.; Tarantino, G.; Severgnini, M.; Baginska, J.; Giobbie-Hurder, A.; Weirather, J.L.; Manos, M.; Russell, J.D.; Pfaff, K.L.; Rodig, S.J.; et al. Circulating cytokine associations with clinical outcomes in melanoma patients treated with combination nivolumab plus ipilimumab. Oncoimmunology 2025, 14, 2432723. [Google Scholar] [CrossRef]
- Reitmajer, M.; Leiter, U.; Nanz, L.; Amaral, T.; Flatz, L.; Garbe, C.; Forschner, A. Long-term survival of stage IV melanoma patients: Evaluation on 640 melanoma patients entering stage IV between 2014 and 2017. J. Cancer Res. Clin. Oncol. 2024, 150, 15. [Google Scholar] [CrossRef]
- Saginala, K.; Barsouk, A.; Aluru, J.S.; Rawla, P.; Barsouk, A. Epidemiology of Melanoma. Med. Sci. 2021, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Curran, M.A.; Montalvo, W.; Yagita, H.; Allison, J.P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 4275–4280. [Google Scholar] [CrossRef]
- Ralli, M.; Botticelli, A.; Visconti, I.C.; Angeletti, D.; Fiore, M.; Marchetti, P.; Lambiase, A.; de Vincentiis, M.; Greco, A. Immunotherapy in the Treatment of Metastatic Melanoma: Current Knowledge and Future Directions. J. Immunol. Res. 2020, 2020, 9235638. [Google Scholar] [CrossRef] [PubMed]
- Heppt, M.V.; Heinzerling, L.; Kahler, K.C.; Forschner, A.; Kirchberger, M.C.; Loquai, C.; Meissner, M.; Meier, F.; Terheyden, P.; Schell, B.; et al. Prognostic factors and outcomes in metastatic uveal melanoma treated with programmed cell death-1 or combined PD-1/cytotoxic T-lymphocyte antigen-4 inhibition. Eur. J. Cancer 2017, 82, 56–65. [Google Scholar] [CrossRef]
- Heppt, M.V.; Steeb, T.; Schlager, J.G.; Rosumeck, S.; Dressler, C.; Ruzicka, T.; Nast, A.; Berking, C. Immune checkpoint blockade for unresectable or metastatic uveal melanoma: A systematic review. Cancer Treat. Rev. 2017, 60, 44–52. [Google Scholar] [CrossRef]
- Shi, H.; Lan, J.; Yang, J. Mechanisms of Resistance to Checkpoint Blockade Therapy. Adv. Exp. Med. Biol. 2020, 1248, 83–117. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, K.M.; Freeman, G.J.; McDermott, D.F. The Next Immune-Checkpoint Inhibitors: PD-1/PD-L1 Blockade in Melanoma. Clin. Ther. 2015, 37, 764–782. [Google Scholar] [CrossRef] [PubMed]
- Kreidieh, F.Y.; Tawbi, H.A. The introduction of LAG-3 checkpoint blockade in melanoma: Immunotherapy landscape beyond PD-1 and CTLA-4 inhibition. Ther. Adv. Med. Oncol. 2023, 15, 17588359231186027. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar] [PubMed]
- Gutic, B.; Bozanovic, T.; Mandic, A.; Dugalic, S.; Todorovic, J.; Stanisavljevic, D.; Dugalic, M.G.; Sengul, D.; Detanac, D.A.; Sengul, I.; et al. Programmed cell death-1 and its ligands: Current knowledge and possibilities in immunotherapy. Clinics 2023, 78, 100177. [Google Scholar] [CrossRef] [PubMed]
- Iwai, Y.; Hamanishi, J.; Chamoto, K.; Honjo, T. Cancer immunotherapies targeting the PD-1 signaling pathway. J. Biomed. Sci. 2017, 24, 26. [Google Scholar] [CrossRef]
- Schachter, J.; Ribas, A.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab for advanced melanoma: Final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 2017, 390, 1853–1862. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Moser, J.C.; Chen, D.; Hu-Lieskovan, S.; Grossmann, K.F.; Patel, S.; Colonna, S.V.; Ying, J.; Hyngstrom, J.R. Real-world survival of patients with advanced BRAF V600 mutated melanoma treated with front-line BRAF/MEK inhibitors, anti-PD-1 antibodies, or nivolumab/ipilimumab. Cancer Med. 2019, 8, 7637–7643. [Google Scholar] [CrossRef]
- Weber, J.; Mandala, M.; Del Vecchio, M.; Gogas, H.J.; Arance, A.M.; Cowey, C.L.; Dalle, S.; Schenker, M.; Chiarion-Sileni, V.; Marquez-Rodas, I.; et al. Adjuvant Nivolumab versus Ipilimumab in Resected Stage III or IV Melanoma. N. Engl. J. Med. 2017, 377, 1824–1835. [Google Scholar] [CrossRef]
- McDermott, D.F.; Atkins, M.B. PD-1 as a potential target in cancer therapy. Cancer Med. 2013, 2, 662–673. [Google Scholar] [CrossRef]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.; Weber, J.S.; et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann. Oncol. 2019, 30, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Luke, J.J.; Rutkowski, P.; Queirolo, P.; Del Vecchio, M.; Mackiewicz, J.; Chiarion-Sileni, V.; de la Cruz Merino, L.; Khattak, M.A.; Schadendorf, D.; Long, G.V.; et al. Pembrolizumab versus placebo as adjuvant therapy in completely resected stage IIB or IIC melanoma (KEYNOTE-716): A randomised, double-blind, phase 3 trial. Lancet 2022, 399, 1718–1729. [Google Scholar] [CrossRef] [PubMed]
- Sansom, D.M. IMMUNOLOGY. Moving CTLA-4 from the trash to recycling. Science 2015, 349, 377–378. [Google Scholar] [CrossRef]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef]
- Azimnasab-Sorkhabi, P.; Soltani-Asl, M.; Kfoury Junior, J.R. Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) as an undetermined tool in tumor cells. Hum. Cell 2023, 36, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Lipson, E.J.; Drake, C.G. Ipilimumab: An anti-CTLA-4 antibody for metastatic melanoma. Clin. Cancer Res. 2011, 17, 6958–6962. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H. Melanoma drug wins US approval. Nature 2011, 471, 561. [Google Scholar] [CrossRef] [PubMed]
- Tarhini, A.A.; Lee, S.J.; Hodi, F.S.; Rao, U.N.M.; Cohen, G.I.; Hamid, O.; Hutchins, L.F.; Sosman, J.A.; Kluger, H.M.; Eroglu, Z.; et al. Phase III Study of Adjuvant Ipilimumab (3 or 10 mg/kg) Versus High-Dose Interferon Alfa-2b for Resected High-Risk Melanoma: North American Intergroup E1609. J. Clin. Oncol. 2020, 38, 567–575. [Google Scholar] [CrossRef]
- Coit, D.G.; Thompson, J.A.; Albertini, M.R.; Barker, C.; Carson, W.E.; Contreras, C.; Daniels, G.A.; DiMaio, D.; Fields, R.C.; Fleming, M.D.; et al. Cutaneous Melanoma, Version 2.2019, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2019, 17, 367–402. [Google Scholar] [CrossRef] [PubMed]
- Fecher, L.A.; Agarwala, S.S.; Hodi, F.S.; Weber, J.S. Ipilimumab and its toxicities: A multidisciplinary approach. Oncologist 2013, 18, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Kefford, R.; Marshall, M.A.; Punt, C.J.; Haanen, J.B.; Marmol, M.; Garbe, C.; Gogas, H.; Schachter, J.; Linette, G.; et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J. Clin. Oncol. 2013, 31, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Castillo Gutiérrez, E.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.L.; Reeves, D.J. Nivolumab/Relatlimab: A Novel Addition to Immune Checkpoint Inhibitor Therapy in Unresectable or Metastatic Melanoma. Ann. Pharmacother. 2023, 57, 738–745. [Google Scholar] [CrossRef] [PubMed]
- Catenacci, D.V.; Rosales, M.; Chung, H.C.; Yoon, H.H.; Shen, L.; Moehler, M.; Kang, Y.K. MAHOGANY: Margetuximab combination in HER2+ unresectable/metastatic gastric/gastroesophageal junction adenocarcinoma. Future Oncol. 2021, 17, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Zettl, M.; Wurm, M.; Schaaf, O.; Mostböck, S.; Tirapu, I.; Apfler, I.; Lorenz, I.C.; Frego, L.; Kenny, C.; Thibodeau, M.; et al. Combination of two novel blocking antibodies, anti-PD-1 antibody ezabenlimab (BI 754091) and anti-LAG-3 antibody BI 754111, leads to increased immune cell responses. Oncoimmunology 2022, 11, 2080328. [Google Scholar] [CrossRef] [PubMed]
- Schöffski, P.; Tan, D.S.W.; Martín, M.; Ochoa-de-Olza, M.; Sarantopoulos, J.; Carvajal, R.D.; Kyi, C.; Esaki, T.; Prawira, A.; Akerley, W.; et al. Phase I/II study of the LAG-3 inhibitor ieramilimab (LAG525) ± anti-PD-1 spartalizumab (PDR001) in patients with advanced malignancies. J. Immunother. Cancer 2022, 10, e003776. [Google Scholar] [CrossRef]
- Justice, J.; Kankaria, R.A.; Johnson, D.B. Immune checkpoint inhibition of metastatic melanoma: Achieving high efficacy in the face of high toxicity. Expert Rev. Clin. Pharmacol. 2024, 21, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Abu-Sbeih, H.; Wang, Y. Immune Checkpoint Inhibitors-Induced Hepatitis. Adv. Exp. Med. Biol. 2018, 995, 159–164. [Google Scholar] [CrossRef]
- Johnson, D.B.; Nebhan, C.A.; Moslehi, J.J.; Balko, J.M. Immune-checkpoint inhibitors: Long-term implications of toxicity. Nat. Rev. Clin. Oncol. 2022, 19, 254–267. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Hossain, S.M.; Eccles, M.R. Phenotype Switching and the Melanoma Microenvironment; Impact on Immunotherapy and Drug Resistance. Int. J. Mol. Sci. 2023, 24, 1601. [Google Scholar] [CrossRef]
- Syrigos, K.N.; Zalonis, A.; Kotteas, E.; Saif, M.W. Targeted therapy for oesophageal cancer: An overview. Cancer Metastasis Rev. 2008, 27, 273–288. [Google Scholar] [CrossRef] [PubMed]
- Bobos, M. Histopathologic classification and prognostic factors of melanoma: A 2021 update. Ital. J. Dermatol. Venerol. 2021, 156, 300–321. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Feng, Y.; Chen, D. Interfering Nuclear Protein Laminb1 Induces DNA Damage and Reduces Vemurafenib Resistance in Melanoma Cells In Vitro. Cancers 2024, 16, 4060. [Google Scholar] [CrossRef]
- Roesch, A.; Vultur, A.; Bogeski, I.; Wang, H.; Zimmermann, K.; Speicher, D.; Krbel, C.; Laschke, M.; Gimotty, P.; Philipp, S.J.C.C. Overcoming Intrinsic Multidrug Resistance in Melanoma by Blocking the Mitochondrial Respiratory Chain of Slow-Cycling JARID1B high Cells. Cancer Cell 2013, 23, 811–825. [Google Scholar] [CrossRef]
- Dudnichenko, O.; Penkov, K.; McKean, M.; Mandala, M.; Kukushkina, M.; Panella, T.; Csoszi, T.; Gerletti, P.; Thakur, M.; Polli, A.; et al. First-line encorafenib plus binimetinib and pembrolizumab for advanced BRAF V600-mutant melanoma: Safety lead-in results from the randomized phase III STARBOARD study. Eur. J. Cancer 2024, 213, 115070. [Google Scholar] [CrossRef] [PubMed]
- Mcarthur, G.A.; Chapman, P.B.; Robert, C.; Larkin, J.; Haanen, J.B.; Dummer, R.; Ribas, A.; Hogg, D.; Hamid, O.; Ascierto, P.A.J.L.O. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): Extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014, 15, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Mandel, V.D.; Medri, M.; Manganoni, A.M.; Pavoni, L.; De Rosa, F.; Ribero, S.; Foca, F.; Andreis, D.; Mazzoni, L.; Magi, S.; et al. Long-term vemurafenib therapy in advanced melanoma patients: Cutaneous toxicity and prognostic implications. J. Dermatol. Treat. 2022, 33, 1368–1375. [Google Scholar] [CrossRef]
- Long, G.V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandalà, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.J.N.E.J.o.M. Adjuvant Dabrafenib plus Trametinib in Stage III BRAF -Mutated Melanoma. eClinicalMedicine 2017, 377, 1813. [Google Scholar]
- Chang, M.T.; Tsai, L.C.; Nakagawa-Goto, K.; Lee, K.H.; Shyur, L.F. Phyto-sesquiterpene lactones DET and DETD-35 induce ferroptosis in vemurafenib sensitive and resistant melanoma via GPX4 inhibition and metabolic reprogramming. Pharmacol. Res. 2022, 178, 106148. [Google Scholar] [CrossRef] [PubMed]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H., Jr.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Sloot, S.; Zager, J.S.; Kudchadkar, R.R.; Messina, J.L.; Benedict, J.J.; Gonzalez, R.J.; Deconti, R.; Turner, L.M.; Mccardle, T.; Smalley, K.S.M.J.M.R. BRAF inhibition for advanced locoregional BRAF V600E mutant melanoma: A potential neoadjuvant strategy. Melanoma Res. 2016, 26, 83–87. [Google Scholar] [CrossRef]
- Si, L.; Zhang, X.; Shin, S.J.; Fan, Y.; Lin, C.C.; Kim, T.M.; Dechaphunkul, A.; Maneechavakajorn, J.; Wong, C.S.; Ilankumaran, P.; et al. Open-label, phase IIa study of dabrafenib plus trametinib in East Asian patients with advanced BRAF V600-mutant cutaneous melanoma. Eur. J. Cancer 2020, 135, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Koelblinger, P.; Thuerigen, O.; Dummer, R.J.C.O.i.O. Development of encorafenib for BRAF-mutated advanced melanoma. Curr. Opin. Oncol. 2018, 30, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.J.L.O. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF -mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Indini, A.; Mandalà, M. Safety and efficacy evaluation of encorafenib plus binimetinib for the treatment of advanced BRAF-mutant melanoma patients. Expert Opin. Drug Saf. 2020, 19, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Marvin, K.; Dana, W.; Rebekka, W.; Marc, S.; Stefan, B.; Christian, P.; Friedegund, M.J.P.R. Immunomodulatory effects of BRAF and MEK Inhibitors: Implications for Melanoma therapy. Pharmacol Res. 2018, 136, 151–159. [Google Scholar]
- Consoli, F.; Bersanelli, M.; Perego, G.; Grisanti, S.; Merelli, B.; Berruti, A.; Petrelli, F. Network indirect comparison of 3 BRAF + MEK inhibitors for the treatment of advanced BRAF mutated melanoma. Clin. Transl. Oncol. 2020, 22, 900–907. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; McArthur, G.A.; Dréno, B.; Larkin, J.; Liszkay, G.; Maio, M.; Mandala, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. coBRIM: A phase 3, double-blind, placebo-controlled study of vemurafenib versus vemurafenib + cobimetinib in previously untreated BRAF V600 mutation–positive patients with unresectable locally advanced or metastatic melanoma (NCT01689519). J. Transl. Med. 2015, 13 (Suppl. S1), O4. [Google Scholar] [CrossRef]
- Kitayama, H.; Tsujimura, T.; Matsumura, I.; Oritani, K.; Ikeda, H.; Ishikawa, J.; Okabe, M.; Suzuki, M.; Yamamura, K.; Matsuzawa, Y.; et al. Neoplastic transformation of normal hematopoietic cells by constitutively activating mutations of c-kit receptor tyrosine kinase. Blood 1996, 88, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.A.; Busam, K.; Pinkel, D.; Bastian, B.C. Somatic activation of KIT in distinct subtypes of melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 4340–4346. [Google Scholar] [CrossRef] [PubMed]
- Tímár, J.; Ladányi, A. Molecular Pathology of Skin Melanoma: Epidemiology, Differential Diagnostics, Prognosis and Therapy Prediction. Int. J. Mol. Sci. 2022, 23, 5384. [Google Scholar] [CrossRef] [PubMed]
- Pham, D.D.M.; Guhan, S.; Tsao, H. KIT and Melanoma: Biological Insights and Clinical Implications. Yonsei Med. J. 2020, 61, 562–571. [Google Scholar] [CrossRef]
- Kong, Y.; Si, L.; Zhu, Y.; Xu, X.; Corless, C.L.; Flaherty, K.T.; Li, L.; Li, H.; Sheng, X.; Cui, C.J.C.C.R. Large-Scale Analysis of KIT Aberrations in Chinese Patients with Melanoma. Clin. Cancer Res. 2011, 17, 1684–1691. [Google Scholar] [CrossRef] [PubMed]
- Coit, D.G.; Andtbacka, R.; Anker, C.J.; Bichakjian, C.K.; Carson, W.E.; Daud, A.; Dimaio, D.; Fleming, M.D.; Guild, V.; Halpern, A.C.; et al. Melanoma, version 2.2013: Featured updates to the NCCN guidelines. J. Natl. Compr. Canc. Netw. 2013, 11, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, X.; Wang, L.; Ma, X.D.; Gong, Z.J.; Zhang, S.S.; Li, Y. Targeting the IDO1 pathway in cancer: From bench to bedside. J. Hematol. Oncol. 2018, 11, 100. [Google Scholar] [CrossRef] [PubMed]
- Merlo, L.M.F.; Peng, W.D.; Mandik-Nayak, L. Impact of IDO1 and IDO2 on the B Cell Immune Response. Front. Immunol. 2022, 13, 886225. [Google Scholar] [CrossRef]
- Mortezaee, K.; Majidpoor, J. Alternative immune checkpoints in immunoregulatory profile of cancer stem cells. Heliyon 2023, 9, e23171. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C.; Malachowski, W.J.; Mondal, A.; Scherle, P.; Muller, A.J. Indoleamine 2,3-Dioxygenase and Its Therapeutic Inhibition in Cancer. Int. Rev. Cel. Mol. Bio. 2018, 336, 175–203. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Kato, S.; Nesline, M.K.; Conroy, J.M.; DePietro, P.; Pabla, S.; Kurzrock, R. Indoleamine 2,3-dioxygenase (IDO) inhibitors and cancer immunotherapy. Cancer Treat. Rev. 2022, 110, 102461. [Google Scholar] [CrossRef] [PubMed]
- Le Naour, J.; Galluzzi, L.; Zitvogel, L.; Kroemer, G.; Vacchelli, E. Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology 2020, 9, 1777625. [Google Scholar] [CrossRef]
- Fox, E.; Oliver, T.; Rowe, M.; Thomas, S.; Zakharia, Y.; Gilman, P.B.; Muller, A.J.; Prendergast, G.C. Indoximod: An Immunometabolic Adjuvant That Empowers T Cell Activity in Cancer. Front. Oncol. 2018, 8, 370. [Google Scholar] [CrossRef] [PubMed]
- Berrong, Z.; Mkrtichyan, M.; Ahmad, S.; Webb, M.; Mohamed, E.; Okoev, G.; Matevosyan, A.; Shrimali, R.; Abu Eid, R.; Hammond, S.; et al. Antigen-Specific Antitumor Responses Induced by OX40 Agonist Are Enhanced by the IDO Inhibitor Indoximod. Cancer Immunol. Res. 2018, 6, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, M.; Furgiuele, A.; Rasini, E.; Legnaro, M.; Ferrari, M.; Luini, A.; Rodrigues-Santos, P.; Caramelo, F.; Marino, F.; Pereira, F.C.; et al. A peripheral blood mononuclear cell-based in vitro model: A tool to explore indoleamine 2, 3-dioxygenase-1 (IDO1). Eur. J. Pharmacol. 2024, 968, 176420. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.F.; Nan, Y.Y.; Liu, C.; Lin, G.Y.; Gu, K.D.; Chen, C.; Zhao, W.L.; Ju, D.W.; Dong, X.C. Design, Synthesis and Biological Evaluation of Novel 1,2,5-Oxadiazol-3-Carboximidamide Derivatives as Indoleamine 2, 3-Dioxygenase 1 (IDO1) Inhibitors. Anticancer Agents Med. Chem. 2020, 20, 1592–1603. [Google Scholar] [CrossRef]
- Brochez, L.; Chevolet, I.; Kruse, V. The rationale of indoleamine 2,3-dioxygenase inhibition for cancer therapy. Eur. J. Cancer 2017, 76, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Dummer, R.; Humid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Chen, F.H.; Zhao, D.M.; Huang, Y.; Wen, X.; Feng, S.C. Synergetic impact of combined navoximod with cisplatin mitigates chemo-immune resistance via blockading IDO1+CAFs-secreted Kyn/AhR/ IL-6 and pol ζ-prevented CIN in human oral squamous cell carcinoma. Life Sci. 2023, 335, 122239. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.H.; Ueng, S.H.; Tseng, C.T.; Hung, M.S.; Song, J.S.; Wu, J.S.; Liao, F.Y.; Fan, Y.S.; Wu, M.H.; Hsiao, W.C.; et al. Important Hydrogen Bond Networks in Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor Design Revealed by Crystal Structures of Imidazoleisoindole Derivatives with IDO1. J. Med. Chem. 2016, 59, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Ebata, T.; Shimizu, T.; Fujiwara, Y.; Tamura, K.; Kondo, S.; Iwasa, S.; Yonemori, K.; Shimomura, A.; Kitano, S.; Koyama, T.; et al. Phase I study of the indoleamine 2,3-dioxygenase 1 inhibitor navoximod (GDC-0919) as monotherapy and in combination with the PD-L1 inhibitor atezolizumab in Japanese patients with advanced solid tumours. Invest. New Drugs 2020, 38, 468–477. [Google Scholar] [CrossRef]
- Cherney, E.C.; Zhang, L.; Nara, S.; Zhu, X.; Gullo-Brown, J.; Maley, D.; Lin, T.A.; Hunt, J.T.; Huang, C.; Yang, Z.; et al. Discovery and Preclinical Evaluation of BMS-986242, a Potent, Selective Inhibitor of Indoleamine-2,3-dioxygenase 1. ACS Med. Chem. Lett. 2021, 12, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Balog, A.; Lin, T.A.; Maley, D.; Gullo-Brown, J.; Kandoussi, E.H.; Zeng, J.; Hunt, J.T. Preclinical Characterization of Linrodostat Mesylate, a Novel, Potent, and Selective Oral Indoleamine 2,3-Dioxygenase 1 Inhibitor. Mol. Cancer Ther. 2021, 20, 467–476. [Google Scholar] [CrossRef]
- Ye, Z.; Yue, L.; Shi, J.; Shao, M.; Wu, T. Role of IDO and TDO in Cancers and Related Diseases and the Therapeutic Implications. J. Cancer 2019, 10, 2771–2782. [Google Scholar] [CrossRef] [PubMed]
- Zaman, K.; Driscoll, R.; Hahn, D.; Werffeli, P.; Goodman, S.L.; Bauer, J.; Leyvraz, S.; Lejeune, F.; Stupp, R.; Rüegg, C. Monitoring multiple angiogenesis-related molecules in the blood of cancer patients shows a correlation between VEGF-A and MMP-9 levels before treatment and divergent changes after surgical vs. conservative therapy. Int. J. Cancer 2006, 118, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Frank, N.Y.; Schatton, T.; Kim, S.; Zhan, Q.; Wilson, B.J.; Ma, J.; Saab, K.R.; Osherov, V.; Widlund, H.R.; Gasser, M.; et al. VEGFR-1 expressed by malignant melanoma-initiating cells is required for tumor growth. Cancer Res. 2011, 71, 1474–1485. [Google Scholar] [CrossRef]
- Sobczuk, P.; Cholewinski, M.; Rutkowski, P. Recent advances in tyrosine kinase inhibitors VEGFR 1-3 for the treatment of advanced metastatic melanoma. Expert Opin. Pharmacother. 2024, 25, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Malekan, M.; Haass, N.K.; Rokni, G.R.; Gholizadeh, N.; Ebrahimzadeh, M.A.; Kazeminejad, A. VEGF/VEGFR axis and its signaling in melanoma: Current knowledge toward therapeutic targeting agents and future perspectives. Life Sci. 2024, 345, 122563. [Google Scholar] [CrossRef] [PubMed]
- Cook, N.; Basu, B.; Biswas, S.; Kareclas, P.; Mann, C.; Palmer, C.; Thomas, A.; Nicholson, S.; Morgan, B.; Lomas, D.; et al. A phase 2 study of vatalanib in metastatic melanoma patients. Eur. J. Cancer 2010, 46, 2671–2673. [Google Scholar] [CrossRef] [PubMed]
- Fruehauf, J.; Lutzky, J.; McDermott, D.; Brown, C.K.; Meric, J.B.; Rosbrook, B.; Shalinsky, D.R.; Liau, K.F.; Niethammer, A.G.; Kim, S.; et al. Multicenter, phase II study of axitinib, a selective second-generation inhibitor of vascular endothelial growth factor receptors 1, 2, and 3, in patients with metastatic melanoma. Clin. Cancer Res. 2011, 17, 7462–7469. [Google Scholar] [CrossRef] [PubMed]
- Algazi, A.P.; Cha, E.; Ortiz-Urda, S.M.; McCalmont, T.; Bastian, B.C.; Hwang, J.; Pampaloni, M.H.; Behr, S.; Chong, K.; Cortez, B.; et al. The combination of axitinib followed by paclitaxel/carboplatin yields extended survival in advanced BRAF wild-type melanoma: Results of a clinical/correlative prospective phase II clinical trial. Br. J. Cancer 2015, 112, 1326–1331. [Google Scholar] [CrossRef]
- Larkin, J.; Hodi, F.S.; Wolchok, J.D. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 1270–1271. [Google Scholar] [CrossRef]
- Schmitt, A.M.; Dumas, L.; Larkin, J. Atezolizumab, cobimetinib, and vemurafenib as first-line treatment for unresectable metastatic BRAF V600 mutated melanoma. Expert Rev. Anticancer Ther. 2022, 22, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Rozeman, E.A.; Menzies, A.M.; van Akkooi, A.C.J.; Adhikari, C.; Bierman, C.; van de Wiel, B.A.; Scolyer, R.A.; Krijgsman, O.; Sikorska, K.; Eriksson, H.; et al. Identification of the optimal combination dosing schedule of neoadjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma (OpACIN-neo): A multicentre, phase 2, randomised, controlled trial. Lancet Oncol. 2019, 20, 948–960. [Google Scholar] [CrossRef]
- ASCO. Oral Abstract Session 9508; ASCO: Alexandria, VA, USA, 2022. [Google Scholar]
- Nathan, P.; Dummer, R.; Long, G.V.; Ascierto, P.A.; Tawbi, H.A.; Robert, C.; Rutkowski, P.; Leonov, O.; Dutriaux, C.; Mandala’, M.; et al. Spartalizumab plus dabrafenib and trametinib in patients with previously untreated BRAF V600–mutant unresectable or metastatic melanoma: Results from the randomized part 3 of the Phase III COMBI-i trial. Ann. Oncol. 2020, 31, S1172. [Google Scholar] [CrossRef]
- Kraehenbuehl, L.; Holland, A.; Armstrong, E.; O’Shea, S.; Mangarin, L.; Chekalil, S.; Johnston, A.; Bomalaski, J.S.; Erinjeri, J.P.; Barker, C.A.; et al. Pilot Trial of Arginine Deprivation Plus Nivolumab and Ipilimumab in Patients with Metastatic Uveal Melanoma. Cancers 2022, 14, 2638. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).