
A Multi-Omics-Based Exploration of the Predictive Role of MSMB in Prostate Cancer Recurrence: A Study Using Bayesian Inverse Convolution and 10 Machine Learning Combinations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Study Design
2.2. Data Collection and Preprocessing
2.3. SMR Analysis and Single-Cell Analysis
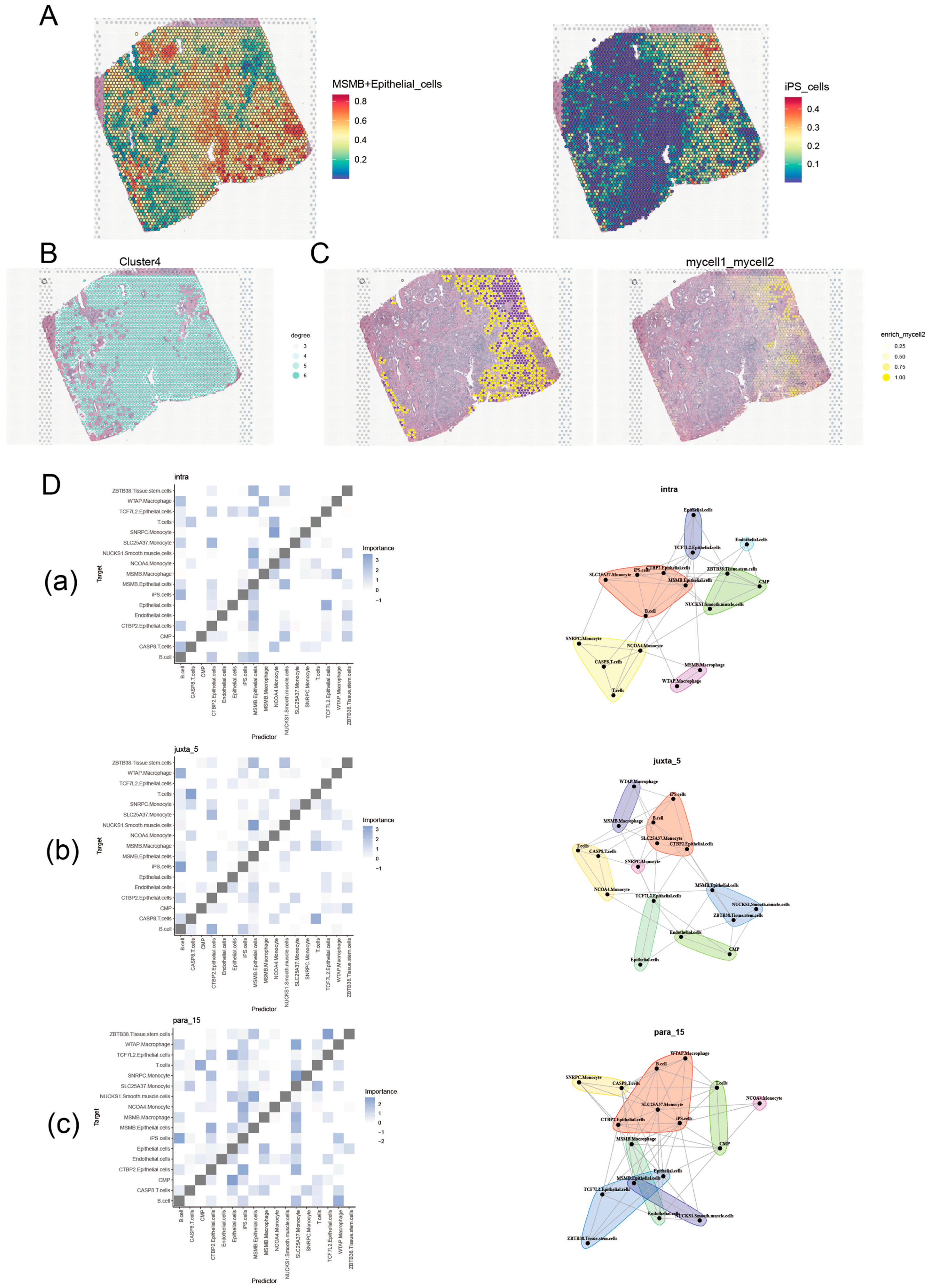
2.4. Spatial Transcriptome Analysis
2.5. BayesPrism Inverse Convolution Analysis
2.6. Weighted Gene Co-Expression Network Analysis (WGCNA) of Convoluted Cells
2.7. Construction of PCa DFI Model Based on Machine Learning
2.8. Acquisition of Published Signatures
2.9. Enrichment Analysis
2.10. Prognostic Analysis of Prognostic Genes
2.11. Correlation Analysis Between Prognosis-Related Genes and Convoluted Cells
2.12. Statistical Analysis
3. Results
3.1. SMR Analysis
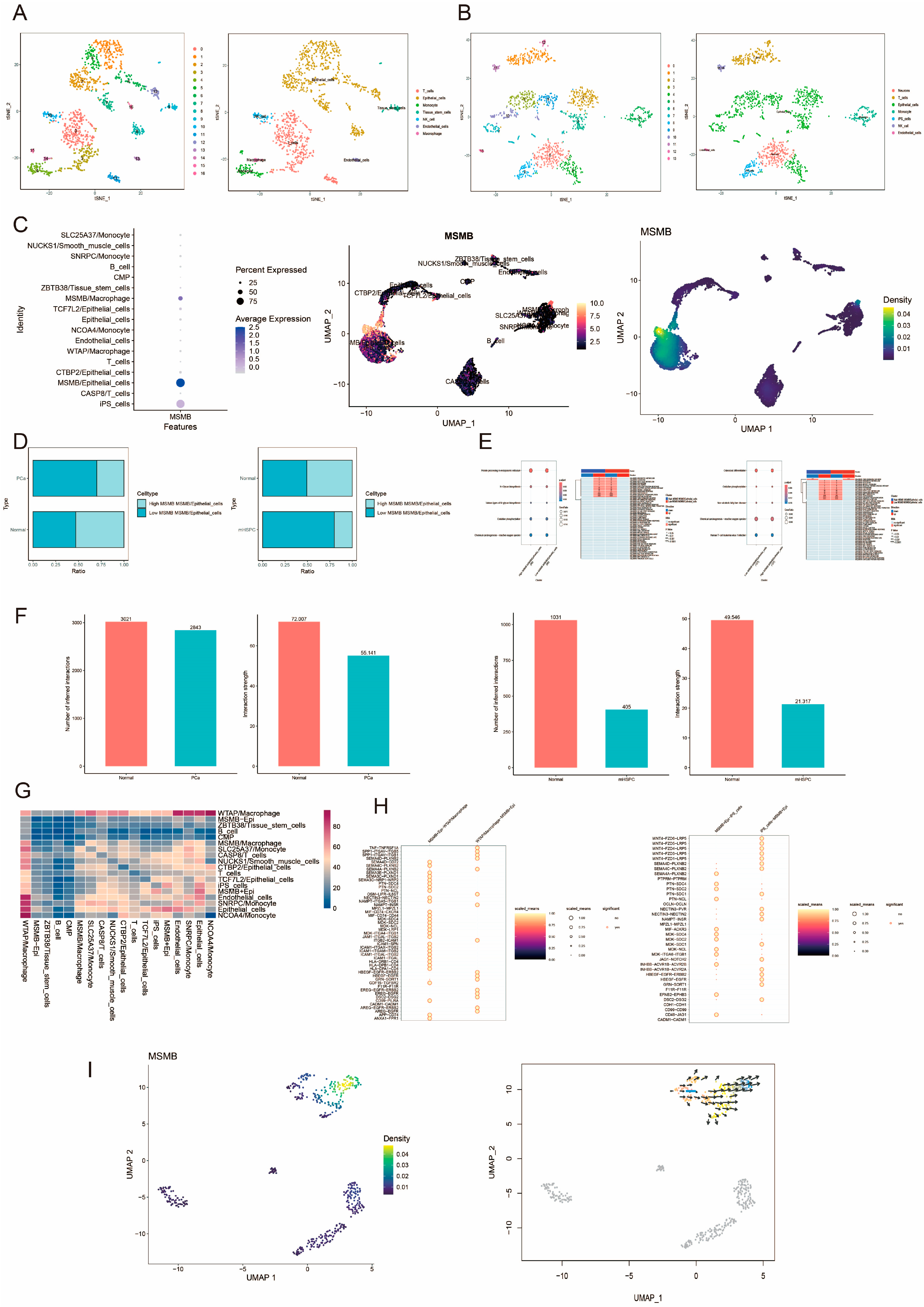
3.2. Verify the Expression Characteristics of Gene MSMB in PCa by Single-Cell and Spatial Transcriptome
3.3. scPagwas Screening for Trait-Associated Genes and Cell Populations
3.4. Bayesian Inverse Convolution Analysis of the Prognostic Properties of Convoluted Cells
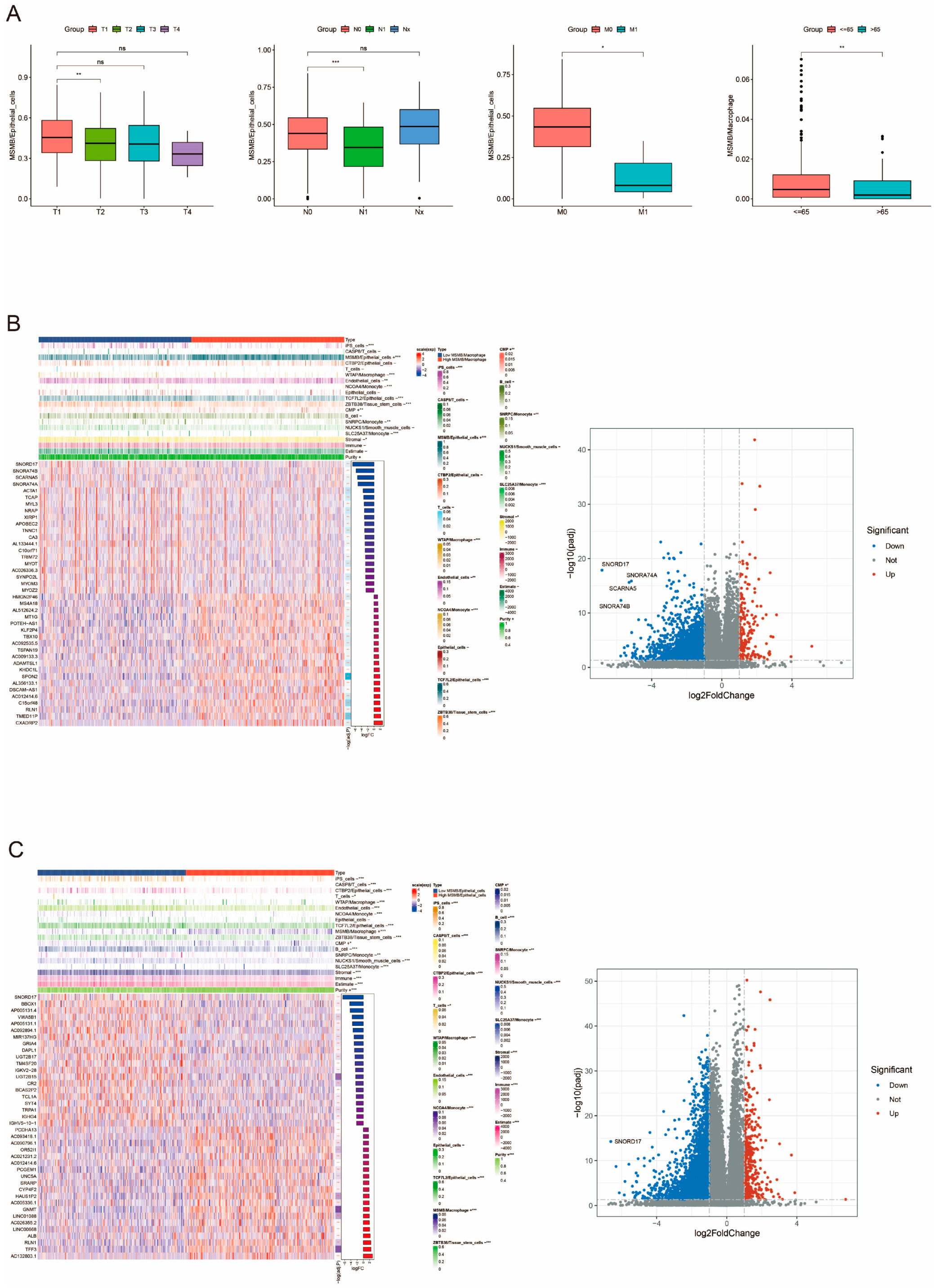
3.5. Clinical Subgroup Analysis of Convolutional Cells
3.6. Correlation Analysis of MSMB/Macrophage with the Remaining Convoluted Cells and Tumor Microenvironment (TME)
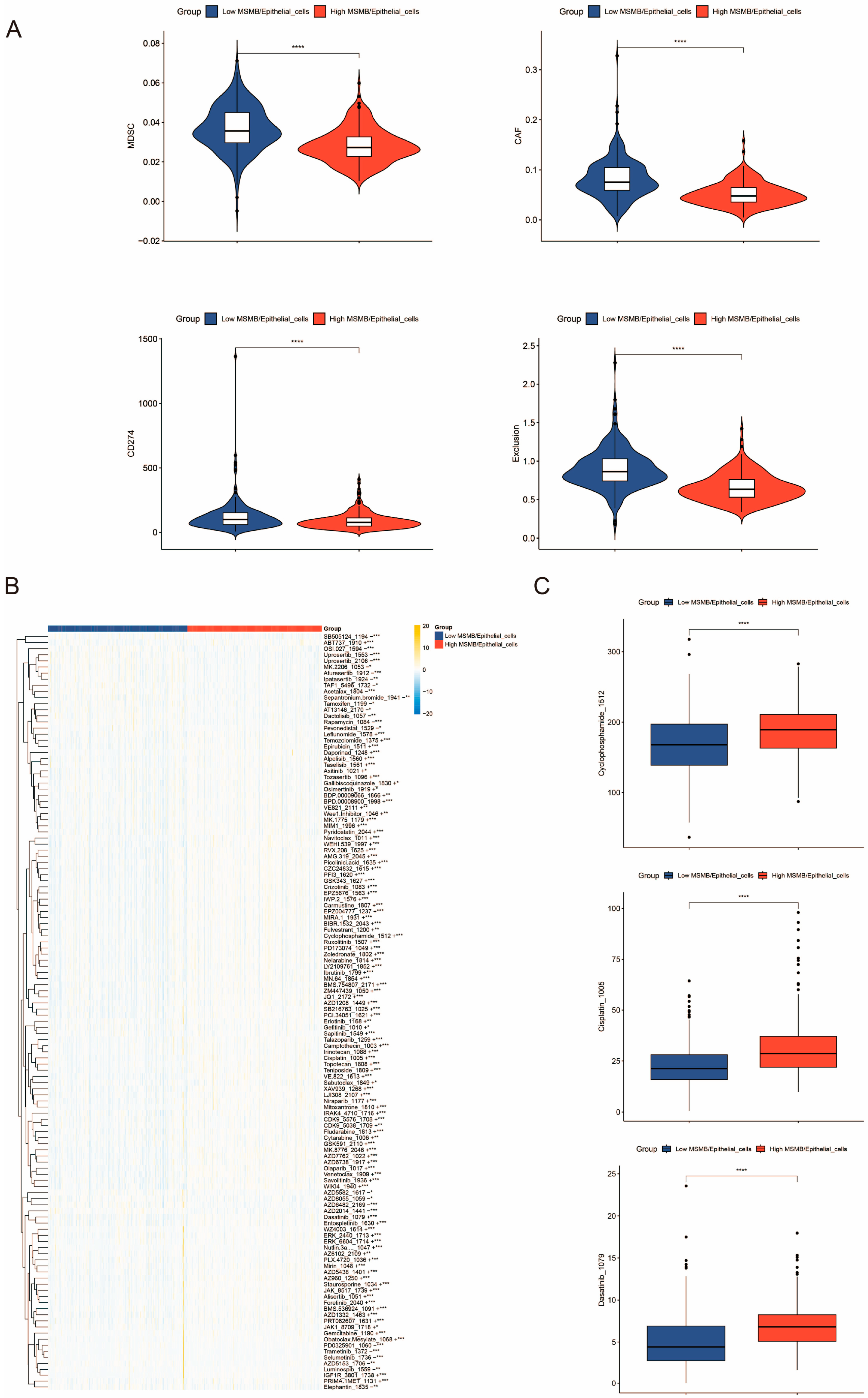
3.7. Immunocorrelation Analysis of Convoluted Cells
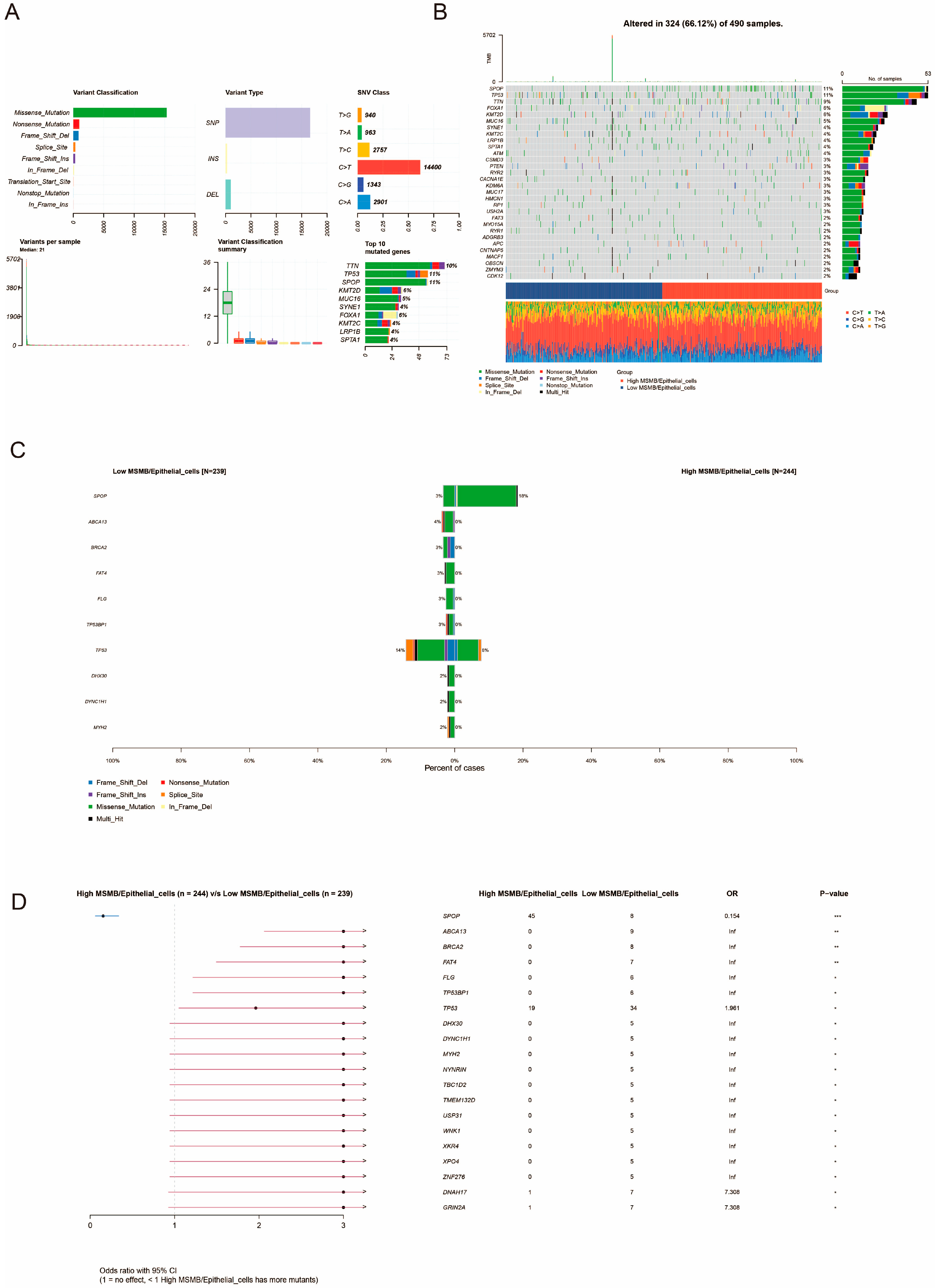
3.8. Unique Genetic Variants Associated with MSMB/Epithelial_Cells Subgroups
3.9. WGCNA Identification of Key Genes in Convoluted Cells
3.10. Construction and Evaluation of PCa DFI Prognostic Models
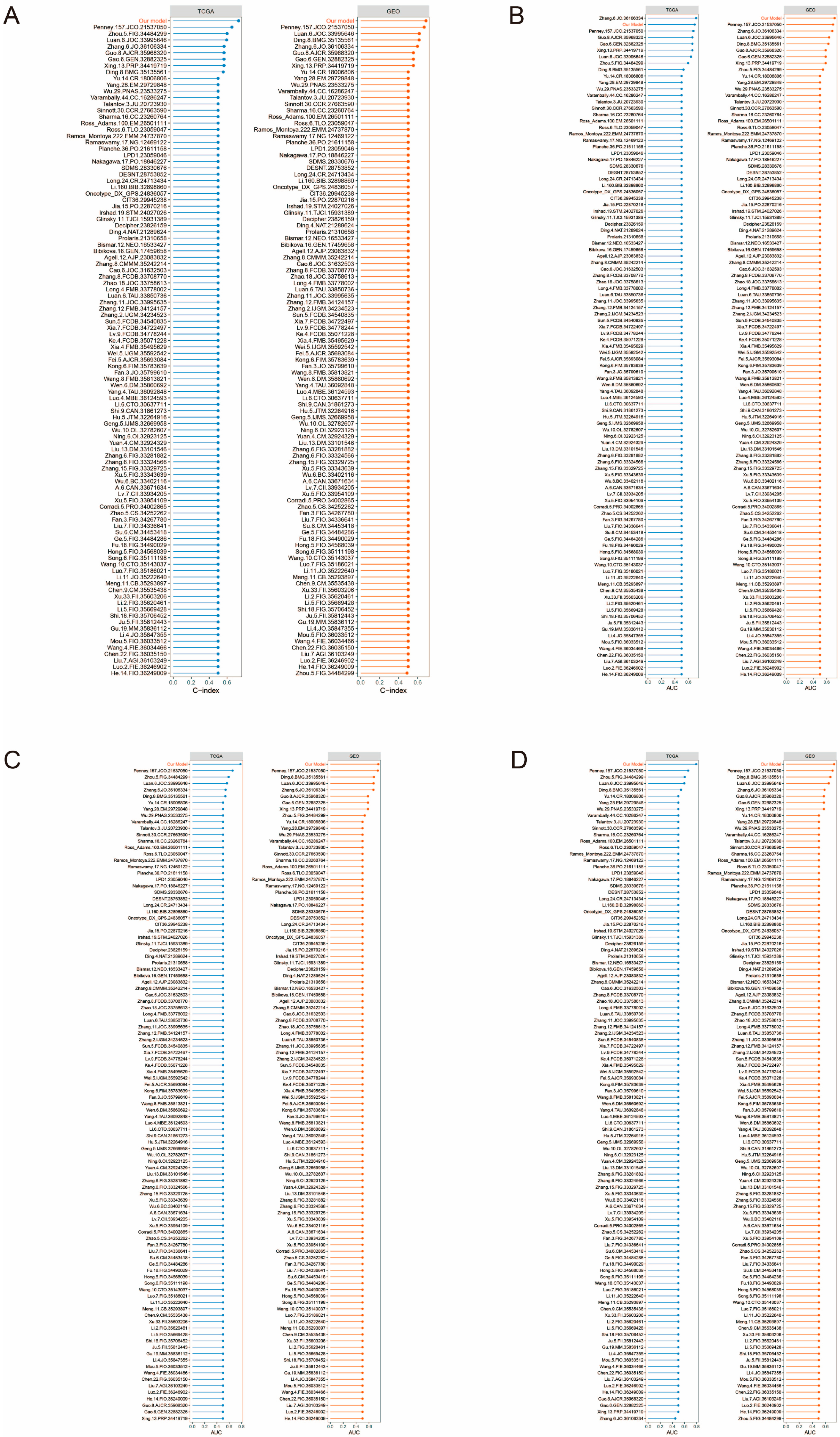
3.11. Comparison of PCa DFI Prognostic Models with 102 Published Models
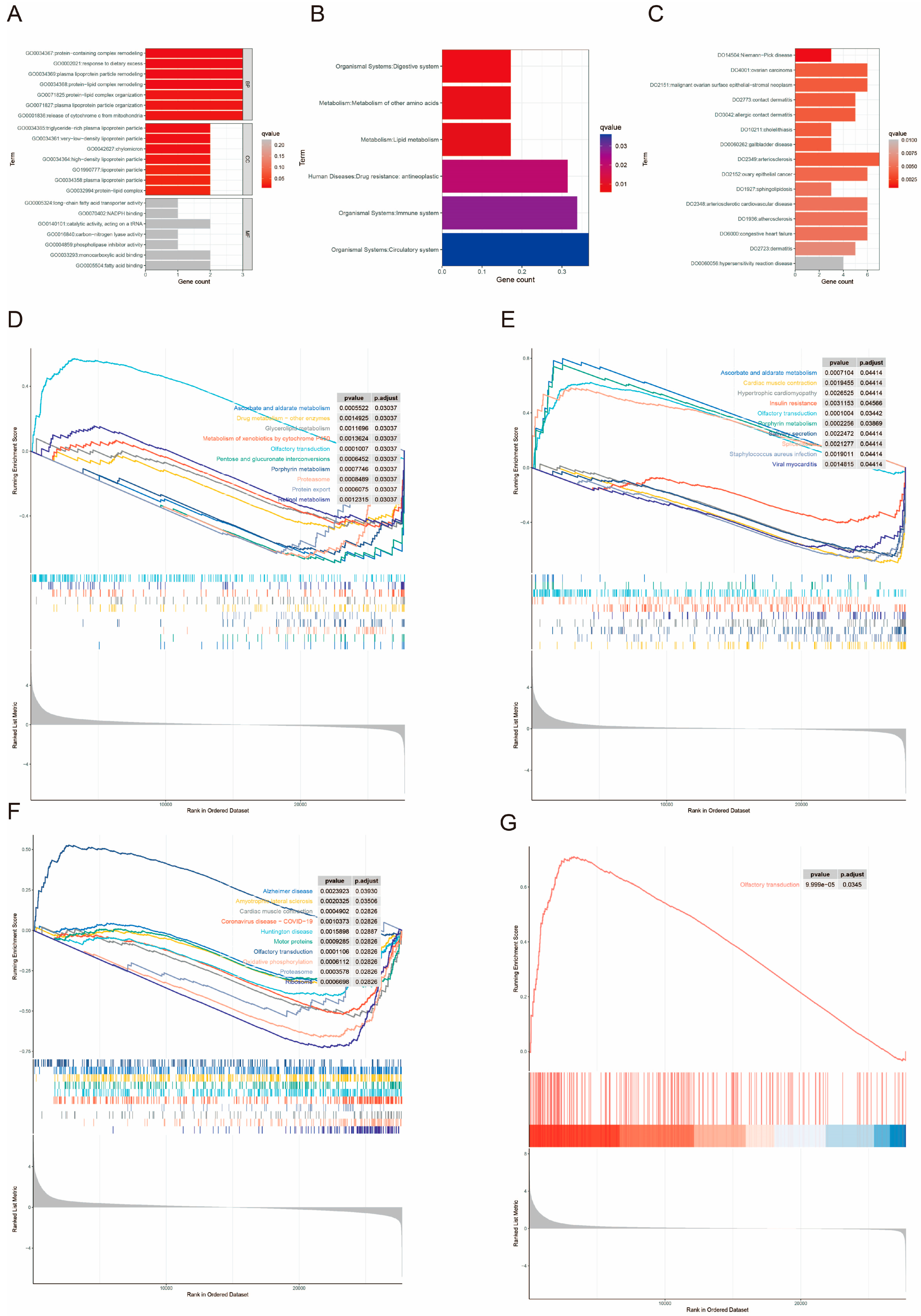
3.12. Enrichment Analysis
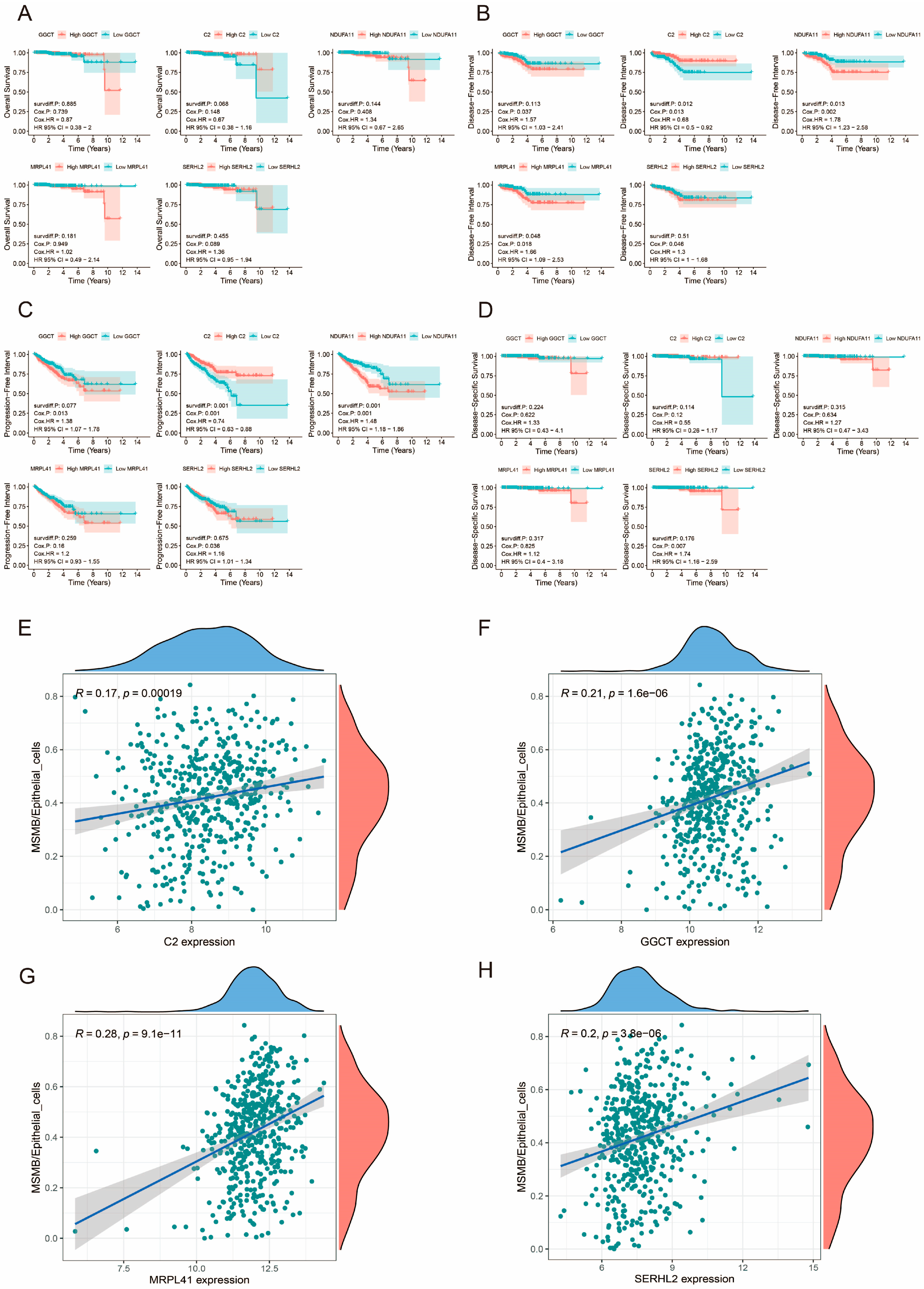
3.13. Prognostic Analysis of Prognosis-Related Genes
3.14. Relationship Between Prognosis-Related Genes and Convoluted Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PCa | prostate cancer |
| DFI | disease-free interval |
| BCR | biochemical recurrence |
| RP | radical prostatectomy |
| RT | radiotherapy |
| PSA | prostate-specific antigen |
| tSNE | t-distributed Stochastic Neighbor Embedding |
| UMAP | uniform manifold approximation and projection |
| eQTL | expression quantitative trait loci |
| pQTL | protein quantitative trait loci |
| SMR | summary-data-based Mendelian randomization |
| CNV | copy number variation |
| WGCNA | weighted gene co-expression network analysis |
| HPA | Human Protein Atlas |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| DO | disease ontology |
| DSS | disease-specific survival |
| PFS | progression-free survival |
| OS | overall survival |
| GSEA | gene set enrichment analysis |
| mHSPC | metastatic hormone-sensitive prostate cancer |
| TRS | trait-relevant score |
| RSF | Randomized Survival Forest |
| Survival-SVM | Survival-Support Vector Machine |
| Lasso | Least Absolute Shrinkage and Selection Operator |
| GBM | Generalized Boosted Regression Modeling |
| Enet | Elastic Networks |
| plsRcox | partial least squares regression for Cox |
| SuperPC | Supervised Principal Components |
| GDSC | Genome of Drug Sensitivity in Cancer |
References
- Shafi, A.A.; Yen, A.E.; Weigel, N.L. Androgen receptors in hormone-dependent and castration-resistant prostate cancer. Pharmacol. Ther. 2013, 140, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef] [PubMed]
- Ruan, X.-H.; Stacia Chun, T.T.; Huang, D.; Wong, H.-L.; Ho, B.S.-H.; Tsang, C.-F.; Lai, T.C.-T.; Ng, A.T.-L.; Na, R.; Tsu, J.H.-L. Outcomes of radical prostatectomy in a 20-year localized prostate cancer single institution series in China. Asian J. Androl. 2023, 25, 345–349. [Google Scholar] [CrossRef]
- Reijnen, C.; Brunenberg, E.J.L.; Kerkmeijer, L.G.W. Advancing the treatment of localized prostate cancer with MR-guided radiotherapy. Prostate Cancer Prostatic Dis. 2023, 26, 50–52. [Google Scholar] [CrossRef]
- Freedland, S.J.; Humphreys, E.B.; Mangold, L.A.; Eisenberger, M.; Dorey, F.J.; Walsh, P.C.; Partin, A.W. Risk of prostate cancer-specific mortality following biochemical recurrence after radical prostatectomy. JAMA 2005, 294, 433–439. [Google Scholar] [CrossRef]
- Zaorsky, N.G.; Raj, G.V.; Trabulsi, E.J.; Lin, J.; Den, R.B. The dilemma of a rising prostate-specific antigen level after local therapy: What are our options? Semin. Oncol. 2013, 40, 322–336. [Google Scholar] [CrossRef]
- Lin, X.; Kapoor, A.; Gu, Y.; Chow, M.J.; Xu, H.; Major, P.; Tang, D. Assessment of biochemical recurrence of prostate cancer (Review). Int. J. Oncol. 2019, 55, 1194–1212. [Google Scholar] [CrossRef]
- Kupelian, P.A.; Mahadevan, A.; Reddy, C.A.; Reuther, A.M.; Klein, E.A. Use of different definitions of biochemical failure after external beam radiotherapy changes conclusions about relative treatment efficacy for localized prostate cancer. Urology 2006, 68, 593–598. [Google Scholar] [CrossRef]
- Roach, M.; Hanks, G.; Thames, H.; Schellhammer, P.; Shipley, W.U.; Sokol, G.H.; Sandler, H. Defining biochemical failure following radiotherapy with or without hormonal therapy in men with clinically localized prostate cancer: Recommendations of the RTOG-ASTRO Phoenix Consensus Conference. Int. J. Radiat. Oncol. Biol. Phys. 2006, 65, 965–974. [Google Scholar] [CrossRef]
- Moul, J.W. Prostate specific antigen only progression of prostate cancer. J. Urol. 2000, 163, 1632–1642. [Google Scholar] [CrossRef] [PubMed]
- Van den Broeck, T.; van den Bergh, R.C.N.; Arfi, N.; Gross, T.; Moris, L.; Briers, E.; Cumberbatch, M.; De Santis, M.; Tilki, D.; Fanti, S.; et al. Prognostic Value of Biochemical Recurrence Following Treatment with Curative Intent for Prostate Cancer: A Systematic Review. Eur. Urol. 2019, 75, 967–987. [Google Scholar] [CrossRef] [PubMed]
- Cackowski, F.C.; Heath, E.I. Prostate cancer dormancy and recurrence. Cancer Lett. 2022, 524, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Hoey, C.; Ahmed, M.; Fotouhi Ghiam, A.; Vesprini, D.; Huang, X.; Commisso, K.; Commisso, A.; Ray, J.; Fokas, E.; Loblaw, D.A.; et al. Circulating miRNAs as non-invasive biomarkers to predict aggressive prostate cancer after radical prostatectomy. J. Transl. Med. 2019, 17, 173. [Google Scholar] [CrossRef]
- Rodrigues-Ferreira, S.; Nahmias, C. Predictive biomarkers for personalized medicine in breast cancer. Cancer Lett. 2022, 545, 215828. [Google Scholar] [CrossRef]
- D’Orsi, L.; Capasso, B.; Lamacchia, G.; Pizzichini, P.; Ferranti, S.; Liverani, A.; Fontana, C.; Panunzi, S.; De Gaetano, A.; Lo Presti, E. Recent Advances in Artificial Intelligence to Improve Immunotherapy and the Use of Digital Twins to Identify Prognosis of Patients with Solid Tumors. Int. J. Mol. Sci. 2024, 25, 11588. [Google Scholar] [CrossRef]
- Chu, T.; Wang, Z.; Pe’er, D.; Danko, C.G. Cell type and gene expression deconvolution with BayesPrism enables Bayesian integrative analysis across bulk and single-cell RNA sequencing in oncology. Nat. Cancer 2022, 3, 505–517. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Barrett, T.; Troup, D.B.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; et al. NCBI GEO: Archive for functional genomics data sets—10 years on. Nucleic Acids Res. 2011, 39, D1005–D1010. [Google Scholar] [CrossRef]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef]
- Zheng, G.X.Y.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef] [PubMed]
- Grün, D.; Lyubimova, A.; Kester, L.; Wiebrands, K.; Basak, O.; Sasaki, N.; Clevers, H.; van Oudenaarden, A. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature 2015, 525, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Gou, X.; Lai, G.; Li, K.; Zhu, X.; Liu, N.; Kuang, Y.; Ren, K.; Xie, Y.; Xu, Y.; et al. Single-nucleus sequencing unveils heterogeneity in renal cell carcinomas microenvironment: Insights into pathogenic origins and treatment-responsive cellular subgroups. Cancer Lett. 2024, 604, 217259. [Google Scholar] [CrossRef]
- Wang, G.; Wu, S.; Xiong, Z.; Qu, H.; Fang, X.; Bao, Y. CROST: A comprehensive repository of spatial transcriptomics. Nucleic Acids Res. 2024, 52, D882–D890. [Google Scholar] [CrossRef]
- Guo, Y.; Xu, T.; Luo, J.; Jiang, Z.; Chen, W.; Chen, H.; Qi, T.; Yang, J. SMR-Portal: An online platform for integrative analysis of GWAS and xQTL data to identify complex trait genes. Nat. Methods 2024. [Google Scholar] [CrossRef]
- Eeles, R.A.; Kote-Jarai, Z.; Al Olama, A.A.; Giles, G.G.; Guy, M.; Severi, G.; Muir, K.; Hopper, J.L.; Henderson, B.E.; Haiman, C.A.; et al. Identification of seven new prostate cancer susceptibility loci through a genome-wide association study. Nat. Genet. 2009, 41, 1116–1121. [Google Scholar] [CrossRef]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef]
- Jin, S.; Plikus, M.V.; Nie, Q. CellChat for systematic analysis of cell-cell communication from single-cell transcriptomics. Nat. Protoc. 2024, 20, 180–219. [Google Scholar] [CrossRef]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.-H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef]
- Zhang, F.; Li, X.; Tian, W. Unsupervised Inference of Developmental Directions for Single Cells Using VECTOR. Cell Rep. 2020, 32, 108069. [Google Scholar] [CrossRef]
- Tanevski, J.; Flores, R.O.R.; Gabor, A.; Schapiro, D.; Saez-Rodriguez, J. Explainable multiview framework for dissecting spatial relationships from highly multiplexed data. Genome Biol. 2022, 23, 97. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Shan, Q.; Li, Y.; Yuan, K.; Yang, X.; Yang, L.; He, J.-Q. Distinguish active tuberculosis with an immune-related signature and molecule subtypes: A multi-cohort analysis. Sci. Rep. 2024, 14, 29564. [Google Scholar] [CrossRef] [PubMed]
- Ambale-Venkatesh, B.; Yang, X.; Wu, C.O.; Liu, K.; Hundley, W.G.; McClelland, R.; Gomes, A.S.; Folsom, A.R.; Shea, S.; Guallar, E.; et al. Cardiovascular Event Prediction by Machine Learning: The Multi-Ethnic Study of Atherosclerosis. Circ. Res. 2017, 121, 1092–1101. [Google Scholar] [CrossRef]
- Van Belle, V.; Pelckmans, K.; Van Huffel, S.; Suykens, J.A.K. Improved performance on high-dimensional survival data by application of Survival-SVM. Bioinformatics 2011, 27, 87–94. [Google Scholar] [CrossRef]
- Nygård, S.; Borgan, O.; Lingjaerde, O.C.; Størvold, H.L. Partial least squares Cox regression for genome-wide data. Lifetime Data Anal. 2008, 14, 179–195. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, W.; Zhang, Y.; Adegboro, A.A.; Fasoranti, D.O.; Dai, L.; Pan, Z.; Liu, H.; Xiong, Y.; Li, W.; et al. Mime: A flexible machine-learning framework to construct and visualize models for clinical characteristics prediction and feature selection. Comput. Struct. Biotechnol. J. 2024, 23, 2798–2810. [Google Scholar] [CrossRef]
- Li, R.; Zhu, J.; Zhong, W.-D.; Jia, Z. Comprehensive Evaluation of Machine Learning Models and Gene Expression Signatures for Prostate Cancer Prognosis Using Large Population Cohorts. Cancer Res. 2022, 82, 1832–1843. [Google Scholar] [CrossRef]
- Yin, W.; Chen, G.; Li, Y.; Li, R.; Jia, Z.; Zhong, C.; Wang, S.; Mao, X.; Cai, Z.; Deng, J.; et al. Identification of a 9-gene signature to enhance biochemical recurrence prediction in primary prostate cancer: A benchmarking study using ten machine learning methods and twelve patient cohorts. Cancer Lett. 2024, 588, 216739. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Schriml, L.M.; Arze, C.; Nadendla, S.; Chang, Y.-W.W.; Mazaitis, M.; Felix, V.; Feng, G.; Kibbe, W.A. Disease Ontology: A backbone for disease semantic integration. Nucleic Acids Res. 2012, 40, D940–D946. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Deng, C.; Zhou, Y.; Zhang, Y.; Qiu, F.; Jiang, D.; Zheng, G.; Li, J.; Shuai, J.; Zhang, Y.; et al. Polygenic regression uncovers trait-relevant cellular contexts through pathway activation transformation of single-cell RNA sequencing data. Cell Genom. 2023, 3, 100383. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Jiang, W.; Li, H.; Wang, X.; Tian, Z.; Yang, K.; Zhu, Y. Single-cell RNA sequencing reveals that an imbalance in monocyte subsets rather than changes in gene expression patterns is a feature of postmenopausal osteoporosis. J. Bone Miner. Res. 2024, 39, 980–993. [Google Scholar] [CrossRef]
- Ghotra, V.P.S.; He, S.; van der Horst, G.; Nijhoff, S.; de Bont, H.; Lekkerkerker, A.; Janssen, R.; Jenster, G.; van Leenders, G.J.L.H.; Hoogland, A.M.M.; et al. SYK is a candidate kinase target for the treatment of advanced prostate cancer. Cancer Res. 2015, 75, 230–240. [Google Scholar] [CrossRef]
- Singh, S.; Singh, U.P.; Stiles, J.K.; Grizzle, W.E.; Lillard, J.W. Expression and functional role of CCR9 in prostate cancer cell migration and invasion. Clin. Cancer Res. 2004, 10, 8743–8750. [Google Scholar] [CrossRef]
- Engl, T.; Relja, B.; Blumenberg, C.; Müller, I.; Ringel, E.M.; Beecken, W.-D.; Jonas, D.; Blaheta, R.A. Prostate tumor CXC-chemokine profile correlates with cell adhesion to endothelium and extracellular matrix. Life Sci. 2006, 78, 1784–1793. [Google Scholar] [CrossRef]
- Heresi, G.A.; Wang, J.; Taichman, R.; Chirinos, J.A.; Regalado, J.J.; Lichtstein, D.M.; Rosenblatt, J.D. Expression of the chemokine receptor CCR7 in prostate cancer presenting with generalized lymphadenopathy: Report of a case, review of the literature, and analysis of chemokine receptor expression. Urol. Oncol. 2005, 23, 261–267. [Google Scholar] [CrossRef]
- Murphy, C.; McGurk, M.; Pettigrew, J.; Santinelli, A.; Mazzucchelli, R.; Johnston, P.G.; Montironi, R.; Waugh, D.J.J. Nonapical and cytoplasmic expression of interleukin-8, CXCR1, and CXCR2 correlates with cell proliferation and microvessel density in prostate cancer. Clin. Cancer Res. 2005, 11, 4117–4127. [Google Scholar] [CrossRef]
- Shen, H.; Schuster, R.; Lu, B.; Waltz, S.E.; Lentsch, A.B. Critical and opposing roles of the chemokine receptors CXCR2 and CXCR3 in prostate tumor growth. Prostate 2006, 66, 1721–1728. [Google Scholar] [CrossRef]
- Mughees, M.; Kaushal, J.B.; Sharma, G.; Wajid, S.; Batra, S.K.; Siddiqui, J.A. Chemokines and cytokines: Axis and allies in prostate cancer pathogenesis. Semin. Cancer Biol. 2022, 86, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Naik, A.; Thakur, N. Epigenetic regulation of TGF-β and vice versa in cancers—A review on recent developments. Biochim. Biophys. Acta Rev. Cancer 2024, 1879, 189219. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef]
- Weber, R.; Groth, C.; Lasser, S.; Arkhypov, I.; Petrova, V.; Altevogt, P.; Utikal, J.; Umansky, V. IL-6 as a major regulator of MDSC activity and possible target for cancer immunotherapy. Cell Immunol. 2021, 359, 104254. [Google Scholar] [CrossRef]
- Jiang, H.H.; Wang, K.X.; Bi, K.H.; Lu, Z.M.; Zhang, J.Q.; Cheng, H.R.; Zhang, M.Y.; Su, J.J.; Cao, Y.X. Sildenafil might impair maternal-fetal immunotolerance by suppressing myeloid-derived suppressor cells in mice. J. Reprod. Immunol. 2020, 142, 103175. [Google Scholar] [CrossRef]
- ChallaSivaKanaka, S.; Vickman, R.E.; Kakarla, M.; Hayward, S.W.; Franco, O.E. Fibroblast heterogeneity in prostate carcinogenesis. Cancer Lett. 2022, 525, 76–83. [Google Scholar] [CrossRef]
- Liu, Q.; Guan, Y.; Li, S. Programmed death receptor (PD-)1/PD-ligand (L)1 in urological cancers: The “all-around warrior” in immunotherapy. Mol. Cancer 2024, 23, 183. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.-C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef]
- Lv, D.; Wu, X.; Chen, X.; Yang, S.; Chen, W.; Wang, M.; Liu, Y.; Gu, D.; Zeng, G. A novel immune-related gene-based prognostic signature to predict biochemical recurrence in patients with prostate cancer after radical prostatectomy. Cancer Immunol. Immunother. 2021, 70, 3587–3602. [Google Scholar] [CrossRef]
- Boorjian, S.A.; Thompson, R.H.; Tollefson, M.K.; Rangel, L.J.; Bergstralh, E.J.; Blute, M.L.; Karnes, R.J. Long-term risk of clinical progression after biochemical recurrence following radical prostatectomy: The impact of time from surgery to recurrence. Eur. Urol. 2011, 59, 893–899. [Google Scholar] [CrossRef]
- Efstathiou, J.A.; Morgans, A.K.; Bland, C.S.; Shore, N.D. Novel hormone therapy and coordination of care in high-risk biochemically recurrent prostate cancer. Cancer Treat. Rev. 2024, 122, 102630. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Lih, T.-S.M.; Höti, N.; Chen, S.-Y.; Ponce, S.; Partin, A.; Zhang, H. Development of Parallel Reaction Monitoring Assays for the Detection of Aggressive Prostate Cancer Using Urinary Glycoproteins. J. Proteome Res. 2021, 20, 3590–3599. [Google Scholar] [CrossRef] [PubMed]
- Desai, T.A.; Hedman, Å.K.; Dimitriou, M.; Koprulu, M.; Figiel, S.; Yin, W.; Johansson, M.; Watts, E.L.; Atkins, J.R.; Sokolov, A.V.; et al. Identifying proteomic risk factors for overall, aggressive, and early onset prostate cancer using Mendelian Randomisation and tumour spatial transcriptomics. EBioMedicine 2024, 105, 105168. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Li, H.; Shi, W.; Lv, Q.; Zhang, W. Associations between genetically predicted concentrations of plasma proteins and the risk of prostate cancer. BMC Cancer 2024, 24, 905. [Google Scholar] [CrossRef]
- Shi, Y.; Xu, Y.; Xu, Z.; Wang, H.; Zhang, J.; Wu, Y.; Tang, B.; Zheng, S.; Wang, K. TKI resistant-based prognostic immune related gene signature in LUAD, in which FSCN1 contributes to tumor progression. Cancer Lett. 2022, 532, 215583. [Google Scholar] [CrossRef]
- Wu, L.; Chen, Y.; Wan, L.; Wen, Z.; Liu, R.; Li, L.; Song, Y.; Wang, L. Identification of unique transcriptomic signatures and key genes through RNA sequencing and integrated WGCNA and PPI network analysis in HIV infected lung cancer. Cancer Med. 2023, 12, 949–960. [Google Scholar] [CrossRef]
- Kageyama, S.; Ii, H.; Taniguchi, K.; Kubota, S.; Yoshida, T.; Isono, T.; Chano, T.; Yoshiya, T.; Ito, K.; Yoshiki, T.; et al. Mechanisms of Tumor Growth Inhibition by Depletion of γ-Glutamylcyclotransferase (GGCT): A Novel Molecular Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 2054. [Google Scholar] [CrossRef]
- Ii, H.; Yoshiya, T.; Nakata, S.; Taniguchi, K.; Hidaka, K.; Tsuda, S.; Mochizuki, M.; Nishiuchi, Y.; Tsuda, Y.; Ito, K.; et al. A Novel Prodrug of a γ-Glutamylcyclotransferase Inhibitor Suppresses Cancer Cell Proliferation in vitro and Inhibits Tumor Growth in a Xenograft Mouse Model of Prostate Cancer. ChemMedChem 2018, 13, 155–163. [Google Scholar] [CrossRef]
- Taniguchi, K.; Matsumura, K.; Ii, H.; Kageyama, S.; Ashihara, E.; Chano, T.; Kawauchi, A.; Yoshiki, T.; Nakata, S. Depletion of gamma-glutamylcyclotransferase in cancer cells induces autophagy followed by cellular senescence. Am. J. Cancer Res. 2018, 8, 650–661. [Google Scholar]
- Saito, Y.; Taniguchi, K.; Ii, H.; Horinaka, M.; Kageyama, S.; Nakata, S.; Ukimura, O.; Sakai, T. Identification of c-Met as a novel target of γ-glutamylcyclotransferase. Sci. Rep. 2023, 13, 11922. [Google Scholar] [CrossRef]
- Gold, B.; Merriam, J.E.; Zernant, J.; Hancox, L.S.; Taiber, A.J.; Gehrs, K.; Cramer, K.; Neel, J.; Bergeron, J.; Barile, G.R.; et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat. Genet. 2006, 38, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Namgoong, S.; Shin, J.-G.; Cheong, H.S.; Kim, L.H.; Kim, J.O.; Seo, J.Y.; Shin, H.D.; Kim, Y.J. Genetic association of complement component 2 variants with chronic hepatitis B in a Korean population. Liver Int. 2018, 38, 1576–1582. [Google Scholar] [CrossRef] [PubMed]
- Cerhan, J.R.; Novak, A.J.; Fredericksen, Z.S.; Wang, A.H.; Liebow, M.; Call, T.G.; Dogan, A.; Witzig, T.E.; Ansell, S.M.; Habermann, T.M.; et al. Risk of non-Hodgkin lymphoma in association with germline variation in complement genes. Br. J. Haematol. 2009, 145, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, H.; Sun, L.; Shen, S.; Zhou, Q.; Yuan, Y.; Xing, C. Epigenetic Alternations of MicroRNAs and DNA Methylation Contribute to Liver Metastasis of Colorectal Cancer. Dig. Dis. Sci. 2019, 64, 1523–1534. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tsukamoto, Y.; Hori, M.; Iha, H. Disulfidptosis: A Novel Prognostic Criterion and Potential Treatment Strategy for Diffuse Large B-Cell Lymphoma (DLBCL). Int. J. Mol. Sci. 2024, 25, 7156. [Google Scholar] [CrossRef]
- Mao, W.; Xiong, G.; Wu, Y.; Wang, C.; St Clair, D.; Li, J.-D.; Xu, R. RORα Suppresses Cancer-Associated Inflammation by Repressing Respiratory Complex I-Dependent ROS Generation. Int. J. Mol. Sci. 2021, 22, 10665. [Google Scholar] [CrossRef]
- Jiang, C.; Xiao, Y.; Xu, D.; Huili, Y.; Nie, S.; Li, H.; Guan, X.; Cao, F. Prognosis Prediction of Disulfidptosis-Related Genes in Bladder Cancer and a Comprehensive Analysis of Immunotherapy. Crit. Rev. Eukaryot. Gene Expr. 2023, 33, 73–86. [Google Scholar] [CrossRef]
- Kim, T.W.; Kim, B.; Kim, J.H.; Kang, S.; Park, S.-B.; Jeong, G.; Kang, H.-S.; Kim, S.J. Nuclear-encoded mitochondrial MTO1 and MRPL41 are regulated in an opposite epigenetic mode based on estrogen receptor status in breast cancer. BMC Cancer 2013, 13, 502. [Google Scholar] [CrossRef]
- Xin, C.; Lai, Y.; Ji, L.; Wang, Y.; Li, S.; Hao, L.; Zhang, W.; Meng, R.; Xu, J.; Hong, Y.; et al. A novel 9-gene signature for the prediction of postoperative recurrence in stage II/III colorectal cancer. Front. Genet. 2022, 13, 1097234. [Google Scholar] [CrossRef]
- Gu, X.-J.; Su, W.-M.; Dou, M.; Jiang, Z.; Duan, Q.-Q.; Yin, K.-F.; Cao, B.; Wang, Y.; Li, G.-B.; Chen, Y.-P. Expanding causal genes for Parkinson’s disease via multi-omics analysis. NPJ Parkinsons Dis. 2023, 9, 146. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, S.; Yin, H. A Multi-Omics-Based Exploration of the Predictive Role of MSMB in Prostate Cancer Recurrence: A Study Using Bayesian Inverse Convolution and 10 Machine Learning Combinations. Biomedicines 2025, 13, 487. https://doi.org/10.3390/biomedicines13020487
Huang S, Yin H. A Multi-Omics-Based Exploration of the Predictive Role of MSMB in Prostate Cancer Recurrence: A Study Using Bayesian Inverse Convolution and 10 Machine Learning Combinations. Biomedicines. 2025; 13(2):487. https://doi.org/10.3390/biomedicines13020487
Chicago/Turabian StyleHuang, Shan, and Hang Yin. 2025. "A Multi-Omics-Based Exploration of the Predictive Role of MSMB in Prostate Cancer Recurrence: A Study Using Bayesian Inverse Convolution and 10 Machine Learning Combinations" Biomedicines 13, no. 2: 487. https://doi.org/10.3390/biomedicines13020487
APA StyleHuang, S., & Yin, H. (2025). A Multi-Omics-Based Exploration of the Predictive Role of MSMB in Prostate Cancer Recurrence: A Study Using Bayesian Inverse Convolution and 10 Machine Learning Combinations. Biomedicines, 13(2), 487. https://doi.org/10.3390/biomedicines13020487