
Immune Clustering Reveals Molecularly Distinct Subtypes of Lung Adenocarcinoma

 , , ,
, , ,
Abstract
:1. Introduction
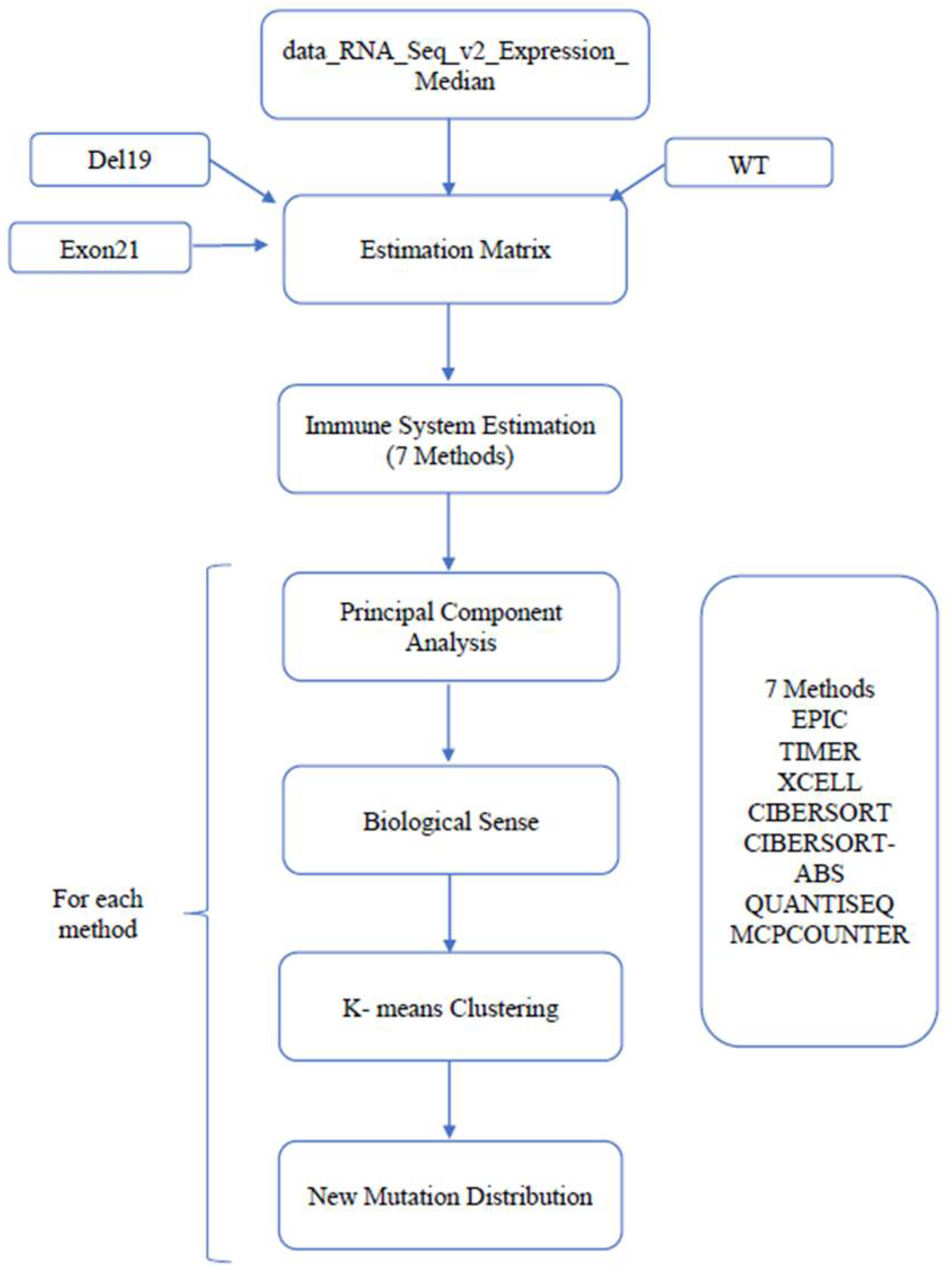
2. Materials and Methods
2.1. Study Population
2.2. Data
2.3. Statistics
2.4. Immune Inference from Bulk Expression Profiles
2.5. Overall Survival
2.6. Differential Expression Analysis
2.7. Clustering
3. Results
3.1. Characteristics of the Study Population
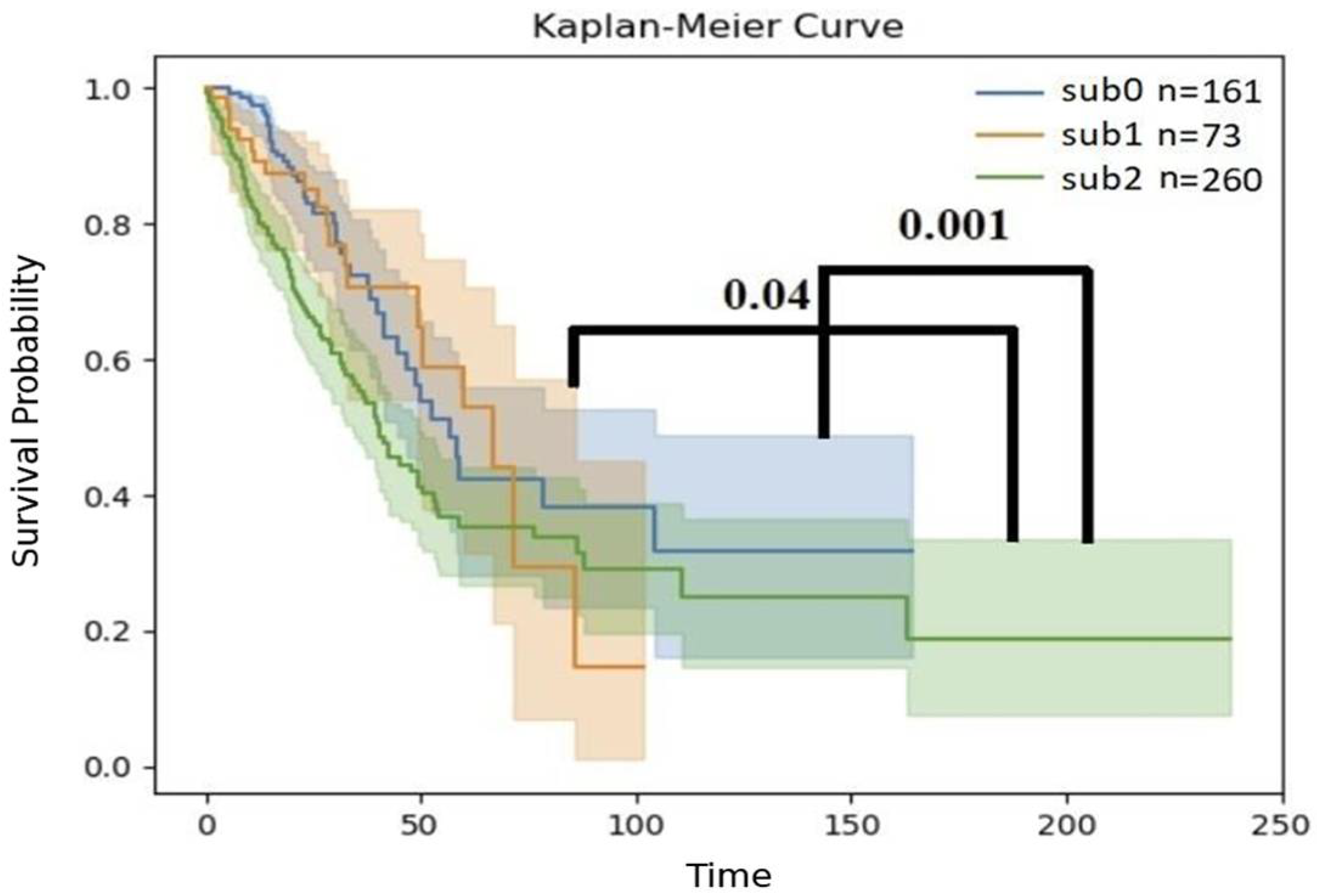
3.2. Immune Inference and Unsupervised Learning Detect New Groups
3.3. Differential Expression of Immune Checkpoints Between Immune Cell Clusters
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer Statistics for the Year 2020: An Overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Zappa, C.; Mousa, S.A. Non-Small Cell Lung Cancer: Current Treatment and Future Advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Zhang, L.; Ren, Y.; Ma, Q. The Genomic Alterations of Lung Adenocarcinoma and Lung Squamous Cell Carcinoma Can Explain the Differences of Their Overall Survival Rates. J. Cell Physiol. 2019, 234, 10918–10925. [Google Scholar] [CrossRef] [PubMed]
- Unni, A.M.; Lockwood, W.W.; Zejnullahu, K.; Lee-Lin, S.-Q.; Varmus, H. Evidence That Synthetic Lethality Underlies the Mutual Exclusivity of Oncogenic KRAS and EGFR Mutations in Lung Adenocarcinoma. eLife 2015, 4, e06907. [Google Scholar] [CrossRef]
- Shen, M.; Qi, R.; Ren, J.; Lv, D.; Yang, H. Characterization with KRAS Mutant Is a Critical Determinant in Immunotherapy and Other Multiple Therapies for Non-Small Cell Lung Cancer. Front. Oncol. 2022, 11, 780655. [Google Scholar] [CrossRef]
- Ferrer, I.; Zugazagoitia, J.; Herbertz, S.; John, W.; Paz-Ares, L.; Schmid-Bindert, G. KRAS-Mutant Non-Small Cell Lung Cancer: From Biology to Therapy. Lung Cancer 2018, 124, 53–64. [Google Scholar] [CrossRef]
- Gu, M.; Xu, T.; Chang, P. KRAS/LKB1 and KRAS/TP53 Co-Mutations Create Divergent Immune Signatures in Lung Adenocarcinomas. Ther. Adv. Med. Oncol. 2021, 13, 17588359211006950. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, H.S.; Kim, B.J. Oncotarget Prognostic Value of KRAS Mutation in Advanced Non-Small-Cell Lung Cancer Treated with Immune Checkpoint Inhibitors: A Meta-Analysis and Review. Oncotarget 2017, 8, 48248. [Google Scholar] [CrossRef]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef]
- Russo, A.; Franchina, T.; Ricciardi, G.; Battaglia, A.; Picciotto, M.; Adamo, V. Heterogeneous Responses to Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Inhibitors (TKIs) in Patients with Uncommon EGFR Mutations: New Insights and Future Perspectives in This Complex Clinical Scenario. Int. J. Mol. Sci. 2019, 20, 1431. [Google Scholar] [CrossRef]
- Baek, J.H.; Sun, J.M.; Min, Y.J.; Cho, E.K.; Cho, B.C.; Kim, J.H.; Ahn, M.J.; Park, K. Efficacy of EGFR Tyrosine Kinase Inhibitors in Patients with EGFR-Mutated Non-Small Cell Lung Cancer except Both Exon 19 Deletion and Exon 21 L858R: A Retrospective Analysis in Korea. Lung Cancer 2015, 87, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Wei, T.; Meng, H.; Luo, P.; Zhang, J. Role of the Dynamic Tumor Microenvironment in Controversies Regarding Immune Checkpoint Inhibitors for the Treatment of Non-Small Cell Lung Cancer (NSCLC) with EGFR Mutations. Mol. Cancer 2019, 18, 139. [Google Scholar] [PubMed]
- Langer, C.J. Roles of EGFR and KRAS Mutations in the Treatment of Patients with Non–Small-Cell Lung Cancer. Pharm. Ther. 2011, 36, 263. [Google Scholar]
- Cucurull, M.; Notario, L.; Sanchez-Cespedes, M.; Hierro, C.; Estival, A.; Carcereny, E.; Saigí, M. Targeting KRAS in Lung Cancer Beyond KRAS G12C Inhibitors: The Immune Regulatory Role of KRAS and Novel Therapeutic Strategies. Front. Oncol. 2022, 11, 793121. [Google Scholar] [CrossRef]
- Takamochi, K.; Oh, S.; Suzuki, K. Differences in EGFR and KRAS Mutation Spectra in Lung Adenocarcinoma of Never and Heavy Smokers. Oncol. Lett. 2013, 6, 1207–1212. [Google Scholar] [CrossRef]
- Aran, D.; Hu, Z.; Butte, A.J. XCell: Digitally Portraying the Tissue Cellular Heterogeneity Landscape. Genome Biol. 2017, 18, 114165. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, H.; Mao, B.; Zhou, Y.; Shi, X.; Tang, L.; Jiang, H.; Wang, G.; Zhuang, W. Transcriptional Characterization of the Tumor Immune Microenvironment and Its Prognostic Value for Locally Advanced Lung Adenocarcinoma in a Chinese Population. Cancer Manag. Res. 2019, 11, 9165–9173. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef]
- Ghosh, D.; Vogt, A. Outliers: An Evaluation of Methodologies. Jt. Stat. Meet. 2012, 12, 3455–3460. [Google Scholar]
- MuTect2 Variant Aggregation and Masking. Available online: https://xenabrowser.net/datapages/?dataset=TCGA-LUAD.mutect2_snv.tsv%20&host=https%3A%2F%2Fgdc.xenahubs.net&removeHub=https%3A%2F%2Fxena.treehouse.gi.ucsc.edu%3A443 (accessed on 24 February 2022).
- Satopää, V.; Albrecht, J.; Irwin, D.; Raghavan, B. Finding a “Kneedle” in a Haystack: Detecting Knee Points in System Behavior. In Proceedings of the 2011 31st International Conference on Distributed Computing Systems Workshops, Minneapolis, MN, USA, 20–24 June 2011; pp. 166–171. [Google Scholar] [CrossRef]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and Interpreting Cancer Genomics Data via the Xena Platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021; Available online: https://www.R-Project.org/ (accessed on 27 February 2022).
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Li, B.; Severson, E.; Pignon, J.C.; Zhao, H.; Li, T.; Novak, J.; Jiang, P.; Shen, H.; Aster, J.C.; Rodig, S.; et al. Comprehensive Analyses of Tumor Immunity: Implications for Cancer Immunotherapy. Genome Biol. 2016, 17, 174. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Sander, C.; Stuart, J.M.; Chang, K.; Creighton, C.J.; et al. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Graham, R.P.; Treece, A.L.; Lindeman, N.I.; Vasalos, P.; Shan, M.; Jennings, L.J.; Rimm, D.L. Worldwide Frequency of Commonly Detected EGFR Mutations. Arch. Pathol. Lab. Med. 2018, 142, 163–167. [Google Scholar] [CrossRef]
- Likas, A.; Vlassis, N.; Verbeek, J.J. The Global K-Means Clustering Algorithm. Pattern Recognit. 2003, 36, 451. [Google Scholar]
- Koletsi, D.; Pandis, N. Survival Analysis, Part 2: Kaplan-Meier Method and the Log-Rank Test. Am. J. Orthod. Dentofac. Orthop. 2017, 152, 569–571. [Google Scholar]
- To, K.K.W.; Fong, W.; Cho, W.C.S. Immunotherapy in Treating EGFR-Mutant Lung Cancer: Current Challenges and New Strategies. Front. Oncol. 2021, 11, 635007. [Google Scholar] [CrossRef]
- Hastings, K.; Yu, H.A.; Wei, W.; Sanchez-Vega, F.; Deveaux, M.; Choi, J.; Rizvi, H.; Lisberg, A.; Truini, A.; Lydon, C.A.; et al. EGFR Mutation Subtypes and Response to Immune Checkpoint Blockade Treatment in Non-Small-Cell Lung Cancer. Ann. Oncol. 2019, 30, 1311–1320. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. Available online: http://scikit-learn.sourceforge.net (accessed on 27 February 2022).
- Deng, M.; Brägelmann, J.; Kryukov, I.; Saraiva-Agostinho, N.; Perner, S. FirebrowseR: An R client to the Broad Institute’s Firehose Pipeline. Database 2017, 2017, baw160. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Bagaev, A.; Kotlov, N.; Nomie, K.; Svekolkin, V.; Gafurov, A.; Isaeva, O.; Osokin, N.; Kozlov, I.; Frenkel, F.; Gancharova, O.; et al. Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell 2021, 39, 845–865.e7. [Google Scholar] [CrossRef]
- Luo, W.; Tian, P.; Wang, Y.; Xu, H.; Chen, L.; Tang, C.; Shu, Y.; Zhang, S.; Wang, Z.; Zhang, J.; et al. Characteristics of Genomic Alterations of Lung Adenocarcinoma in Young Never-Smokers. Int. J. Cancer 2018, 143, 1696–1705. [Google Scholar] [CrossRef]
- Chen, Y.; Jin, L.; Jiang, Z.; Liu, S.; Feng, W. Identifying and Validating Potential Biomarkers of Early Stage Lung Adenocarcinoma Diagnosis and Prognosis. Front. Oncol. 2021, 11, 644426. [Google Scholar] [CrossRef]
- Toedt, G.; Barbus, S.; Wolter, M.; Felsberg, J.; Tews, B.; Blond, F.; Sabel, M.C.; Hofmann, S.; Becker, N.; Hartmann, C.; et al. Molecular Signatures Classify Astrocytic Gliomas by IDH1 Mutation Status. Int. J. Cancer 2011, 128, 1095–1103. [Google Scholar] [CrossRef]
- Plath, M.; Gass, J.; Hlevnjak, M.; Li, Q.; Feng, B.; Hostench, X.P.; Bieg, M.; Schroeder, L.; Holzinger, D.; Zapatka, M.; et al. Unraveling Most Abundant Mutational Signatures in Head and Neck Cancer. Int. J. Cancer 2021, 148, 115–127. [Google Scholar] [CrossRef]
- Liu, X.; Chen, W.; Fang, Y.; Yang, S.; Chang, L.; Chen, X.; Ye, H.; Tang, X.; Zhong, S.; Zhang, W.; et al. ADEIP: An Integrated Platform of Age-Dependent Expression and Immune Profiles across Human Tissues. Brief. Bioinform. 2021, 22, bbab274. [Google Scholar] [CrossRef]
- Alldredge, J.; Randall, L.; de Robles, G.; Agrawal, A.; Mercola, D.; Liu, M.; Randhawa, P.; Edwards, R.; McClelland, M.; Rahmatpanah, F. Transcriptome Analysis of Ovarian and Uterine Clear Cell Malignancies. Front. Oncol. 2020, 10, 598579. [Google Scholar] [CrossRef]
- Li, J.; Chen, H.; Guo, H.; Qiu, M.; Yang, F. Characterization of Gene Expression Profiles of Esophageal Cancer Patients with Different Nonsynonymous Tumor Mutation Burden. Thorac. Cancer 2020, 11, 2270–2278. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Han, X.; Lin, L.; Chen, J.; Wang, F.; Ding, Q.; Hao, L.; Wang, L.; Wei, J.; Wang, Y.; et al. Unraveling the Expression Patterns of Immune Checkpoints Identifies New Subtypes and Emerging Therapeutic Indicators in Lung Adenocarcinoma. Oxid. Med. Cell Longev. 2022, 2022, 3583985. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, M.; Yang, M.; Yang, Y.; Song, F.; Zhang, W.; Li, X.; Chen, K. Analysis of Immune-Related Signatures of Lung Adenocarcinoma Identified Two Distinct Subtypes: Implications for Immune Checkpoint Blockade Therapy. Aging 2020, 12, 3312–3339. [Google Scholar] [CrossRef] [PubMed]
- Doroshow, D.B.; Bhalla, S.; Beasley, M.B.; Sholl, L.M.; Kerr, K.M.; Gnjatic, S.; Wistuba, I.I.; Rimm, D.L.; Tsao, M.S.; Hirsch, F.R. PD-L1 as a Biomarker of Response to Immune-Checkpoint Inhibitors. Nat. Rev. Clin. Oncol. 2021, 18, 345–362. [Google Scholar] [CrossRef]
- Gustafson, M.P.; Lin, Y.; LaPlant, B.; Liwski, C.J.; Maas, M.L.; League, S.C.; Bauer, P.R.; Abraham, R.S.; Tollefson, M.K.; Kwon, E.D.; et al. Immune Monitoring Using the Predictive Power of Immune Profiles. J. Immunother. Cancer 2013, 1, 1–11. [Google Scholar] [CrossRef]
- Michaud, D.S.; Houseman, E.A.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. Understanding the Role of the Immune System in the Development of Cancer: New Opportunities for Population-Based Research. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1811–1819. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | EGFR Status | p-Val WT-Mut | ||
|---|---|---|---|---|
| WT | Mut | |||
| Tumor grade | Population (n) | 492 | 66 | |
| Grade I | 247 | 29 | 0.65 | |
| Grade II | 105 | 14 | 0.89 | |
| Grade III | 70 | 13 | 0.42 | |
| Grade IV | 21 | 6 | 0.19 | |
| Not available | 49 | 4 | ||
| Smoking | Yes | 386 | 34 | 0.07 |
| No | 46 | 27 | ||
| Not available | 60 | 5 | ||
| Diagnosis age (years) | ≤60 | 140 | 17 | 0.84 |
| >60 | 289 | 42 | 0.78 | |
| Not available | 63 | 7 | ||
| Gender | Female | 230 | 43 | 0.14 |
| Male | 215 | 19 | 0.16 | |
| Not available | 47 | 4 | ||
| Race | White | 336 | 46 | 0.99 |
| Black | 46 | 6 | 0.87 | |
| Asian | 5 | 3 | 0.1 | |
| Other | 1 | 0 | 1 | |
| Not available | 104 | 11 | ||
| Sub0 | Sub1 | Sub2 | |
|---|---|---|---|
| EGFR-WT | 131 | 71 | 239 |
| EGFR-mutated | 33 (20.1%) | 4 (5.3%) | 25 (9.5%) |
| KRAS-WT | 178 | 59 | 116 |
| KRAS-mutated | 86 (32.6%) | 16 (21.3%) | 48 (29%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lender, Y.; Givton, O.; Bornshten, R.; Azar, M.; Moscona, R.; Yarden, Y.; Rubin, E. Immune Clustering Reveals Molecularly Distinct Subtypes of Lung Adenocarcinoma. Biomedicines 2025, 13, 849. https://doi.org/10.3390/biomedicines13040849
Lender Y, Givton O, Bornshten R, Azar M, Moscona R, Yarden Y, Rubin E. Immune Clustering Reveals Molecularly Distinct Subtypes of Lung Adenocarcinoma. Biomedicines. 2025; 13(4):849. https://doi.org/10.3390/biomedicines13040849
Chicago/Turabian StyleLender, Yan, Ofer Givton, Ruth Bornshten, Meitar Azar, Roy Moscona, Yosef Yarden, and Eitan Rubin. 2025. "Immune Clustering Reveals Molecularly Distinct Subtypes of Lung Adenocarcinoma" Biomedicines 13, no. 4: 849. https://doi.org/10.3390/biomedicines13040849
APA StyleLender, Y., Givton, O., Bornshten, R., Azar, M., Moscona, R., Yarden, Y., & Rubin, E. (2025). Immune Clustering Reveals Molecularly Distinct Subtypes of Lung Adenocarcinoma. Biomedicines, 13(4), 849. https://doi.org/10.3390/biomedicines13040849