

Exploring Immune-Related Ferroptosis Genes in Thyroid Cancer: A Comprehensive Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset Acquisition and Preprocessing
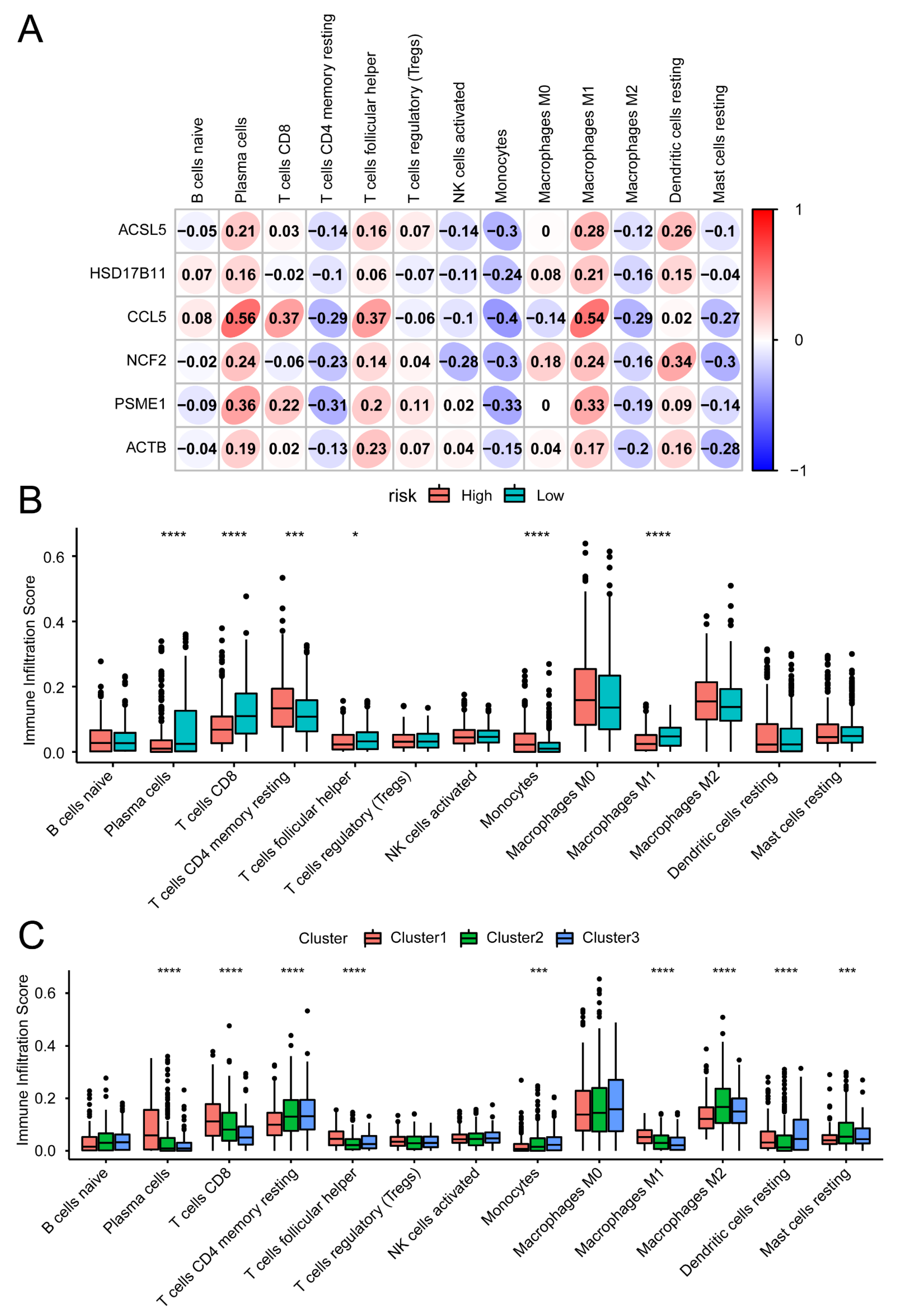
2.2. Estimated Immune Cell Type Fractions
2.3. WGCNA
2.4. Differential Expression Analysis
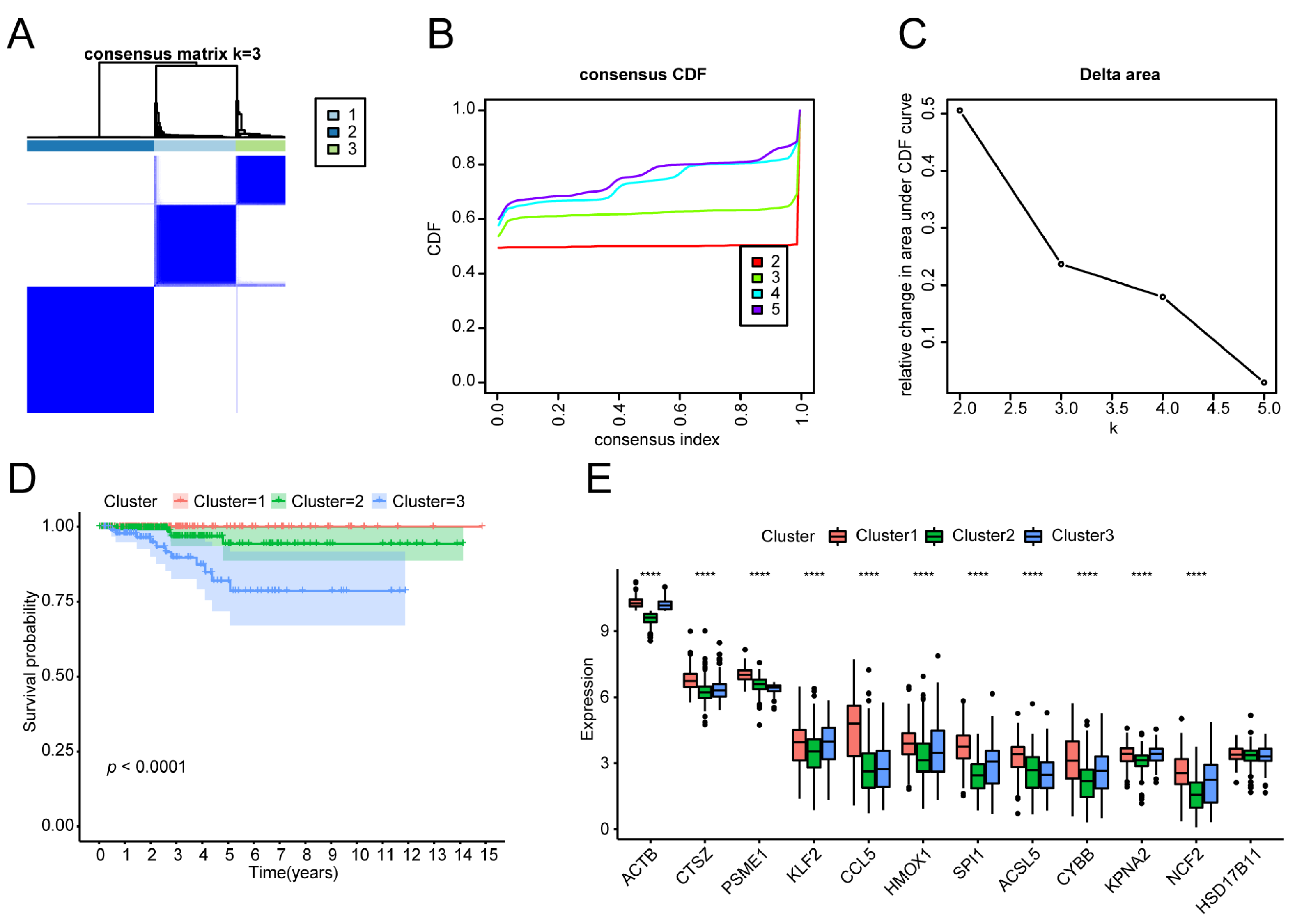
2.5. Molecular Typing
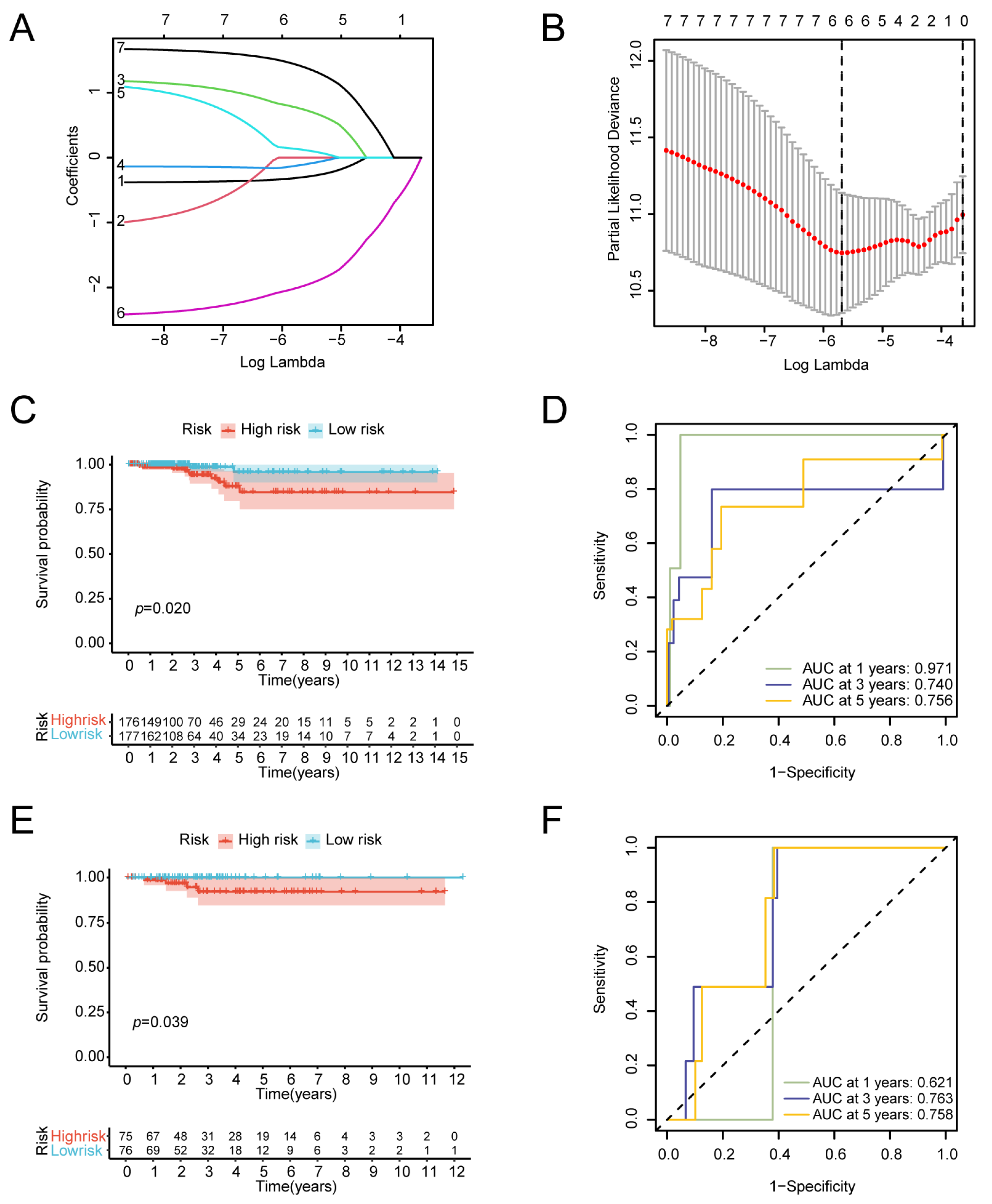
2.6. Construction of the Prognostic Model
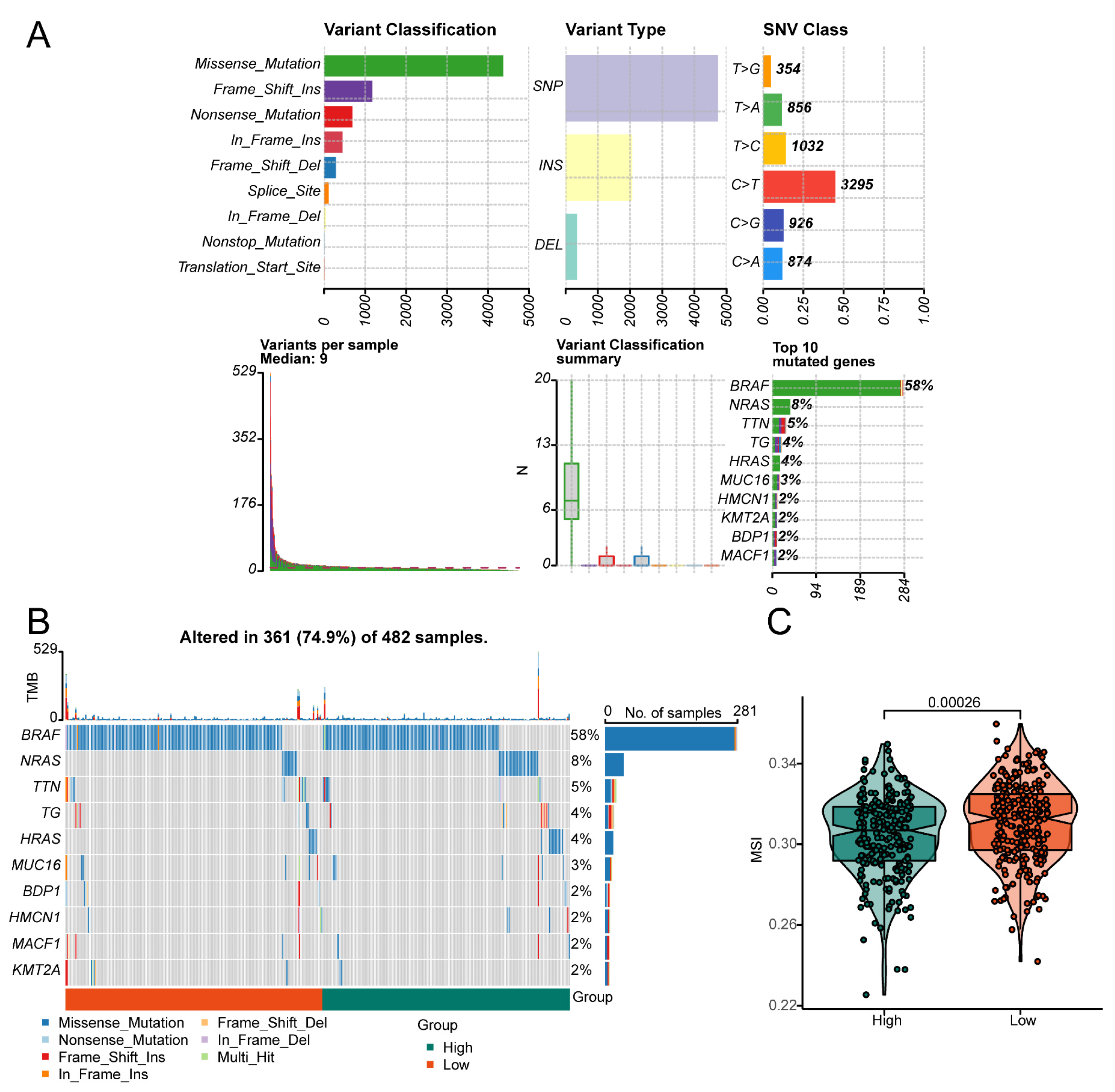
2.7. Mutation Analysis
2.8. Construction of the Clinical Prediction Model
2.9. GSVA Analysis
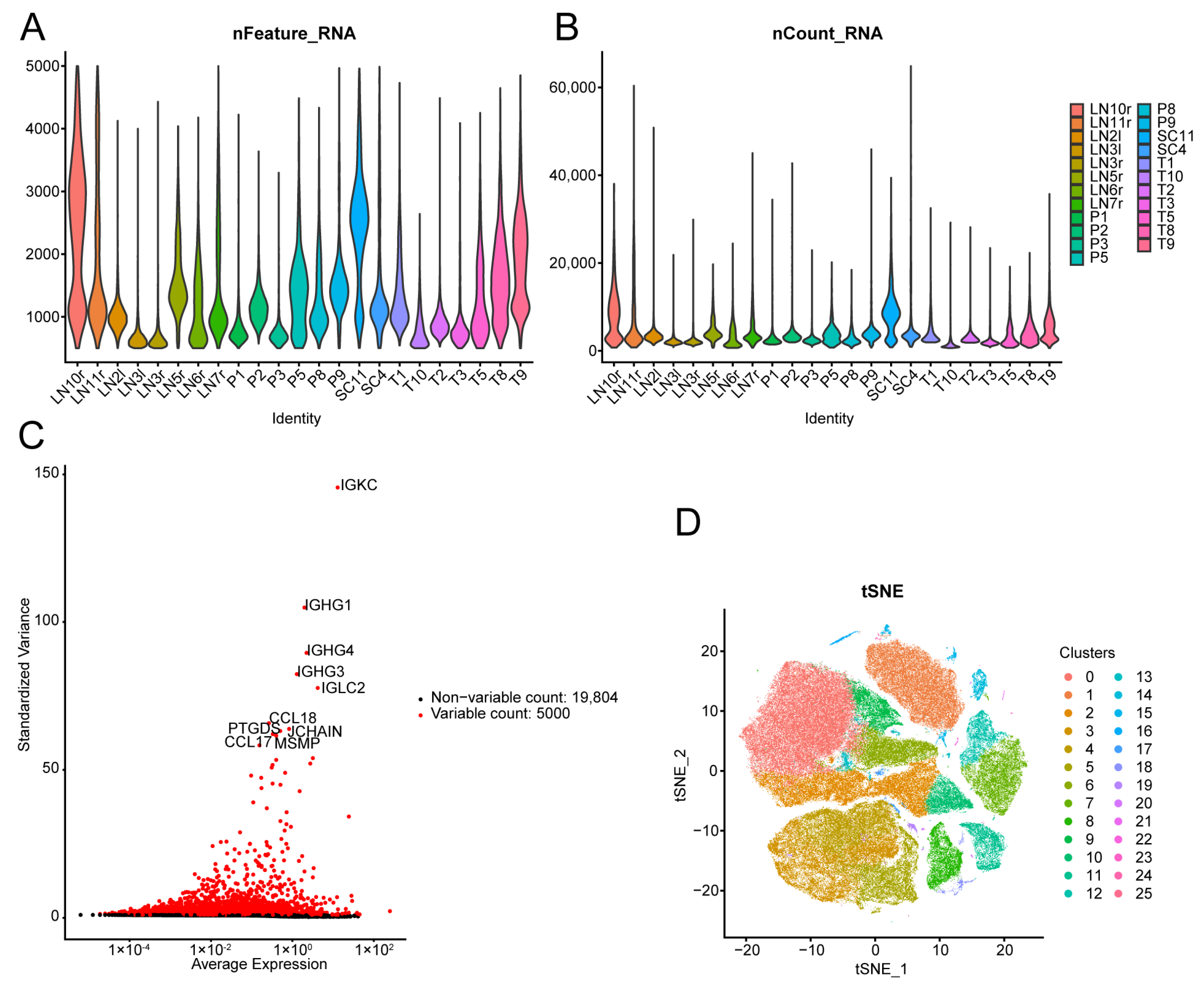
2.10. Quality Control of Single-Cell Data
2.11. Cluster Analysis and Annotation
2.12. Cel Communication
2.13. RNA Sequencing (RNA-Seq) Analysis
2.14. Quantitative Real-Time PCR (qRT–PCR)
2.14.1. Clinical Samples
2.14.2. qRT–PCR
2.15. Statistical Methods
3. Results
3.1. Identification of Immune-Related Modules Using WGCNA
3.2. Expression Analysis of IRFGs
3.3. Molecular Typing of the THCA Tumour Samples
3.4. Construction of the Prognostic Gene Model
3.5. Immune Signature Correlation Analysis
3.6. Effect of the Risk Score on Genomic Alterations
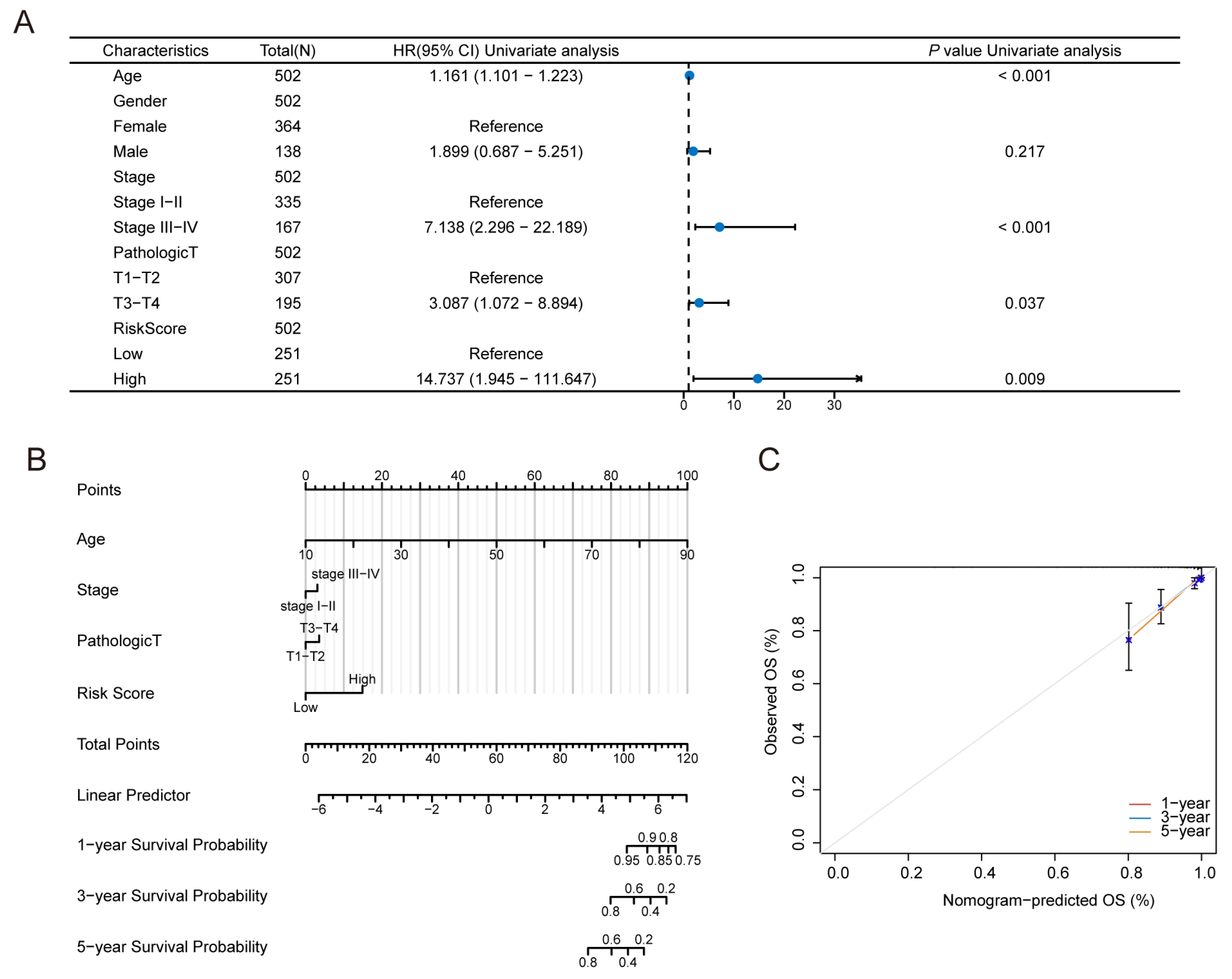
3.7. Construction of the Predictive Nomogram Model
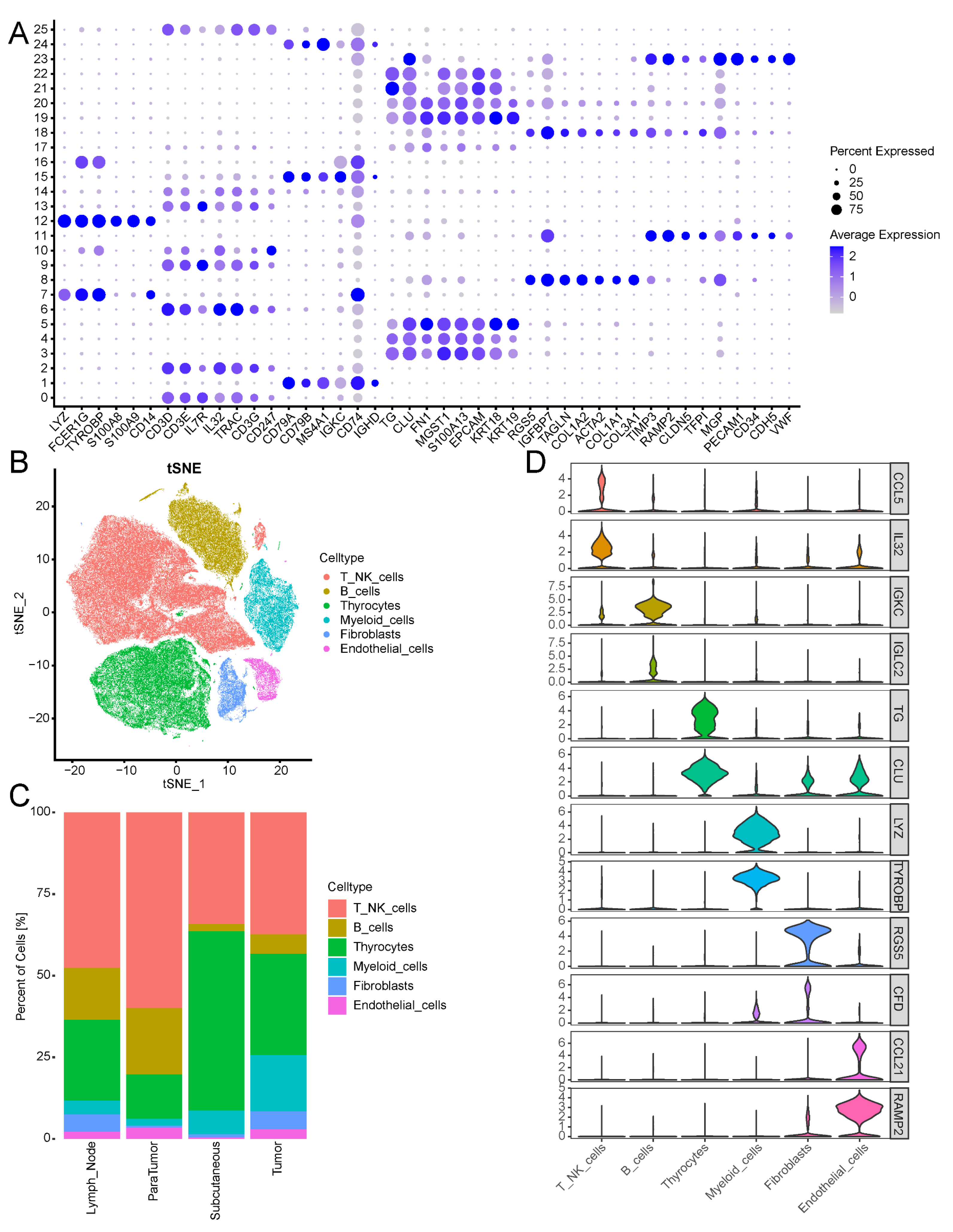
3.8. Single-Cell Data Revealed the Cellular Heterogeneity of Thyroid Cancer Cells
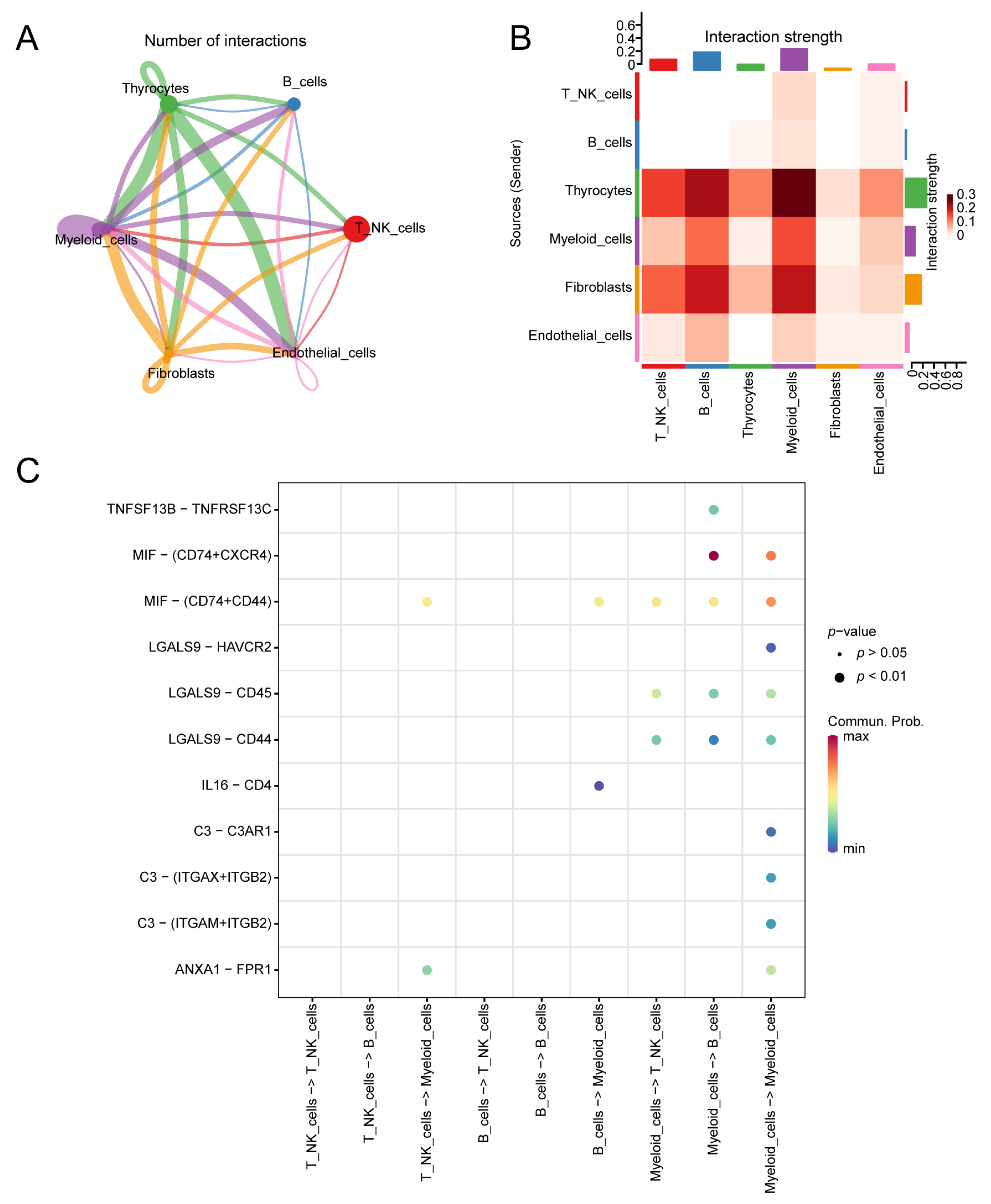
3.9. Cell Communication
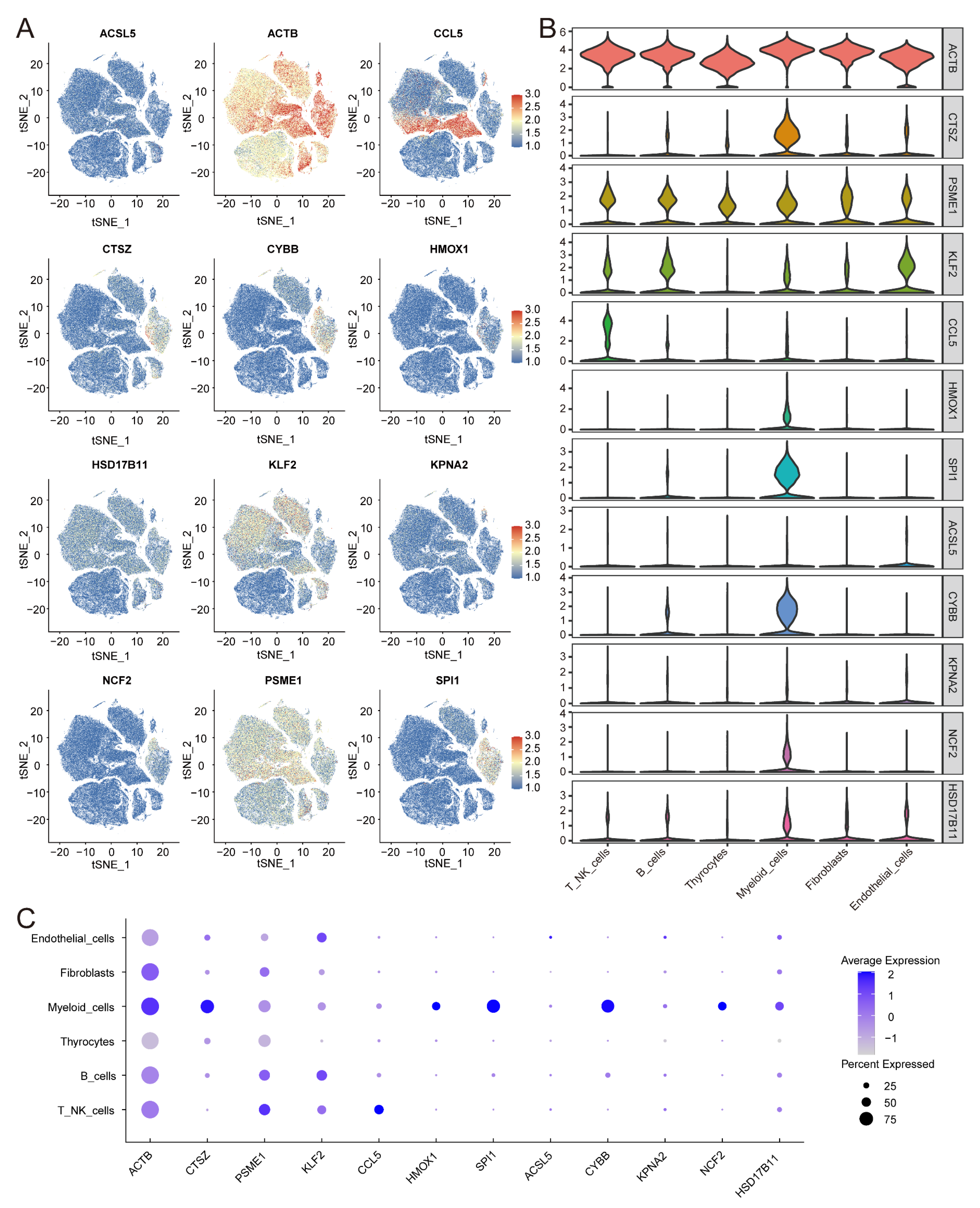
3.10. Expression of IRFGs in Single-Cell Datasets
3.11. Expression of IRFGs Genes in the Tumour Tissues
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, J.; Gosnell, J.E.; Roman, S.A. Geographic Influences in the Global Rise of Thyroid Cancer. Nat. Rev. Endocrinol. 2020, 16, 17–29. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Adolescents and Young Adults with Cancer: Types of Cancer in Adolescents and Young Adults (AYAs). Available online: https://www.cancer.gov/types/aya#types-of-cancer-in-adolescents-and-young-adults-ayas (accessed on 10 October 2024).
- Boucai, L.; Zafereo, M.; Cabanillas, M.E. Thyroid Cancer: A Review. JAMA 2024, 331, 425. [Google Scholar] [CrossRef] [PubMed]
- Filetti, S.; Durante, C.; Hartl, D.; Leboulleux, S.; Locati, L.D.; Newbold, K.; Papotti, M.G.; Berruti, A. Thyroid Cancer: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2019, 30, 1856–1883. [Google Scholar] [CrossRef] [PubMed]
- Schlumberger, M.; Brose, M.; Elisei, R.; Leboulleux, S.; Luster, M.; Pitoia, F.; Pacini, F. Definition and Management of Radioactive Iodine-Refractory Differentiated Thyroid Cancer. Lancet Diabetes Endocrinol. 2014, 2, 356–358. [Google Scholar] [CrossRef]
- Brose, M.S.; Smit, J.; Capdevila, J.; Elisei, R.; Nutting, C.; Pitoia, F.; Robinson, B.; Schlumberger, M.; Shong, Y.K.; Takami, H. Regional Approaches to the Management of Patients with Advanced, Radioactive Iodine-Refractory Differentiated Thyroid Carcinoma. Expert Rev. Anticancer. Ther. 2012, 12, 1137–1147. [Google Scholar] [CrossRef]
- Zhang, Q.-Y.; Ye, X.-P.; Zhou, Z.; Zhu, C.-F.; Li, R.; Fang, Y.; Zhang, R.-J.; Li, L.; Liu, W.; Wang, Z.; et al. Lymphocyte Infiltration and Thyrocyte Destruction Are Driven by Stromal and Immune Cell Components in Hashimoto’s Thyroiditis. Nat. Commun. 2022, 13, 775. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Hernández, R.; Sánchez De La Blanca, N.; Sacristán-Gómez, P.; Serrano-Somavilla, A.; Muñoz De Nova, J.L.; Sánchez Cabo, F.; Heyn, H.; Sampedro-Núñez, M.; Marazuela, M. Unraveling the Molecular Architecture of Autoimmune Thyroid Diseases at Spatial Resolution. Nat. Commun. 2024, 15, 5895. [Google Scholar] [CrossRef]
- Subbiah, V.; Kreitman, R.J.; Wainberg, Z.A.; Gazzah, A.; Lassen, U.; Stein, A.; Wen, P.Y.; Dietrich, S.; De Jonge, M.J.A.; Blay, J.-Y.; et al. Dabrafenib plus Trametinib in BRAFV600E-Mutated Rare Cancers: The Phase 2 ROAR Trial. Nat. Med. 2023, 29, 1103–1112. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, J.R.; Dadu, R.; Busaidy, N.L.; Xu, L.; Learned, K.O.; Chasen, N.N.; Vu, T.; Maniakas, A.; Eguia, A.A.; et al. Surgery After BRAF-Directed Therapy Is Associated with Improved Survival in BRAFV600E Mutant Anaplastic Thyroid Cancer: A Single-Center Retrospective Cohort Study. Thyroid 2023, 33, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Liu, Y.; Gu, W. P53 in Ferroptosis Regulation: The New Weapon for the Old Guardian. Cell Death Differ. 2022, 29, 895–910. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Li, H.; Graham, E.T.; Deik, A.A.; Eaton, J.K.; Wang, W.; Sandoval-Gomez, G.; Clish, C.B.; Doench, J.G.; Schreiber, S.L. Cytochrome P450 Oxidoreductase Contributes Tophospholipid Peroxidation in Ferroptosis. Nat. Chem. Biol. 2020, 16, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Floros, K.V.; Cai, J.; Jacob, S.; Kurupi, R.; Fairchild, C.K.; Shende, M.; Coon, C.M.; Powell, K.M.; Belvin, B.R.; Hu, B.; et al. MYCN-Amplified Neuroblastoma Is Addicted to Iron and Vulnerable to Inhibition of the System Xc-/Glutathione Axis. Cancer Res. 2021, 81, 1896–1908. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Daher, B.; Parks, S.K.; Durivault, J.; Cormerais, Y.; Baidarjad, H.; Tambutte, E.; Pouysségur, J.; Vučetić, M. Genetic Ablation of the Cystine Transporter xCT in PDAC Cells Inhibits mTORC1, Growth, Survival, and Tumor Formation via Nutrient and Oxidative Stresses. Cancer Res. 2019, 79, 3877–3890. [Google Scholar] [CrossRef] [PubMed]
- Arensman, M.D.; Yang, X.S.; Leahy, D.M.; Toral-Barza, L.; Mileski, M.; Rosfjord, E.C.; Wang, F.; Deng, S.; Myers, J.S.; Abraham, R.T.; et al. Cystine–Glutamate Antiporter xCT Deficiency Suppresses Tumor Growth While Preserving Antitumor Immunity. Proc. Natl. Acad. Sci. USA 2019, 116, 9533–9542. [Google Scholar] [CrossRef]
- Gong, Z.; Xue, L.; Li, H.; Fan, S.; Van Hasselt, C.A.; Li, D.; Zeng, X.; Tong, M.C.F.; Chen, G.G. Targeting Nrf2 to Treat Thyroid Cancer. Biomed. Pharmacother. 2024, 173, 116324. [Google Scholar] [CrossRef]
- Huang, T.; Guan, S.; Wang, C. CERS6-AS1 Facilitates Oncogenesis and Restrains Ferroptosis in Papillary Thyroid Carcinoma by Serving as a ceRNA through miR-497-5p/LASP1 Axis. Ann. Clin. Lab. Sci. 2022, 52, 426–438. [Google Scholar]
- Paget, S. The Distribution of Secondary Growths in Cancer of the Breast. 1889. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar] [PubMed]
- Liao, P.; Wang, W.; Wang, W.; Kryczek, I.; Li, X.; Bian, Y.; Sell, A.; Wei, S.; Grove, S.; Johnson, J.K.; et al. CD8+ T Cells and Fatty Acids Orchestrate Tumor Ferroptosis and Immunity via ACSL4. Cancer Cell 2022, 40, 365–378.e6. [Google Scholar] [CrossRef]
- Wang, W.; Green, M.; Choi, J.E.; Gijón, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8+ T Cells Regulate Tumour Ferroptosis during Cancer Immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Kapralov, A.A.; Yang, Q.; Dar, H.H.; Tyurina, Y.Y.; Anthonymuthu, T.S.; Kim, R.; St. Croix, C.M.; Mikulska-Ruminska, K.; Liu, B.; Shrivastava, I.H.; et al. Redox Lipid Reprogramming Commands Susceptibility of Macrophages and Microglia to Ferroptotic Death. Nat. Chem. Biol. 2020, 16, 278–290. [Google Scholar] [CrossRef]
- Drijvers, J.M.; Gillis, J.E.; Muijlwijk, T.; Nguyen, T.H.; Gaudiano, E.F.; Harris, I.S.; LaFleur, M.W.; Ringel, A.E.; Yao, C.-H.; Kurmi, K.; et al. Pharmacologic Screening Identifies Metabolic Vulnerabilities of CD8+ T Cells. Cancer Immunol. Res. 2021, 9, 184–199. [Google Scholar] [CrossRef]
- Chen, C.; Wang, L.; Cui, X.-F.; Shang, X.-Y.; Bai, S.-H.; Li, L.; Wang, N.; Han, Z.-G. SCARA5 Deficiency Inhibits Ferroptosis via Regulating Iron Homeostasis and Results in Sorafenib Resistance in Hepatocellular Carcinoma. Cell. Signal. 2025, 129, 111656. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Autophagy Promotes Ferroptosis by Degradation of Ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Xiao, Y.; Ding, J.-H.; Jin, X.; Ma, D.; Li, D.-Q.; Shi, J.-X.; Huang, W.; Wang, Y.-P.; Jiang, Y.-Z.; et al. Ferroptosis Heterogeneity in Triple-Negative Breast Cancer Reveals an Innovative Immunotherapy Combination Strategy. Cell Metab. 2023, 35, 84–100.e8. [Google Scholar] [CrossRef] [PubMed]
- Pu, W.; Shi, X.; Yu, P.; Zhang, M.; Liu, Z.; Tan, L.; Han, P.; Wang, Y.; Ji, D.; Gan, H.; et al. Single-Cell Transcriptomic Analysis of the Tumor Ecosystems Underlying Initiation and Progression of Papillary Thyroid Carcinoma. Nat. Commun. 2021, 12, 6058. [Google Scholar] [CrossRef]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar] [CrossRef]
- Zhou, C.; Feng, M.; Chen, Y.; Lv, S.; Zhang, Y.; Chen, J.; Zhang, R.; Huang, X. Unraveling Immunotherapeutic Targets for Endometriosis: A Transcriptomic and Single-Cell Analysis. Front. Immunol. 2023, 14, 1288263. [Google Scholar] [CrossRef]
- Chen, W.; Chen, Y.; Wu, L.; Gao, Y.; Zhu, H.; Li, Y.; Ji, X.; Wang, Z.; Wang, W.; Han, L.; et al. Identification of Cell Death-Related Biomarkers and Immune Infiltration in Ischemic Stroke between Male and Female Patients. Front. Immunol. 2023, 14, 1164742. [Google Scholar] [CrossRef]
- Zhou, N.; Bao, J. FerrDb: A Manually Curated Resource for Regulators and Markers of Ferroptosis and Ferroptosis-Disease Associations. Database 2020, 2020, baaa021. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- The Gene Ontology Consortium The Gene Ontology Resource: 20 Years and Still GOing Strong. Nucleic Acids Res. 2019, 47, D330–D338. [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A Class Discovery Tool with Confidence Assessments and Item Tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef]
- Berlyand, Y.; Weintraub, D.; Xie, S.X.; Mellis, I.A.; Doshi, J.; Rick, J.; McBride, J.; Davatzikos, C.; Shaw, L.M.; Hurtig, H.; et al. An Alzheimer’s Disease-Derived Biomarker Signature Identifies Parkinson’s Disease Patients with Dementia. PLoS ONE 2016, 11, e0147319. [Google Scholar] [CrossRef] [PubMed]
- Ünlü, R.; Xanthopoulos, P. Estimating the Number of Clusters in a Dataset via Consensus Clustering. Expert Syst. Appl. 2019, 125, 33–39. [Google Scholar] [CrossRef]
- Moro, F.; Albanese, M.; Boldrini, L.; Chiappa, V.; Lenkowicz, J.; Bertolina, F.; Mascilini, F.; Moroni, R.; Gambacorta, M.A.; Raspagliesi, F.; et al. Developing and Validating Ultrasound-Based Radiomics Models for Predicting High-Risk Endometrial Cancer. Ultrasound Obs. Gynecol. 2022, 60, 256–268. [Google Scholar] [CrossRef]
- Hou, X.-Z.; Wu, Q.; Lv, Q.-Y.; Yang, Y.-T.; Li, L.-L.; Ye, X.-J.; Yang, C.-Y.; Lv, Y.-F.; Wang, S.-H. Development and External Validation of a Risk Prediction Model for Depression in Patients with Coronary Heart Disease. J. Affect. Disord. 2024, 367, 137–147. [Google Scholar] [CrossRef]
- Vijayalakshmi, K.; Shankar, V.; Bain, R.M.; Nolley, R.; Sonn, G.A.; Kao, C.-S.; Zhao, H.; Tibshirani, R.; Zare, R.N.; Brooks, J.D. Identification of Diagnostic Metabolic Signatures in Clear Cell Renal Cell Carcinoma Using Mass Spectrometry Imaging. Int. J. Cancer 2020, 147, 256–265. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression Shrinkage and Selection Via the Lasso. J. R. Stat. Soc. Ser. B Stat. Methodol. 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Yan, C.; Niu, Y.; Ma, L.; Tian, L.; Ma, J. System Analysis Based on the Cuproptosis-Related Genes Identifies LIPT1 as a Novel Therapy Target for Liver Hepatocellular Carcinoma. J. Transl. Med. 2022, 20, 452. [Google Scholar] [CrossRef]
- George, B.; Seals, S.; Aban, I. Survival Analysis and Regression Models. J. Nucl. Cardiol. 2014, 21, 686–694. [Google Scholar] [CrossRef]
- Blanche, P.; Dartigues, J.; Jacqmin-Gadda, H. Estimating and Comparing Time-dependent Areas under Receiver Operating Characteristic Curves for Censored Event Times with Competing Risks. Stat. Med. 2013, 32, 5381–5397. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.-C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and Comprehensive Analysis of Somatic Variants in Cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene Set Variation Analysis for Microarray and RNA-Seq Data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Andorf, S.; Gomes, L.; Dunn, P.; Schaefer, H.; Pontius, J.; Berger, P.; Desborough, V.; Smith, T.; Campbell, J.; et al. ImmPort: Disseminating Data to the Public for the Future of Immunology. Immunol. Res. 2014, 58, 234–239. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated Analysis of Multimodal Single-Cell Data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.-H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and Analysis of Cell-Cell Communication Using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef]
- Han, J.; Chen, M.; Wang, Y.; Gong, B.; Zhuang, T.; Liang, L.; Qiao, H. Identification of Biomarkers Based on Differentially Expressed Genes in Papillary Thyroid Carcinoma. Sci. Rep. 2018, 8, 9912. [Google Scholar] [CrossRef]
- Zhang, L.; Li, S.; Choi, Y.-L.; Lee, J.; Gong, Z.; Liu, X.; Pei, Y.; Jiang, A.; Ye, M.; Mao, M.; et al. Systematic Identification of Cancer-Related Long Noncoding RNAs and Aberrant Alternative Splicing of Quintuple-Negative Lung Adenocarcinoma through RNA-Seq. Lung Cancer 2017, 109, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; He, M.; Lin, R.; Zhan, M.; Xu, S.; Huang, X.; Xu, C.; Chen, W.; Yao, Y.; Mohan, M.; et al. PLEK2 Promotes Gallbladder Cancer Invasion and Metastasis through EGFR/CCL2 Pathway. J. Exp. Clin. Cancer Res. 2019, 38, 247. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B.; André, T.; Atreya, C.E.; Schellens, J.H.M.; Yoshino, T.; Bendell, J.C.; Hollebecque, A.; McRee, A.J.; Siena, S.; Middleton, G.; et al. Combined BRAF, EGFR, and MEK Inhibition in Patients with BRAFV600E-Mutant Colorectal Cancer. Cancer Discov. 2018, 8, 428–443. [Google Scholar] [CrossRef]
- Chen, X.; Chen, X.; Xie, W.; Ge, H.; He, H.; Zhang, A.; Zheng, J. BRAF-Activated ARSI Suppressed EREG-Mediated Ferroptosis to Promote BRAFV600E (Mutant) Papillary Thyroid Carcinoma Progression and Sorafenib Resistance. Int. J. Biol. Sci. 2025, 21, 128–142. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Yang, S.; Zhang, Y.; Li, J.; Wang, M.; Yeerkenbieke, P.; Liao, Q.; Liu, Q. Modulating Ferroptosis Sensitivity: Environmental and Cellular Targets within the Tumor Microenvironment. J. Exp. Clin. Cancer Res. 2024, 43, 19. [Google Scholar] [CrossRef]
- Lupica-Tondo, G.L.; Arner, E.N.; Mogilenko, D.A.; Voss, K. Immunometabolism of Ferroptosis in the Tumor Microenvironment. Front. Oncol. 2024, 14, 1441338. [Google Scholar] [CrossRef]
- Simchen, C.; Lehmann, I.; Sittig, D.; Steinert, M.; Aust, G. Expression and Regulation of Regulated on Activation, Normal T Cells Expressed and Secreted in Thyroid Tissue of Patients with Graves’ Disease and Thyroid Autonomy and in Thyroid-Derived Cell Populations. J. Clin. Endocrinol. Metab. 2000, 85, 4758–4764. [Google Scholar] [PubMed]
- Sekhar, K.R.; Hanna, D.N.; Cyr, S.; Baechle, J.J.; Kuravi, S.; Balusu, R.; Rathmell, K.; Baregamian, N. Glutathione Peroxidase 4 Inhibition Induces Ferroptosis and mTOR Pathway Suppression in Thyroid Cancer. Sci. Rep. 2022, 12, 19396. [Google Scholar] [CrossRef]
- Wu, J.; Liang, J.; Liu, R.; Lv, T.; Fu, K.; Jiang, L.; Ma, W.; Pan, Y.; Tan, Z.; Liu, Q.; et al. Autophagic Blockade Potentiates Anlotinib-Mediated Ferroptosis in Anaplastic Thyroid Cancer. Endocr.-Relat. Cancer 2023, 30, e230036. [Google Scholar] [CrossRef]
- Hou, T.; Tian, Y.; Cao, Z.; Zhang, J.; Feng, T.; Tao, W.; Sun, H.; Wen, H.; Lu, X.; Zhu, Q.; et al. Cytoplasmic SIRT6-Mediated ACSL5 Deacetylation Impedes Nonalcoholic Fatty Liver Disease by Facilitating Hepatic Fatty Acid Oxidation. Mol. Cell 2022, 82, 4099–4115.e9. [Google Scholar] [CrossRef]
- Zhang, Q.; Xiong, L.; Wei, T.; Liu, Q.; Yan, L.; Chen, J.; Dai, L.; Shi, L.; Zhang, W.; Yang, J.; et al. Hypoxia-Responsive PPARGC1A/BAMBI/ACSL5 Axis Promotes Progression and Resistance to Lenvatinib in Hepatocellular Carcinoma. Oncogene 2023, 42, 1509–1523. [Google Scholar] [CrossRef]
- Li, M.; Sun, X.; Zhao, J.; Xia, L.; Li, J.; Xu, M.; Wang, B.; Guo, H.; Yu, C.; Gao, Y.; et al. CCL5 Deficiency Promotes Liver Repair by Improving Inflammation Resolution and Liver Regeneration through M2 Macrophage Polarization. Cell Mol. Immunol. 2020, 17, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Santana-Hernández, S.; Suarez-Olmos, J.; Servitja, S.; Berenguer-Molins, P.; Costa-Garcia, M.; Comerma, L.; Rea, A.; Perera-Bel, J.; Menendez, S.; Arpí, O.; et al. NK Cell-Triggered CCL5/IFNγ-CXCL9/10 Axis Underlies the Clinical Efficacy of Neoadjuvant Anti-HER2 Antibodies in Breast Cancer. J. Exp. Clin. Cancer Res. 2024, 43, 10. [Google Scholar] [CrossRef]
- Cabo, M.; Santana-Hernández, S.; Costa-Garcia, M.; Rea, A.; Lozano-Rodríguez, R.; Ataya, M.; Balaguer, F.; Juan, M.; Ochoa, M.C.; Menéndez, S.; et al. CD137 Costimulation Counteracts TGFβ Inhibition of NK-Cell Antitumor Function. Cancer Immunol. Res. 2021, 9, 1476–1490. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.; Koo, J.S. Cell Component and Function of Tumor Microenvironment in Thyroid Cancer. Int. J. Mol. Sci. 2022, 23, 12578. [Google Scholar] [CrossRef]
- Wang, T.; Shi, J.; Li, L.; Zhou, X.; Zhang, H.; Zhang, X.; Wang, Y.; Liu, L.; Sheng, L. Single-Cell Transcriptome Analysis Reveals Inter-Tumor Heterogeneity in Bilateral Papillary Thyroid Carcinoma. Front. Immunol. 2022, 13, 840811. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, C.; Shah, S.; Lu, W.-C.; Ray, H.; Wang, J.; Hockaden, N.; Sandusky, G.; Nephew, K.P.; Lu, X.; Cao, S.; et al. HSF1 Inhibits Antitumor Immune Activity in Breast Cancer by Suppressing CCL5 to Block CD8+ T-Cell Recruitment. Cancer Res. 2024, 84, 276–290. [Google Scholar] [CrossRef]
- Tarazona-Santos, E.; Machado, M.; Magalhaes, W.C.S.; Chen, R.; Lyon, F.; Burdett, L.; Crenshaw, A.; Fabbri, C.; Pereira, L.; Pinto, L.; et al. Evolutionary Dynamics of the Human NADPH Oxidase Genes CYBB, CYBA, NCF2, and NCF4: Functional Implications. Mol. Biol. Evol. 2013, 30, 2157–2167. [Google Scholar] [CrossRef]
- Zhong, F.; Zeng, Y.; Yan, Y.; Guo, L.; Guo, Q.; Liu, W.; Liu, C. Comprehensive Multi-Omics Analysis of the Prognostic Value and Immune Signature of NCF2 in Pan-Cancer and Its Relationship with Acute Myeloid Leukemia. Int. Immunopharmacol. 2024, 143, 113364. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Zhang, J.; Kuang, S.; Li, Z.; Zhao, H.; Wu, J.; Liu, M.; Wang, L. Role of NCF2 as a Potential Prognostic Factor and Immune Infiltration Indicator in Hepatocellular Carcinoma. Cancer Med. 2023, 12, 8991–9004. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.; Erlander, M.; Leturcq, D.; Peterson, P.A.; Früh, K.; Yang, Y. Characterization of the Proteasome Regulator PA28. J. Biol. Chem. 1996, 271, 18237–18242. [Google Scholar] [CrossRef]
- Ravi, A.; Hellmann, M.D.; Arniella, M.B.; Holton, M.; Freeman, S.S.; Naranbhai, V.; Stewart, C.; Leshchiner, I.; Kim, J.; Akiyama, Y.; et al. Genomic and Transcriptomic Analysis of Checkpoint Blockade Response in Advanced Non-Small Cell Lung Cancer. Nat. Genet. 2023, 55, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Bednarz-Misa, I.; Neubauer, K.; Zacharska, E.; Kapturkiewicz, B.; Krzystek-Korpacka, M. Whole Blood ACTB, B2M and GAPDH Expression Reflects Activity of Inflammatory Bowel Disease, Advancement of Colorectal Cancer, and Correlates with Circulating Inflammatory and Angiogenic Factors: Relevance for Real Time Quantitative PCR. Adv. Clin. Exp. Med. 2020, 29, 547–556. [Google Scholar] [CrossRef]
- Gu, Y.; Tang, S.; Wang, Z.; Cai, L.; Lian, H.; Shen, Y.; Zhou, Y. A Pan-Cancer Analysis of the Prognostic and Immunological Role of β-Actin (ACTB) in Human Cancers. Bioengineered 2021, 12, 6166–6185. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhang, L.; You, W.; Xu, J.; Dai, J.; Hua, D.; Zhang, R.; Yao, F.; Zhou, S.; Huang, W.; et al. PRDM1/BLIMP1 Induces Cancer Immune Evasion by Modulating the USP22-SPI1-PD-L1 Axis in Hepatocellular Carcinoma Cells. Nat. Commun. 2022, 13, 7677. [Google Scholar] [CrossRef] [PubMed]
- Jha, P.; Das, H. KLF2 in Regulation of NF-κB-Mediated Immune Cell Function and Inflammation. Int. J. Mol. Sci. 2017, 18, 2383. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.S.H.; Tay, T.K.Y.; Ong, E.H.W.; Fehlings, M.; Tan, D.S.-W.; Sukma, N.B.; Chen, E.X.; Sng, J.-H.; Yip, C.S.P.; Lim, K.H.; et al. Combinatorial Hypofractionated Radiotherapy and Pembrolizumab in Anaplastic Thyroid Cancer. Eur. Thyroid J. 2024, 13, e230144. [Google Scholar] [CrossRef]
- Dierks, C.; Seufert, J.; Aumann, K.; Ruf, J.; Klein, C.; Kiefer, S.; Rassner, M.; Boerries, M.; Zielke, A.; La Rosee, P.; et al. Combination of Lenvatinib and Pembrolizumab Is an Effective Treatment Option for Anaplastic and Poorly Differentiated Thyroid Carcinoma. Thyroid 2021, 31, 1076–1085. [Google Scholar] [CrossRef] [PubMed]
- Vedvyas, Y.; McCloskey, J.E.; Yang, Y.; Min, I.M.; Fahey, T.J.; Zarnegar, R.; Hsu, Y.-M.S.; Hsu, J.-M.; Van Besien, K.; Gaudet, I.; et al. Manufacturing and Preclinical Validation of CAR T Cells Targeting ICAM-1 for Advanced Thyroid Cancer Therapy. Sci. Rep. 2019, 9, 10634. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Liu, Z.; Gao, Z.; Luo, Y.; Li, F.; Yang, C.; Wang, T.; Meng, X.; Chen, H.; Li, J.; et al. KLF5 Inhibition Potentiates Anti-PD1 Efficacy by Enhancing CD8+ T-Cell-Dependent Antitumor Immunity. Theranostics 2023, 13, 1381–1400. [Google Scholar] [CrossRef]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and Transcriptomic Hallmarks of Poorly Differentiated and Anaplastic Thyroid Cancers. J. Clin. Investig. 2016, 126, 1052–1066. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Peng, F.; Xu, J.; Wang, G.; Zhao, Y. Increased Expression of GPX4 Promotes the Tumorigenesis of Thyroid Cancer by Inhibiting Ferroptosis and Predicts Poor Clinical Outcomes. Aging 2023, 15, 230–245. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Qian, C.; Cao, H.; Wang, Z.; Luo, T.; Liang, C. Upregulation of the Solute Carrier Family 7 Genes Is Indicative of Poor Prognosis in Papillary Thyroid Carcinoma. World J. Surg. Oncol. 2018, 16, 235. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, T.; Wu, X.; Tian, H.; Gao, P.; Chen, Q.; Chen, C.; Zhang, Y.; Wang, S.; Qi, X.; et al. Construction of a Ferroptosis-Based Prognostic Model for Breast Cancer Helps to Discriminate High/Low Risk Groups and Treatment Priority. Front. Immunol. 2023, 14, 1264206. [Google Scholar] [CrossRef]
- Ren, X.; Du, H.; Cheng, W.; Wang, Y.; Xu, Y.; Yan, S.; Gao, Y. Construction of a Ferroptosis-Related Eight Gene Signature for Predicting the Prognosis and Immune Infiltration of Thyroid Cancer. Front. Endocrinol. 2022, 13, 997873. [Google Scholar] [CrossRef]
- Goswami, S.; Anandhan, S.; Raychaudhuri, D.; Sharma, P. Myeloid Cell-Targeted Therapies for Solid Tumours. Nat. Rev. Immunol. 2023, 23, 106–120. [Google Scholar] [CrossRef]
- Lasser, S.A.; Ozbay Kurt, F.G.; Arkhypov, I.; Utikal, J.; Umansky, V. Myeloid-Derived Suppressor Cells in Cancer and Cancer Therapy. Nat. Rev. Clin. Oncol. 2024, 21, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Yakoub, M.A.; Chandler, A.; Christ, A.B.; Yang, G.; Ouerfelli, O.; Rajasekhar, V.K.; Yoshida, A.; Kondo, H.; Hata, T.; et al. CSF1/CSF1R Signaling Inhibitor Pexidartinib (PLX3397) Reprograms Tumor-Associated Macrophages and Stimulates T-Cell Infiltration in the Sarcoma Microenvironment. Mol. Cancer Ther. 2021, 20, 1388–1399. [Google Scholar] [CrossRef] [PubMed]
- Larroquette, M.; Guegan, J.-P.; Besse, B.; Cousin, S.; Brunet, M.; Le Moulec, S.; Le Loarer, F.; Rey, C.; Soria, J.-C.; Barlesi, F.; et al. Spatial Transcriptomics of Macrophage Infiltration in Non-Small Cell Lung Cancer Reveals Determinants of Sensitivity and Resistance to Anti-PD1/PD-L1 Antibodies. J. Immunother. Cancer 2022, 10, e003890. [Google Scholar] [CrossRef] [PubMed]
- Laureano, R.S.; Sprooten, J.; Vanmeerbeerk, I.; Borras, D.M.; Govaerts, J.; Naulaerts, S.; Berneman, Z.N.; Beuselinck, B.; Bol, K.F.; Borst, J.; et al. Trial Watch: Dendritic Cell (DC)-Based Immunotherapy for Cancer. OncoImmunology 2022, 11, 2096363. [Google Scholar] [CrossRef] [PubMed]
- Heras-Murillo, I.; Adán-Barrientos, I.; Galán, M.; Wculek, S.K.; Sancho, D. Dendritic Cells as Orchestrators of Anticancer Immunity and Immunotherapy. Nat. Rev. Clin. Oncol. 2024, 21, 257–277. [Google Scholar] [CrossRef] [PubMed]
- Greene, S.; Robbins, Y.; Mydlarz, W.K.; Huynh, A.P.; Schmitt, N.C.; Friedman, J.; Horn, L.A.; Palena, C.; Schlom, J.; Maeda, D.Y.; et al. Inhibition of MDSC Trafficking with SX-682, a CXCR1/2 Inhibitor, Enhances NK-Cell Immunotherapy in Head and Neck Cancer Models. Clin. Cancer Res. 2020, 26, 1420–1431. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ru, Z.; Li, S.; Wang, M.; Ni, Y.; Qiao, H. Exploring Immune-Related Ferroptosis Genes in Thyroid Cancer: A Comprehensive Analysis. Biomedicines 2025, 13, 903. https://doi.org/10.3390/biomedicines13040903
Ru Z, Li S, Wang M, Ni Y, Qiao H. Exploring Immune-Related Ferroptosis Genes in Thyroid Cancer: A Comprehensive Analysis. Biomedicines. 2025; 13(4):903. https://doi.org/10.3390/biomedicines13040903
Chicago/Turabian StyleRu, Zixuan, Siwei Li, Minnan Wang, Yanan Ni, and Hong Qiao. 2025. "Exploring Immune-Related Ferroptosis Genes in Thyroid Cancer: A Comprehensive Analysis" Biomedicines 13, no. 4: 903. https://doi.org/10.3390/biomedicines13040903
APA StyleRu, Z., Li, S., Wang, M., Ni, Y., & Qiao, H. (2025). Exploring Immune-Related Ferroptosis Genes in Thyroid Cancer: A Comprehensive Analysis. Biomedicines, 13(4), 903. https://doi.org/10.3390/biomedicines13040903