
Contribution of Sorting Nexin 3 in the Cytomegalovirus Assembly

 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Viruses and Infection Conditions
2.3. Antibodies and Reagents
2.4. Immunofluorescence and Confocal Microscopy
2.5. Image Analysis
2.6. Small Interfering RNA (siRNA)
2.7. Small Hairpin RNA (shRNA) and Cell Line Development
2.8. Flow Cytometry
2.8.1. Detection of Cells Infected with C3X-GFP-MCMV
2.8.2. Cell Surface M55/gB Protein Expression
2.9. Western Blot
2.10. RNA Preparation and Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
2.11. Plaque Assay and Virus Growth
2.12. Data Presentation and Statistical Analysis
3. Results
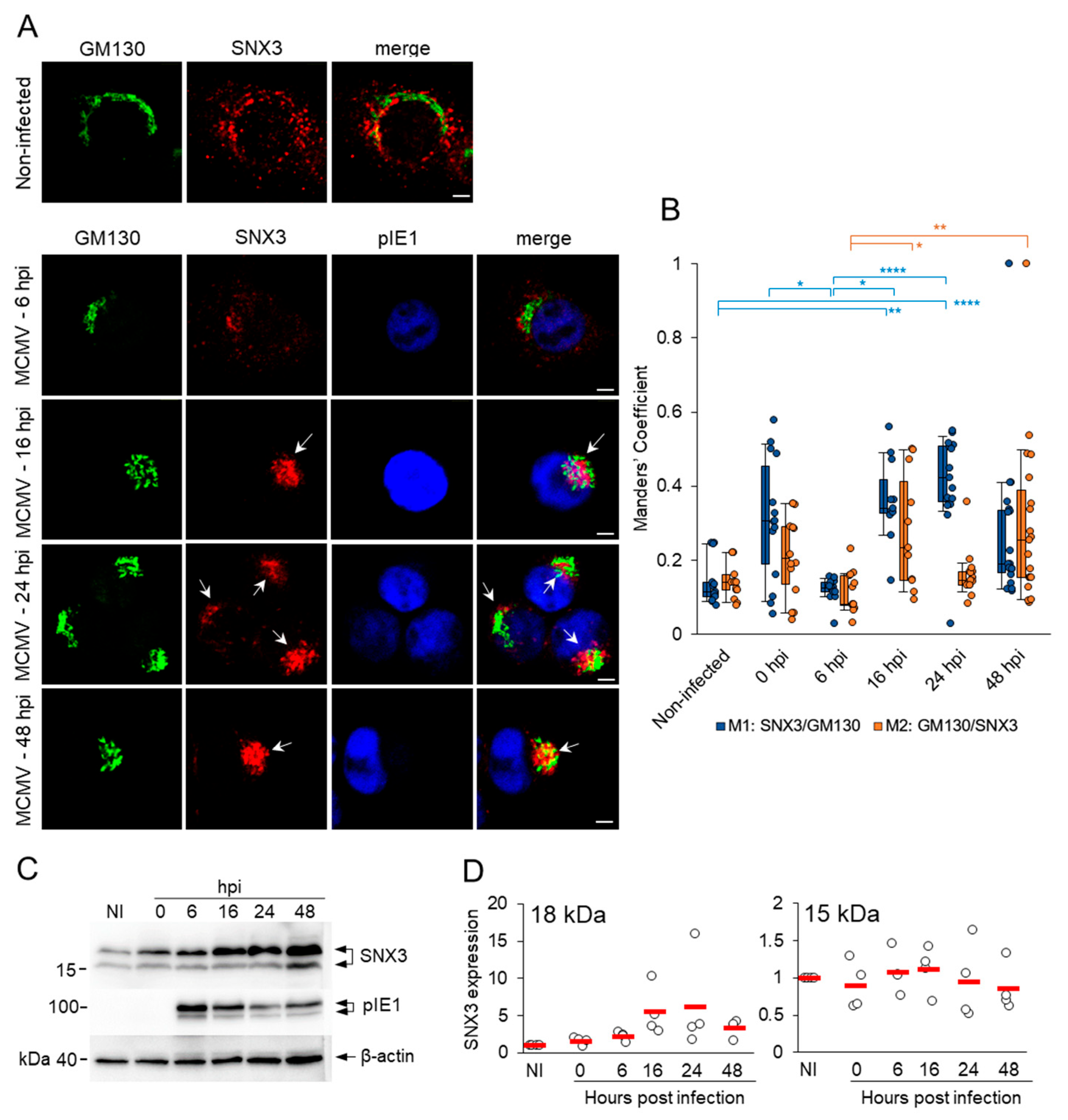
3.1. SNX3 Accumulates in the Inner Pre-AC and the Peripheral Membrane System of Infected Cells in the E Phase of Infection and in the AC During the L Phase
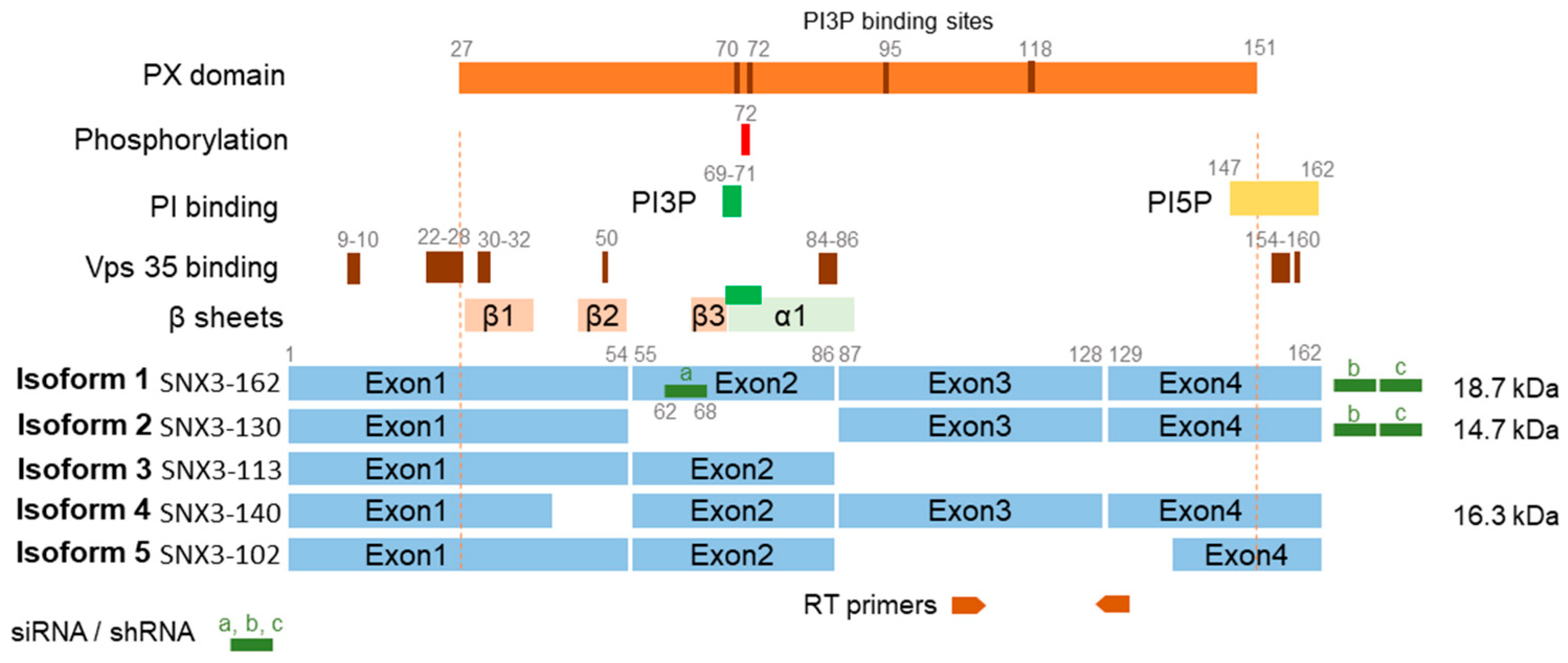
3.2. SNX3 Localizes at the SNX27-Dependent Rab10-PD

3.3. SNX3 Contributes to the Expansion of Rab10-PD
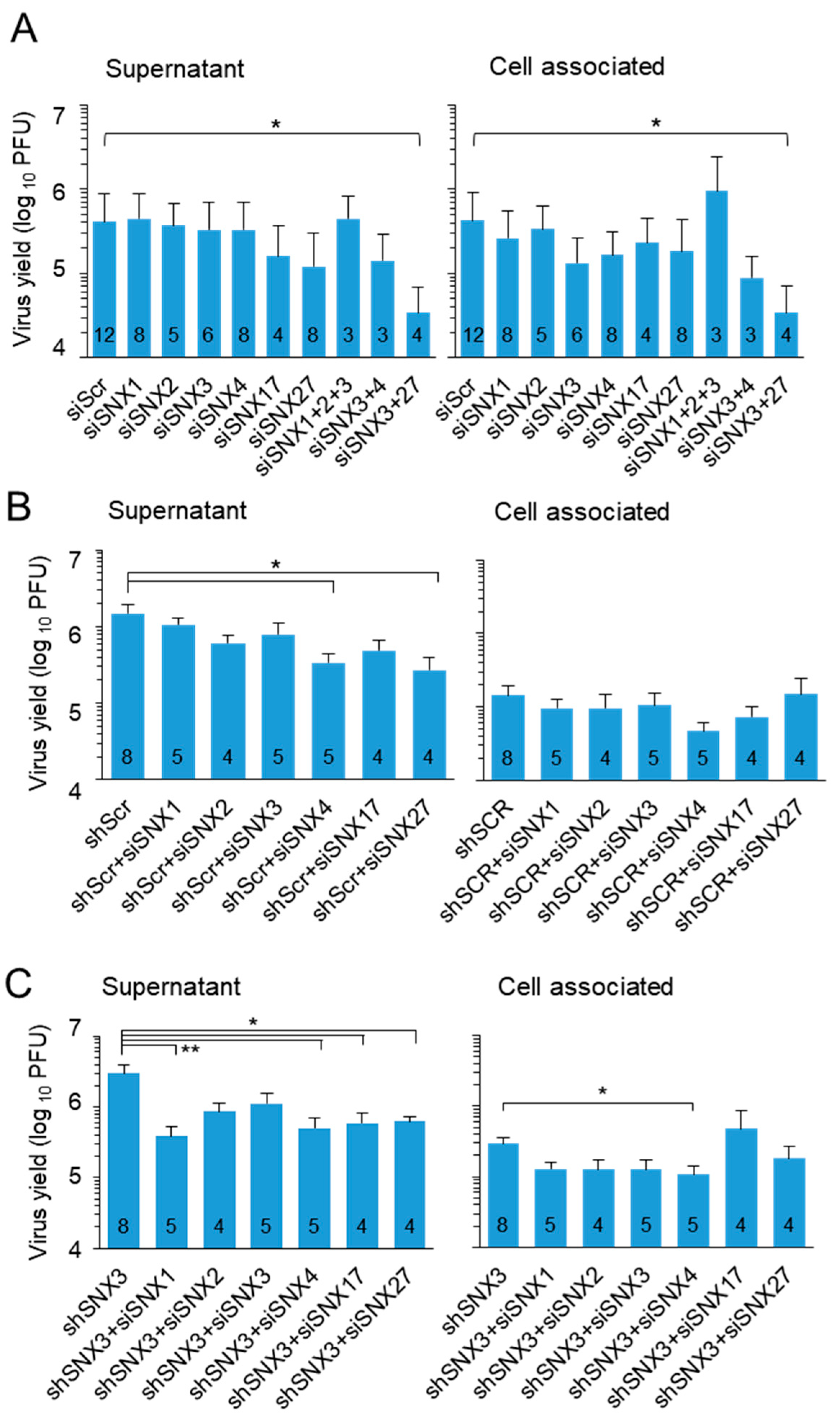
3.4. SNX3 Depletion Impairs Virion Assembly but Does Not Reduce Virus Yield
3.5. SNX3 Contributes to a Mechanism That Controls Virion Assembly and Egress
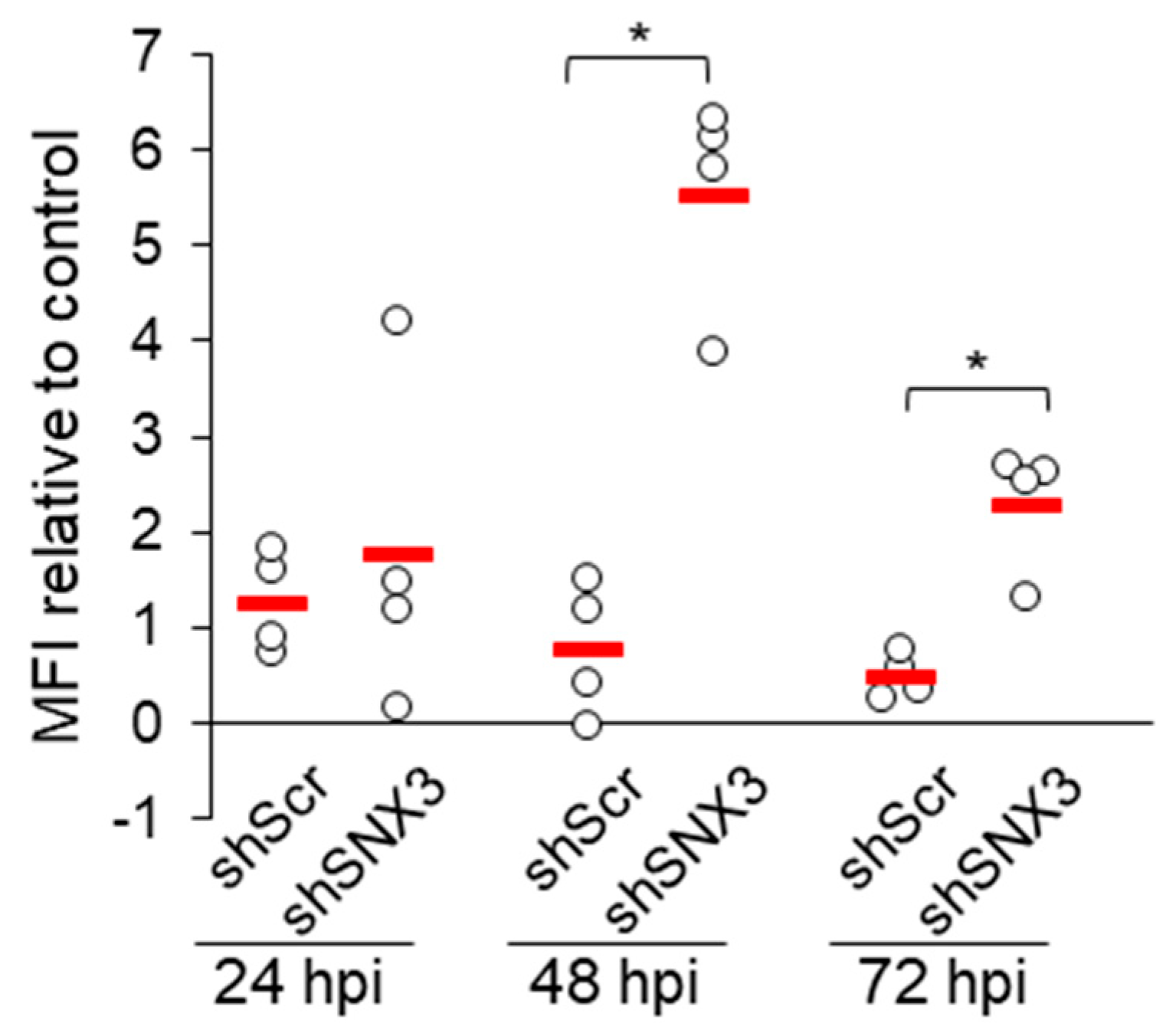
3.6. Depletion of SNX3 with shRNA Relocalizes gB
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gugliesi, F.; Coscia, A.; Griffante, G.; Galitska, G.; Pasquero, S.; Albano, C.; Biolatti, M. Where Do We Stand after Decades of Studying Human Cytomegalovirus? Microorganisms 2020, 8, 685. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, P.; Reeves, M. Pathogenesis of Human Cytomegalovirus in the Immunocompromised Host. Nat. Rev. Microbiol. 2021, 19, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Kabani, N.; Ross, S.A. Congenital Cytomegalovirus Infection. J. Infect. Dis. 2020, 221, S9–S14. [Google Scholar] [CrossRef] [PubMed]
- Rozman, B.; Nachshon, A.; Levi Samia, R.; Lavi, M.; Schwartz, M.; Stern-Ginossar, N. Temporal Dynamics of HCMV Gene Expression in Lytic and Latent Infections. Cell Rep. 2022, 39, 110653. [Google Scholar] [CrossRef]
- Stern-Ginossar, N.; Weisburd, B.; Michalski, A.; Le, V.T.K.; Hein, M.Y.; Huang, S.X.; Ma, M.; Shen, B.; Qian, S.B.; Hengel, H.; et al. Decoding Human Cytomegalovirus. Science 2012, 338, 1088–1093. [Google Scholar] [CrossRef]
- Lodha, M.; Muchsin, I.; Jürges, C.; Juranic Lisnic, V.; L’Hernault, A.; Rutkowski, A.J.; Prusty, B.K.; Grothey, A.; Milic, A.; Hennig, T.; et al. Decoding Murine Cytomegalovirus. PLoS Pathog. 2023, 19, e1010992. [Google Scholar] [CrossRef]
- Fisher, M.A.; Lloyd, M.L. A Review of Murine Cytomegalovirus as a Model for Human Cytomegalovirus Disease—Do Mice Lie? Int. J. Mol. Sci. 2021, 22, 214. [Google Scholar] [CrossRef]
- Turner, D.L.; Mathias, R.A. The Human Cytomegalovirus Decathlon: Ten Critical Replication Events Provide Opportunities for Restriction. Front. Cell Dev. Biol. 2022, 10, 1053139. [Google Scholar] [CrossRef]
- Wofford, A.S.; McCusker, I.; Green, J.C.; Vensko, T.A.; Pellett, P.E. Betaherpesvirus Assembly and Egress: Recent Advances Illuminate the Path. Adv. Virus Res. 2020, 108, 337–392. [Google Scholar] [CrossRef]
- Das, S.; Pellett, P.E. Spatial Relationships between Markers for Secretory and Endosomal Machinery in Human Cytomegalovirus-Infected Cells versus Those in Uninfected Cells. J. Virol. 2011, 85, 5864–5879. [Google Scholar] [CrossRef]
- Das, S.; Vasanji, A.; Pellett, P.E. Three-Dimensional Structure of the Human Cytomegalovirus Cytoplasmic Virion Assembly Complex Includes a Reoriented Secretory Apparatus. J. Virol. 2007, 81, 11861–11869. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Ortiz, D.A.; Gurczynski, S.J.; Khan, F.; Pellett, P.E. Identification of Human Cytomegalovirus Genes Important for Biogenesis of the Cytoplasmic Virion Assembly Complex. J. Virol. 2014, 88, 9086–9099. [Google Scholar] [CrossRef] [PubMed]
- Lučin, P.; Jug Vučko, N.; Karleuša, L.; Mahmutefendić Lučin, H.; Blagojević Zagorac, G.; Lisnić, B.; Pavišić, V.; Marcelić, M.; Grabušić, K.; Brizić, I.; et al. Cytomegalovirus Generates Assembly Compartment in the Early Phase of Infection by Perturbation of Host-Cell Factors Recruitment at the Early Endosome/Endosomal Recycling Compartment/Trans-Golgi Interface. Front. Cell Dev. Biol. 2020, 8, 914. [Google Scholar] [CrossRef] [PubMed]
- Lučin, P.; Kareluša, L.; Blagojević Zagorac, G.; Mahmutefendić Lučin, H.; Pavišić, V.; Jug Vučko, N.; Lukanović Jurić, S.; Marcelić, M.; Lisnić, B.; Jonjić, S. Cytomegaloviruses Exploit Recycling Rab Proteins in the Sequential Establishment of the Assembly Compartment. Front. Cell Dev. Biol. 2018, 6, 165. [Google Scholar] [CrossRef]
- Štimac, I.; Vučko, N.J.; Zagorac, G.B.; Marcelić, M.; Lučin, H.M.; Lučin, P. Dynamin Inhibitors Prevent the Establishment of the Cytomegalovirus Assembly Compartment in the Early Phase of Infection. Life 2021, 11, 876. [Google Scholar] [CrossRef]
- Karleuša, L.; Mahmutefendić, H.; Tomaš, M.I.M.I.; Zagorac, G.B.G.B.; Lučin, P. Landmarks of Endosomal Remodeling in the Early Phase of Cytomegalovirus Infection. Virology 2018, 515, 108–122. [Google Scholar] [CrossRef]
- Hook, L.M.; Grey, F.; Grabski, R.; Tirabassi, R.; Doyle, T.; Hancock, M.; Landais, I.; Jeng, S.; McWeeney, S.; Britt, W.; et al. Cytomegalovirus MiRNAs Target Secretory Pathway Genes to Facilitate Formation of the Virion Assembly Compartment and Reduce Cytokine Secretion. Cell Host Microbe 2014, 15, 363–373. [Google Scholar] [CrossRef]
- Štimac, I.; Marcelić, M.; Radić, B.; Viduka, I.; Blagojević Zagorac, G.; Lukanović Jurić, S.; Rožmanić, C.; Messerle, M.; Brizić, I.; Lučin, P.; et al. SNX27:Retromer:ESCPE-1-Mediated Early Endosomal Tubulation Impacts Cytomegalovirus Replication. Front. Cell. Infect. Microbiol. 2024, 14, 1399761. [Google Scholar] [CrossRef]
- Pavišić, V.; Lučin, H.M.; Zagorac, G.B.; Lučin, P. Arf GTPases Are Required for the Establishment of the Pre-Assembly Compartment in the Early Phase of Cytomegalovirus Infection. Life 2021, 11, 867. [Google Scholar] [CrossRef]
- Tomaš, M.I.; Kučić, N.; Mahmutefendić, H.; Blagojević, G.; Lučin, P. Murine Cytomegalovirus Perturbs Endosomal Trafficking of Major Histocompatibility Complex Class I Molecules in the Early Phase of Infection. J. Virol. 2010, 84, 1101–1112. [Google Scholar] [CrossRef]
- D’Souza, R.S.; Semus, R.; Billings, E.A.; Meyer, C.B.; Conger, K.; Casanova, J.E. Rab4 Orchestrates a Small GTPase Cascade for Recruitment of Adaptor Proteins to Early Endosomes. Curr. Biol. 2014, 24, 1187–1198. [Google Scholar] [CrossRef] [PubMed]
- Farmer, T.; Xie, S.; Naslavsky, N.; Stöckli, J.; James, D.E.; Caplan, S. Defining the Protein and Lipid Constituents of Tubular Recycling Endosomes. J. Biol. Chem. 2021, 296, 100190. [Google Scholar] [CrossRef] [PubMed]
- Mahmutefendić Lučin, H.; Štimac, I.; Marcelić, M.; Skočaj, M.; Lisnić, B.; Omerović, A.; Viduka, I.; Radić, B.; Karleuša, L.; Blagojević Zagorac, G.; et al. Rab10-Associated Tubulation as an Early Marker for Biogenesis of the Assembly Compartment in Cytomegalovirus-Infected Cells. Front. Cell Dev. Biol. 2025, 12, 1517236. [Google Scholar] [CrossRef] [PubMed]
- Babbey, C.M.; Ahktar, N.; Wang, E.; Chen, C.C.H.; Grant, B.D.; Dunn, K.W. Rab10 Regulates Membrane Transport through Early Endosomes of Polarized Madin-Darby Canine Kidney Cells. Mol. Biol. Cell 2006, 17, 3156–3175. [Google Scholar] [CrossRef]
- Chandra, M.; Collins, B.M. The Phox Homology (PX) Domain. Adv. Exp. Med. Biol. 2019, 1111, 1–17. [Google Scholar] [CrossRef]
- Overduin, M.; Bhat, R. Recognition and Remodeling of Endosomal Zones by Sorting Nexins. Biochim. Biophys. Acta Biomembr. 2024, 1866, 184305. [Google Scholar] [CrossRef]
- Overduin, M.; Kervin, T.A. The Phosphoinositide Code Is Read by a Plethora of Protein Domains. Expert. Rev. Proteom. 2021, 18, 483–502. [Google Scholar] [CrossRef]
- Simonetti, B.; Guo, Q.; Gimenez-Andres, M.; Chen, K.E.; Moody, E.R.R.; Evans, A.J.; Chandra, M.; Danson, C.M.; Williams, T.A.; Collins, B.M.; et al. SNX27-Retromer Directly Binds ESCPE-1 to Transfer Cargo Proteins during Endosomal Recycling. PLoS Biol. 2022, 20, e3001601. [Google Scholar] [CrossRef]
- Leneva, N.; Kovtun, O.; Morado, D.R.; Briggs, J.A.G.; Owen, D.J. Architecture and Mechanism of Metazoan Retromer:SNX3 Tubular Coat Assembly. Sci. Adv. 2021, 7, eabf8598. [Google Scholar] [CrossRef]
- Chandra, M.; Kendall, A.K.; Jackson, L.P. Toward Understanding the Molecular Role of SNX27/Retromer in Human Health and Disease. Front. Cell Dev. Biol. 2021, 9, 642378. [Google Scholar] [CrossRef]
- Yong, X.; Zhao, L.; Deng, W.; Sun, H.; Zhou, X.; Mao, L.; Hu, W.; Shen, X.; Sun, Q.; Billadeau, D.D.; et al. Mechanism of Cargo Recognition by Retromer-Linked SNX-BAR Proteins. PLoS Biol. 2020, 18, e3000631. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Kang, Q.; Shi, X.; Wang, Y.; Zhang, N.; Ye, H.; Xu, Q.; Xu, T.; Zhang, R. SNX-3 Mediates Retromer-Independent Tubular Endosomal Recycling by Opposing EEA-1-Facilitated Trafficking. PLoS Genet. 2021, 17, e1009607. [Google Scholar] [CrossRef] [PubMed]
- Lenoir, M.; Ustunel, C.; Rajesh, S.; Kaur, J.; Moreau, D.; Gruenberg, J.; Overduin, M. Phosphorylation of Conserved Phosphoinositide Binding Pocket Regulates Sorting Nexin Membrane Targeting. Nat. Commun. 2018, 9, 993. [Google Scholar] [CrossRef] [PubMed]
- Harterink, M.; Port, F.; Lorenowicz, M.J.; McGough, I.J.; Silhankova, M.; Betist, M.C.; Van Weering, J.R.T.; Van Heesbeen, R.G.H.P.; Middelkoop, T.C.; Basler, K.; et al. A SNX3-Dependent Retromer Pathway Mediates Retrograde Transport of the Wnt Sorting Receptor Wntless and Is Required for Wnt Secretion. Nat. Cell Biol. 2011, 13, 914–923. [Google Scholar] [CrossRef]
- Xu, Y.; Hortsman, H.; Seet, L.; Wong, S.H.; Hong, W. SNX3 Regulates Endosomal Function through Its PX-Domain-Mediated Interaction with Ptdlns(3)P. Nat. Cell Biol. 2001, 3, 658–666. [Google Scholar] [CrossRef]
- Chua, R.Y.R.; Wong, S.H. SNX3 Recruits to Phagosomes and Negatively Regulates Phagocytosis in Dendritic Cells. Immunology 2013, 139, 30–47. [Google Scholar] [CrossRef]
- Gillooly, D.J.; Simonsen, A.; Stenmark, H. Cellular Functions of Phosphatidylinositol 3-Phosphate and FYVE Domain Proteins. Biochem. J. 2001, 355, 249–258. [Google Scholar] [CrossRef]
- Crnković-Mertens, I.; Messerle, M.; Milotić, I.; Szepan, U.; Kučić, N.; Krmpotić, A.; Jonjić, S.; Koszinowski, U.H. Virus Attenuation after Deletion of the Cytomegalovirus Fc Receptor Gene Is Not Due to Antibody Control. J. Virol. 1998, 72, 1377–1382. [Google Scholar] [CrossRef]
- Marcelić, M.; Mahmutefendić Lučin, H.; Jurak Begonja, A.; Blagojević Zagorac, G.; Juranić Lisnić, V.; Lučin, P. Endosomal Phosphatidylinositol-3-Phosphate-Associated Functions Are Dispensable for Establishment of the Cytomegalovirus Pre-Assembly Compartment but Essential for the Virus Growth. Life 2021, 11, 859. [Google Scholar] [CrossRef]
- Angulo, A.; Ghazal, P.; Messerle, M. The Major Immediate-Early Gene Ie3 of Mouse Cytomegalovirus Is Essential for Viral Growth. J. Virol. 2000, 74, 11129–11136. [Google Scholar] [CrossRef]
- Bosse, J.B.; Bauerfeind, R.; Popilka, L.; Marcinowski, L.; Taeglich, M.; Jung, C.; Striebinger, H.; von Einem, J.; Gaul, U.; Walther, P.; et al. A Beta-Herpesvirus with Fluorescent Capsids to Study Transport in Living Cells. PLoS ONE 2012, 7, e40585. [Google Scholar] [CrossRef] [PubMed]
- Brizić, I.; Lisnić, B.; Brune, W.; Hengel, H.; Jonjić, S. Cytomegalovirus Infection: Mouse Model. Curr. Protoc. Immunol. 2018, 122, e51. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, A.M.; Cai, M.; Hoyer-Fender, S. Heterogeneity of the NIH3T3 Fibroblast Cell Line. Cells 2022, 11, 2677. [Google Scholar] [CrossRef] [PubMed]
- Bolte, S.; Cordelières, F.P. A Guided Tour into Subcellular Colocalization Analysis in Light Microscopy. J. Microsc. 2006, 224, 213–232. [Google Scholar] [CrossRef]
- McCloy, R.A.; Rogers, S.; Caldon, C.E.; Lorca, T.; Castro, A.; Burgess, A. Partial Inhibition of Cdk1 in G2 Phase Overrides the SAC and Decouples Mitotic Events. Cell Cycle 2014, 13, 1400–1412. [Google Scholar] [CrossRef]
- Marcelić, M.; Lučin, H.M.; Begonja, A.J.; Zagorac, G.B.; Lučin, P. Early Endosomal Vps34-Derived Phosphatidylinositol-3-Phosphate Is Indispensable for the Biogenesis of the Endosomal Recycling Compartment. Cells 2022, 11, 962. [Google Scholar] [CrossRef]
- Lemmermann, N.A.W.; Podlech, J.; Seckert, C.K.; Kropp, K.A.; Grzimek, N.K.A.; Reddehase, M.J.; Holtappels, R. CD8 T-Cell Immunotherapy of Cytomegalovirus Disease in the Murine Model. In Methods in Microbiology; Academic Press: Cambridge, MA, USA, 2010; pp. 369–420. [Google Scholar]
- Zhou, C.-H.; Zhang, Y.-H.; Xue, F.; Xue, S.-S.; Chen, Y.-C.; Gu, T.; Peng, Z.-W.; Wang, H.-N. Isoflurane Exposure Regulates the Cell Viability and BDNF Expression of Astrocytes via Upregulation of TREK-1. Mol. Med. Rep. 2017, 16, 7305–7314. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Feng, S.; Streets, A.J.; Nesin, V.; Tran, U.; Nie, H.; Onopiuk, M.; Wessely, O.; Tsiokas, L.; Ong, A.C.M. The Sorting Nexin 3 Retromer Pathway Regulates the Cell Surface Localization and Activity of a Wnt-Activated Polycystin Channel Complex. J. Am. Soc. Nephrol. 2017, 28, 2973–2984. [Google Scholar] [CrossRef]
- UniProt Three Protein Coding Sequences for Mouse Isoforms. Available online: https://www.uniprot.org/ (accessed on 17 January 2025).
- Dyer, S.C.; Austine-Orimoloye, O.; Azov, A.G.; Barba, M.; Barnes, I.; Barrera-Enriquez, V.P.; Becker, A.; Bennett, R.; Beracochea, M.; Berry, A.; et al. Ensembl 2025. Nucleic Acids Res. 2025, 53, D948–D957. [Google Scholar] [CrossRef]
- Harrison, M.S.; Hung, C.-S.; Liu, T.; Christiano, R.; Walther, T.C.; Burd, C.G. A Mechanism for Retromer Endosomal Coat Complex Assembly with Cargo. Proc. Natl. Acad. Sci. USA 2014, 111, 267–272. [Google Scholar] [CrossRef] [PubMed]
- AlphaFold Predicted Domain Structure. Available online: https://alphafold.ebi.ac.uk/ (accessed on 17 January 2025).
- Mizutani, R.; Yamauchi, J.; Kusakawa, S.; Nakamura, K.; Sanbe, A.; Torii, T.; Miyamoto, Y.; Tanoue, A. Sorting Nexin 3, a Protein Upregulated by Lithium, Contains a Novel Phosphatidylinositol-Binding Sequence and Mediates Neurite Outgrowth in N1E-115 Cells. Cell Signal 2009, 21, 1586–1594. [Google Scholar] [CrossRef] [PubMed]
- McGough, I.J.; de Groot, R.E.A.; Jellett, A.P.; Betist, M.C.; Varandas, K.C.; Danson, C.M.; Heesom, K.J.; Korswagen, H.C.; Cullen, P.J. SNX3-Retromer Requires an Evolutionary Conserved MON2:DOPEY2:ATP9A Complex to Mediate Wntless Sorting and Wnt Secretion. Nat. Commun. 2018, 9, 3737. [Google Scholar] [CrossRef] [PubMed]
- Cullen, P.J.; Steinberg, F. To Degrade or Not to Degrade: Mechanisms and Significance of Endocytic Recycling. Nat. Rev. Mol. Cell Biol. 2018, 19, 679–696. [Google Scholar] [CrossRef]
- Gopaldass, N.; Chen, K.E.; Collins, B.; Mayer, A. Assembly and Fission of Tubular Carriers Mediating Protein Sorting in Endosomes. Nat. Rev. Mol. Cell Biol. 2024, 25, 765–783. [Google Scholar] [CrossRef]
- Wu, Y.; Prager, A.; Boos, S.; Resch, M.; Brizic, I.; Mach, M.; Wildner, S.; Scrivano, L.; Adler, B. Human Cytomegalovirus Glycoprotein Complex GH/GL/GO Uses PDGFR-α as a Key for Entry. PLoS Pathog. 2017, 13, e1006281. [Google Scholar] [CrossRef]
- Radsak, K.; Eickmann, M.; Mockenhaupt, T.; Bogner, E.; Kern, H.; Eis-Hübinger, A.; Reschke, M. Retrieval of Human Cytomegalovirus Glycoprotein B from the Infected Cell Surface for Virus Envelopment. Arch. Virol. 1996, 141, 557–572. [Google Scholar] [CrossRef]
- Tugizov, S.; Maidji, E.; Xiao, J.; Pereira, L. An Acidic Cluster in the Cytosolic Domain of Human Cytomegalovirus Glycoprotein B Is a Signal for Endocytosis from the Plasma Membrane. J. Virol. 1999, 73, 8677–8688. [Google Scholar] [CrossRef]
- Van Weering, J.R.T.; Verkade, P.; Cullen, P.J. SNX-BAR-Mediated Endosome Tubulation Is Co-Ordinated with Endosome Maturation. Traffic 2012, 13, 94–107. [Google Scholar] [CrossRef]
- Wang, P.; Liu, H.; Wang, Y.; Liu, O.; Zhang, J.; Gleason, A.; Yang, Z.; Wang, H.; Shi, A.; Grant, B.D. RAB-10 Promotes EHBP-1 Bridging of Filamentous Actin and Tubular Recycling Endosomes. PLoS Genet. 2016, 12, e1006093. [Google Scholar] [CrossRef]
- Etoh, K.; Fukuda, M. Rab10 Regulates Tubular Endosome Formation through KIF13A and KIF13B Motors. J. Cell Sci. 2019, 132, jcs226977. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Thapa, N.; Choi, S.; Anderson, R.A. Emerging Roles of PtdIns(4,5)P2—Beyond the Plasma Membrane. J. Cell Sci. 2015, 128, 4047–4056. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.; Kanno, E.; Ishibashi, K.; Itoh, T. Large Scale Screening for Novel Rab Effectors Reveals Unexpected Broad Rab Binding Specificity. Mol. Cell. Proteom. 2008, 7, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, M.A.; Fish, K.N.; Söderberg-Naucler, C.; Streblow, D.N.; Meyers, H.L.; Thomas, G.; Nelson, J.A. Retrieval of Human Cytomegalovirus Glycoprotein B from Cell Surface Is Not Required for Virus Envelopment in Astrocytoma Cells. J. Virol. 2002, 76, 5147–5155. [Google Scholar] [CrossRef]
- Jarvis, M.A.; Jones, T.R.; Drummond, D.D.; Smith, P.P.; Britt, W.J.; Nelson, J.A.; Baldick, C.J. Phosphorylation of Human Cytomegalovirus Glycoprotein B (GB) at the Acidic Cluster Casein Kinase 2 Site (Ser900) Is Required for Localization of GB to the Trans-Golgi Network and Efficient Virus Replication. J. Virol. 2004, 78, 285–293. [Google Scholar] [CrossRef]
- Kropff, B.; Koedel, Y.; Britt, W.; Mach, M. Optimal Replication of Human Cytomegalovirus Correlates with Endocytosis of Glycoprotein GpUL132. J. Virol. 2010, 84, 7039–7052. [Google Scholar] [CrossRef]
- Thomas, G. Furin at the Cutting Edge: From Protein Traffic to Embryogenesis and Disease. Nat. Rev. Mol. Cell Biol. 2002, 3, 753–766. [Google Scholar] [CrossRef]
- Bergner, T.; Rayas, L.C.; Freimann, G.; Read, C.; Von Einem, J. Secondary Envelopment of Human Cytomegalovirus Is a Fast Process Utilizing the Endocytic Compartment as a Major Membrane Source. Biomolecules 2024, 14, 1149. [Google Scholar] [CrossRef]
- Pim, D.; Broniarczyk, J.; Bergant, M.; Playford, M.P.; Banks, L. A Novel PDZ Domain Interaction Mediates the Binding between Human Papillomavirus 16 L2 and Sorting Nexin 27 and Modulates Virion Trafficking. J. Virol. 2015, 89, 10145–10155. [Google Scholar] [CrossRef]
- Lučin, P.; Mahmutefendić Lučin, H.; Blagojević Zagorac, G. Cytomegaloviruses Reorganize Endomembrane System to Intersect Endosomal and Amphisome-like Egress Pathway. Front. Cell Dev. Biol. 2023, 11, 1328751. [Google Scholar] [CrossRef]
- Mahmutefendić Lučin, H.; Blagojević Zagorac, G.; Marcelić, M.; Lučin, P. Host Cell Signatures of the Envelopment Site within Beta-Herpes Virions. Int. J. Mol. Sci. 2022, 23, 9994. [Google Scholar] [CrossRef] [PubMed]
- Tooze, J.; Hollinshead, M.; Reis, B.; Radsak, K.; Kern, H. Progeny Vaccinia and Human Cytomegalovirus Particles Utilize Early Endosomal Cisternae for Their Envelopes. Eur. J. Cell Biol. 1993, 60, 163–178. [Google Scholar] [PubMed]
- Turner, D.L.; Korneev, D.V.; Purdy, J.G.; de Marco, A.; Mathias, R.A. The Host Exosome Pathway Underpins Biogenesis of the Human Cytomegalovirus Virion. Elife 2020, 9, e58288. [Google Scholar] [CrossRef] [PubMed]
- Krzyzaniak, M.A.; Mach, M.; Britt, W.J. HCMV-Encoded Glycoprotein M (UL100) Interacts with Rab11 Effector Protein FIP4. Traffic 2009, 10, 1439–1457. [Google Scholar] [CrossRef]
- Puri, C.; Gratian, M.J.; Rubinsztein, D.C. Mammalian Autophagosomes Form from Finger-like Phagophores. Dev. Cell 2023, 58, 2746–2760.e5. [Google Scholar] [CrossRef]
- Varnum, S.M.; Streblow, D.N.; Monroe, M.E.; Smith, P.; Auberry, K.J.; Pas, L.; Wang, D.; Ii, D.G.C.; Rodland, K.; Wiley, S.; et al. Identification of Proteins in Human Cytomegalovirus (HCMV) particles: The HCMV proteome. J. Virol. 2004, 78, 10960–10966. [Google Scholar] [CrossRef]
- Couté, Y.; Kraut, A.; Zimmermann, C.; Büscher, N.; Hesse, A.M.; Bruley, C.; De Andrea, M.; Wangen, C.; Hahn, F.; Marschall, M.; et al. Mass Spectrometry-Based Characterization of the Virion Proteome, Phosphoproteome, and Associated Kinase Activity of Human Cytomegalovirus. Microorganisms 2020, 8, 820. [Google Scholar] [CrossRef]
- Rieder, F.J.J.; Kastner, M.T.; Hartl, M.; Puchinger, M.G.; Schneider, M.; Majdic, O.; Britt, W.J.; Djinović-Carugo, K.; Steininger, C. Human Cytomegalovirus Phosphoproteins Are Hypophosphorylated and Intrinsically Disordered. J. General. Virol. 2017, 98, 471–485. [Google Scholar] [CrossRef]
- Reyda, S.; Büscher, N.; Tenzer, S.; Plachter, B. Proteomic Analyses of Human Cytomegalovirus Strain AD169 Derivatives Reveal Highly Conserved Patterns of Viral and Cellular Proteins in Infected Fibroblasts. Viruses 2014, 6, 172–188. [Google Scholar] [CrossRef]
- Flomm, F.J.; Soh, T.K.; Schneider, C.; Wedemann, L.; Britt, H.M.; Thalassinos, K.; Pfitzner, S.; Reimer, R.; Grünewald, K.; Bosse, J.B. Intermittent Bulk Release of Human Cytomegalovirus. PLoS Pathog. 2022, 18, e1010575. [Google Scholar] [CrossRef]
- Hein, M.Y.; Weissman, J.S. Functional Single-Cell Genomics of Human Cytomegalovirus Infection. Nat. Biotechnol. 2022, 40, 391–401. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viduka, I.; Štimac, I.; Jurić, S.L.; Gulić, T.; Lisnić, B.; Zagorac, G.B.; Lučin, H.M.; Lučin, P. Contribution of Sorting Nexin 3 in the Cytomegalovirus Assembly. Biomedicines 2025, 13, 936. https://doi.org/10.3390/biomedicines13040936
Viduka I, Štimac I, Jurić SL, Gulić T, Lisnić B, Zagorac GB, Lučin HM, Lučin P. Contribution of Sorting Nexin 3 in the Cytomegalovirus Assembly. Biomedicines. 2025; 13(4):936. https://doi.org/10.3390/biomedicines13040936
Chicago/Turabian StyleViduka, Ivona, Igor Štimac, Silvija Lukanović Jurić, Tamara Gulić, Berislav Lisnić, Gordana Blagojević Zagorac, Hana Mahmutefendić Lučin, and Pero Lučin. 2025. "Contribution of Sorting Nexin 3 in the Cytomegalovirus Assembly" Biomedicines 13, no. 4: 936. https://doi.org/10.3390/biomedicines13040936
APA StyleViduka, I., Štimac, I., Jurić, S. L., Gulić, T., Lisnić, B., Zagorac, G. B., Lučin, H. M., & Lučin, P. (2025). Contribution of Sorting Nexin 3 in the Cytomegalovirus Assembly. Biomedicines, 13(4), 936. https://doi.org/10.3390/biomedicines13040936