
Pt(IV) Complexes as Anticancer Drugs and Their Relationship with Oxidative Stress



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methodology
3. Cancer and OS
4. Pt(IV) Compounds and Their Antitumor Activity
4.1. Monitoring the Reduction of Pt(IV) Complexes: Detection Principles
4.2. Pt(IV) Compounds in the Treatment of Cancer
4.2.1. Pt(IV) Compounds’ Mechanism of Action
4.2.2. Structure and Ligands Depended Pt(IV) Prodrugs Properties
4.2.3. Reduction of Pt(IV) and Anticancer Activity
4.3. Antioxidant Enzymes-like Activity of Pt
5. Pt-Based Compounds, OS, and Cell Death
6. The Influence of GSH in Cancer Development
7. The Influence of AA in Cancer Development
8. Overview of the Reactions of Pt(IV) Compounds with GSH and AA
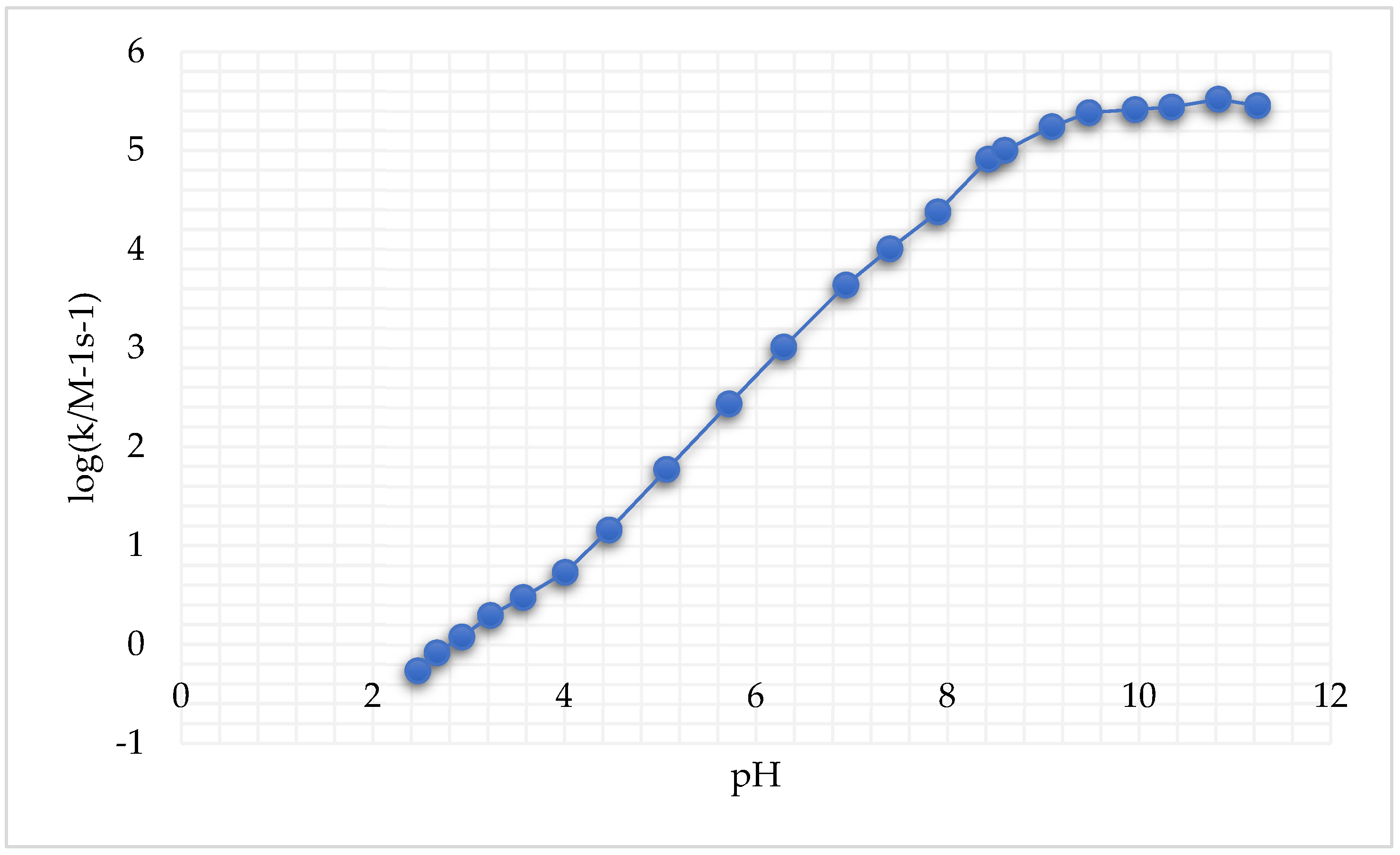
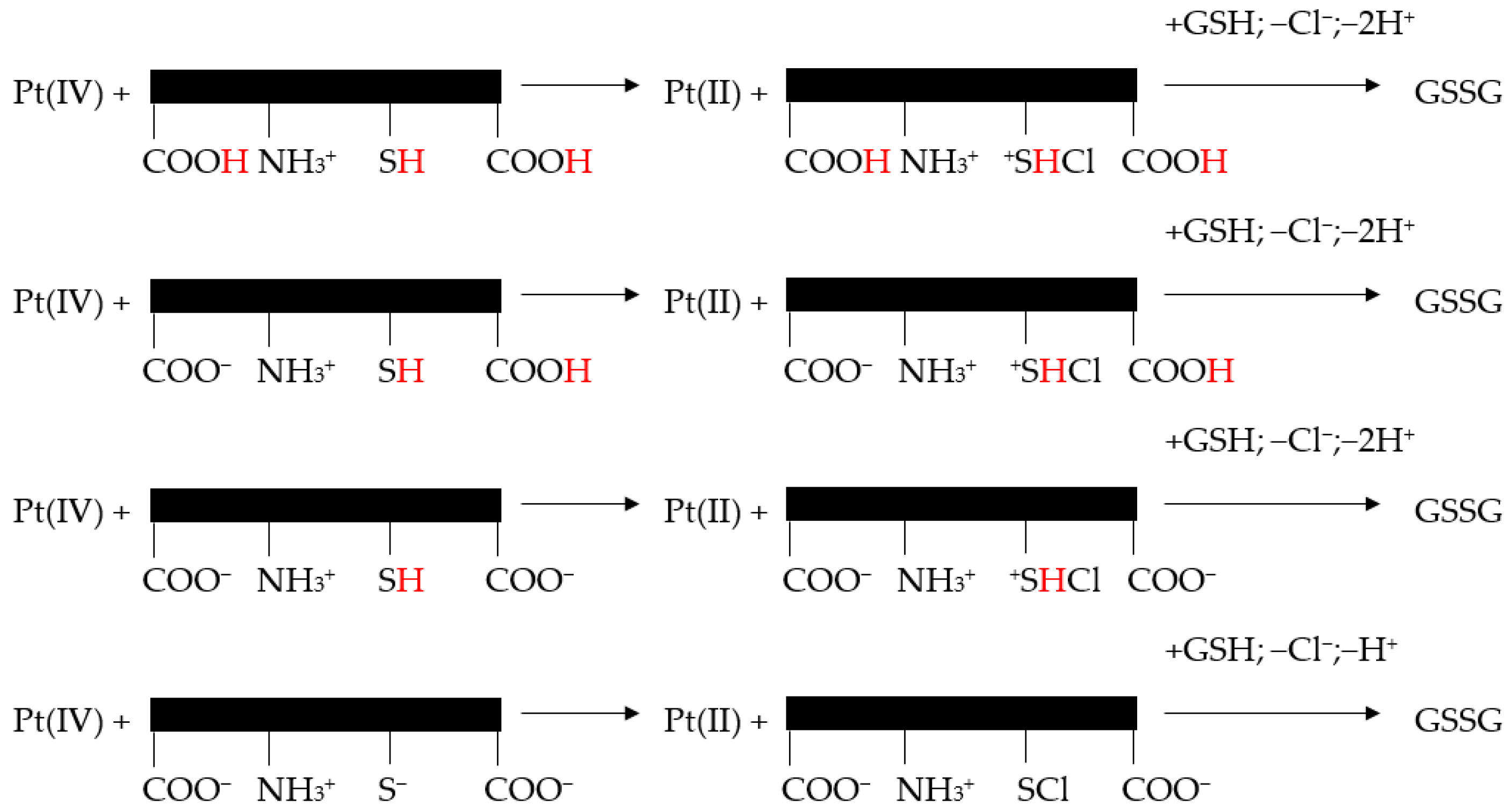
8.1. Reaction of Pt(IV) with GSH
8.2. Reaction of Pt(IV) with AA
9. Conclusions and Prospective
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| Pt | platinum |
| OS | oxidative stress |
| DNA | Deoxyribonucleic acid |
| ncRNAs | non-coding Ribonucleic acid |
| ROS | reactive oxygen species |
| miRNA | micro-Ribonucleic acid |
| FR | free radicals |
| GSH | reduced glutathione |
| AA | ascorbic acid |
| UA | uric acid |
| L-AA | L-ascorbic acid |
| NfkB | Nuclear factor-kappa B |
| MAPK | mitogen-activated protein kinase |
| ERCC | Excision Repair Cross-Complementation group |
| XP | xeroderma pigmentosum complementation group |
| UV-Vis | Ultraviolet-visible spectroscopy |
| LMCT | ligand-to-metal charge transfer |
| 195Pt NMR | platinum-195 nuclear magnetic resonance |
| CAT | catalase |
| SOD | superoxide dismutase |
| GSSG | oxidized form of glutathione |
| PtNPs | platinum nanoparticles |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| Bcl-2 | B-cell leukemia/lymphoma 2 protein |
| PARP | Poly-ADP-ribose-polymerase |
References
- Wu, Z.; Xia, F.; Lin, R. Global burden of cancer and associated risk factors in 204 countries and territories, 1980–2021: A systematic analysis for the GBD 2021. J. Hematol. Oncol. 2024, 17, 119. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Cancer Burden Growing, Amidst Mounting Need for Services. 1 February 2024. Available online: https://www.who.int/news/item/01-02-2024-global-cancer-burden-growing--amidst-mounting-need-for-services (accessed on 1 January 2025).
- Global Surg Collaborative and National Institute for Health Research Global Health Research Unit on Global Surgery. Global variation in postoperative mortality and complications after cancer surgery: A multicentre, prospective cohort study in 82 countries. Lancet 2021, 397, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Corrie, P.G. Cytotoxic chemotherapy: Clinical aspects. Medicine 2008, 36, 24–28. [Google Scholar] [CrossRef]
- Wang, K.; Tepper, J.E. Radiation therapy-associated toxicity: Etiology, management, and prevention. CA Cancer J. Clin. 2021, 71, 437–454. [Google Scholar] [CrossRef] [PubMed]
- Thirumaran, R.; Prendergast, G.C.; Gilman, P.B. Cytotoxic Chemotherapy in Clinical Treatment of Cancer. In Cancer Immunotherapy; Prendergast, G.C., Jaffee, E.M., Eds.; Academic Press: Cambridge, MA, USA, 2007; pp. 101–116. [Google Scholar]
- Xia, Y.; Sun, M.; Huang, H.; Jin, W.L. Drug repurposing for cancer therapy. Signal Transduct. Target. Ther. 2024, 9, 92. [Google Scholar] [CrossRef]
- Gadade, D.D.; Jha, H.; Kumar, C. Unlocking the power of precision medicine: Exploring the role of biomarkers in cancer management. Futur. J. Pharm. Sci. 2024, 10, 5. [Google Scholar] [CrossRef]
- Brawer, M.K. Hormonal therapy for prostate cancer. Rev. Urol. 2006, 8 (Suppl. 2), S35–S47. [Google Scholar]
- Shah, T.T.; Ahmed, H.; Kanthabalan, A.; Lau, B.; Ghei, M.; Maraj, B.; Arya, M. Focal cryotherapy of localized prostate cancer: A systematic review of the literature. Expert Rev. Anticancer Ther. 2014, 14, 1337–1347. [Google Scholar] [CrossRef]
- Cazzaniga, M.E.; Cordani, N.; Capici, S.; Cogliati, V.; Riva, F.; Cerrito, M.G. Metronomic Chemotherapy. Cancers 2021, 13, 2236. [Google Scholar] [CrossRef]
- Bilia, A.R.; Piazzini, V.; Risaliti, L.; Vanti, G.; Casamonti, M.; Wang, M.; Bergonzi, M.C. Nanocarriers: A Successful Tool to Increase Solubility, Stability and Optimise Bioefficacy of Natural Constituents. Curr. Med. Chem. 2019, 26, 4631–4656. [Google Scholar] [CrossRef]
- Maiti, R.; Patel, B.; Patel, N.; Patel, M.; Patel, A.; Dhanesha, N. Antibody drug conjugates as targeted cancer therapy: Past development, present challenges and future opportunities. Arch. Pharm. Res. 2023, 46, 361–388. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Li, D.; Zhu, X. Cancer immunotherapy: Pros, cons and beyond. Biomed. Pharmacother. 2020, 124, 109821. [Google Scholar] [CrossRef] [PubMed]
- Global Burden of Disease 2019 Cancer Collaboration. Cancer Incidence, Mortality, Years of Life Lost, Years Lived with Disability, and Disability-Adjusted Life Years for 29 Cancer Groups From 2010 to 2019: A Systematic Analysis for the Global Burden of Disease Study 2019. JAMA Oncol. 2022, 8, 420–444. [Google Scholar] [CrossRef]
- Hill, B.T. Etiology of Cancer. In Clinical Ophthalmic Oncology, 3rd ed.; Singh, A., Damato, B., Eds.; Springer: Cham, Switzerland, 2019; pp. 11–17. [Google Scholar]
- Sun, L.; Zhang, H.; Gao, P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell 2022, 13, 877–919. [Google Scholar] [CrossRef]
- Lu, Y.; Chan, Y.T.; Tan, H.Y.; Li, S.; Wang, N.; Feng, Y. Epigenetic regulation in human cancer: The potential role of epi-drug in cancer therapy. Mol. Cancer 2020, 19, 79. [Google Scholar] [CrossRef]
- Iqbal, M.J.; Kabeer, A.; Abbas, Z. Interplay of oxidative stress, cellular communication and signaling pathways in cancer. Cell Commun. Signal. 2024, 22, 7. [Google Scholar] [CrossRef]
- Krishnamurthy, H.K.; Rajavelu, I.; Pereira, M.; Jayaraman, V.; Krishna, K.; Wang, T.; Bei, K.; Rajasekaran, J.J. Inside the genome: Understanding genetic influences on oxidative stress. Front. Genet. 2024, 15, 1397352. [Google Scholar] [CrossRef]
- Franco, R.; Schoneveld, O.; Georgakilas, A.G.; Panayiotidis, M.I. Oxidative stress, DNA methylation and carcinogenesis. Cancer Lett. 2008, 266, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Mahalingaiah, P.K.S.; Ponnusamy, L.; Singh, K.P. Oxidative stress-induced epigenetic changes associated with malignant transformation of human kidney epithelial cells. Oncotarget 2017, 8, 11127–11143. [Google Scholar] [CrossRef]
- Yi, X.; Zhu, Q.X.; Wu, X.L.; Tan, T.T.; Jiang, X.J. Histone Methylation and Oxidative Stress in Cardiovascular Diseases. Oxid. Med. Cell Longev. 2022, 2022, 6023710. [Google Scholar] [CrossRef]
- Huang, M.; Wu, Q.; Jiang, Z.H. Epigenetic Alterations under Oxidative Stress in Stem Cells. Oxid. Med. Cell Longev. 2022, 2022, 6439097. [Google Scholar]
- He, J.; Jiang, B.H. Interplay between reactive oxygen species and microRNAs in cancer. Curr. Pharmacol. Rep. 2016, 2, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.Y.; Luo, J.Y.; Wang, L.; Huang, Y. MicroRNAs regulating reactive oxygen species in cardiovascular diseases. Antioxid. Redox Signal. 2018, 29, 1092–1107. [Google Scholar] [CrossRef]
- Lan, J.; Huang, Z.; Han, J.; Shao, J.; Huang, C. Redox regulation of microRNAs in cancer. Cancer Lett. 2018, 418, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H. MicroRNA Networks Modulate Oxidative Stress in Cancer. Int. J. Mol. Sci. 2019, 20, 4497. [Google Scholar] [CrossRef]
- Okada, F. Inflammation and free radicals in tumor development and progression. Redox Rep. 2002, 7, 357–368. [Google Scholar] [CrossRef]
- Sosa, V.; Moliné, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; Leonart, M.E. Oxidative stress and cancer: An overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef]
- Matsuzawa, A.; Ichijo, H. Redox control of cell fate by MAP kinase: Physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochim. Biophys. Acta 2008, 1780, 1325–1336. [Google Scholar] [CrossRef]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef]
- Wiemer, E. Stressed tumor cell, chemosensitized cancer. Nat. Med. 2011, 17, 1552–1554. [Google Scholar] [CrossRef]
- Luo, Y.; Zou, P.; Zou, J.; Wang, J.; Zhou, D.; Liu, L. Autophagy regulates ROS-induced cellular senescence via p21 in a p38 MAPKα dependent manner. Exp. Gerontol. 2011, 46, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Umber, J.; Qasim, M.; Ashraf, S.; Ashfaq, U.A.; Iram, A.; Bhatti, R.; Tariq, M.; Masoud, M.S. Antioxidants Mitigate Oxidative Stress: A General Overview. In The Role of Natural Antioxidants in Brain Disorders, 1st ed.; Imran, A., Hussain, G., Eds.; Springer: Cham, Switzerland, 2023; pp. 149–169. [Google Scholar]
- Moura, F.A.; de Andrade, K.Q.; dos Santos, J.C.F.; Araújo, O.R.P.; Goulart, M.O.F. Antioxidant therapy for treatment of inflammatory bowel disease: Does it work? Redox Biol. 2015, 6, 617–639. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef]
- Jones, D.P. Redefining oxidative stress. Antioxid. Redox Signal. 2006, 8, 1865–1879. [Google Scholar] [CrossRef]
- Jones, D.P. Radical-free biology of oxidative stress. Am. J. Physiol. Cell Physiol. 2008, 295, C849. [Google Scholar] [CrossRef]
- Arfin, S.; Jha, N.K.; Jha, S.K.; Kesari, K.K.; Ruokolainen, J.; Roychoudhury, S.; Rathi, B.; Kumar, D. Oxidative Stress in Cancer Cell Metabolism. Antioxidants 2021, 10, 642. [Google Scholar] [CrossRef]
- Newman, L.A. Breast cancer screening in low and middle-income countries. Best Pract. Res. Clin. Obstet. Gynaecol. 2022, 83, 15–23. [Google Scholar] [CrossRef]
- Gupta, S. Screening for Colorectal Cancer. Hematol. Oncol. Clin. N. Am. 2022, 36, 393–414. [Google Scholar] [CrossRef]
- Schabath, M.B.; Cote, M.L. Cancer Progress and Priorities: Lung Cancer. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1563–1579. [Google Scholar] [CrossRef]
- Carlsson, S.V.; Vickers, A.J. Screening for Prostate Cancer. Med. Clin. N. Am. 2020, 104, 1051–1062. [Google Scholar] [CrossRef]
- Lin, J.S.; Aiello Bowles, E.J.; Williams, S.B.; Morrison, C.C. Screening for Thyroid Cancer: A Systematic Evidence Review for the U.S. Preventive Services Task Force; Agency for Healthcare Research and Quality (US): Rockville, MD, USA, 2017.
- Xia, J.Y.; Aadam, A.A. Advances in screening and detection of gastric cancer. J. Surg. Oncol. 2022, 125, 1104–1109. [Google Scholar] [CrossRef]
- Obeagu, E.I.; Obeagu, G.U. Breast cancer: A review of risk factors and diagnosis. Medicine 2024, 103, e36905. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Zhang, B.; Li, P.; Zhao, Y. Methods and biomarkers for early detection, prediction, and diagnosis of colorectal cancer. Biomed. Pharmacother. 2023, 163, 114786. [Google Scholar] [CrossRef] [PubMed]
- Nooreldeen, R.; Bach, H. Current and Future Development in Lung Cancer Diagnosis. Int. J. Mol. Sci. 2021, 22, 8661. [Google Scholar] [CrossRef] [PubMed]
- Cornford, P.; van den Bergh, R.C.N.; Briers, E.; Van den Broeck, T.; Brunckhorst, O.; Darraugh, J.; Eberli, D.; De Meerleer, G.; De Santis, M.; Farolfi, A.; et al. EAU-EANM-ESTRO-ESUR-ISUP-SIOG Guidelines on Prostate Cancer-2024 Update. Part I: Screening, Diagnosis, and Local Treatment with Curative Intent. Eur. Urol. 2024, 86, 148–163. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.T.; Lee, E.J.; Huang, M.G.; Park, Y.I.; Khullar, A.; Plodkowski, R.A. Diagnosis and treatment of patients with thyroid cancer. Am. Health Drug Benefits 2015, 8, 30–40. [Google Scholar]
- Lordick, F.; Carneiro, F.; Cascinu, S.; Fleitas, T.; Haustermans, K.; Piessen, G.; Vogel, A.; Smyth, E.C. Gastric cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. 2022, 33, 1005–1020. [Google Scholar] [CrossRef]
- Das, S.; Dey, M.K.; Devireddy, R.; Gartia, M.R. Biomarkers in Cancer Detection, Diagnosis, and Prognosis. Sensors 2024, 24, 37. [Google Scholar] [CrossRef]
- Martins, I.; Ribeiro, I.P.; Jorge, J.; Gonçalves, A.C.; Sarmento-Ribeiro, A.B.; Melo, J.B.; Carreira, I.M. Liquid Biopsies: Applications for Cancer Diagnosis and Monitoring. Genes 2021, 12, 349. [Google Scholar] [CrossRef]
- Marín, R.; Abad, C.; Rojas, D.; Chiarello, D.I.; Alejandro, T.G. Biomarkers of oxidative stress and reproductive complications. Adv. Clin. Chem. 2023, 113, 157–233. [Google Scholar] [PubMed]
- Yu, B.P. Cellular defenses against damage from reactive oxygen species. Physiol. Rev. 1994, 74, 139–162. [Google Scholar] [CrossRef] [PubMed]
- Rabenstein, D.L.; Guevremont, R.; Evans, C.A. Glutathione and its metal complexes. In Metal Ions in Biological Systems, 1st ed.; Sigel, H., Ed.; CRC Press: Boca Raton, FL, USA, 1979; Volume 9, pp. 103–141. [Google Scholar]
- Henderson, G.B.; Fairlamb, A.H.; Ulrich, P.; Cerami, A. Substrate specificity of the flavoprotein trypanothione disulfide reductase from Crithidia fasciculata. Biochemistry 1987, 26, 3023–3027. [Google Scholar] [CrossRef]
- Taylor, J.E.; Yan, J.F.; Wang, J. The iron (III)-catalyzed oxidation of cysteine by molecular oxygen in the aqueous phase. An example of a two-thirds-order reaction. J. Am. Chem. Soc. 1966, 88, 1663–1667. [Google Scholar] [CrossRef]
- Bridgart, G.J.; Wilson, I.R. Metal-ion catalysis in some reactions of hexacyanoferrate(III) ions. Part II. The oxidation of cysteine and related thiols in the presence of ethylenediaminetetra-acetic acid. J. Chem. Soc. Dalton Trans. 1973, 12, 1281–1284. [Google Scholar] [CrossRef]
- Ehrenberg, L.; Harms-Ringdahl, M.; Fedorcsak, I.; Granath, F. Kinetics of the Copper- and Iron-Catalysed Oxidation of Cysteine by Dioxygen. Acta Chem. Scand. 1989, 43, 177–187. [Google Scholar] [CrossRef]
- Shi, T.; Berglund, J.; Elding, L.I. Kinetics and mechanism for reduction of trans-dichlorotetracyanoplatinate(IV) by thioglycolic acid, L-cysteine, DL-penicillamine, and glutathione in aqueous solution. Inorg. Chem. 1996, 35, 3498–3503. [Google Scholar] [CrossRef]
- Pelizzetti, E.; Mentasti, E.; Pramauro, E. Kinetics and mechanism of the oxidation of ascorbic acid by tris (1, 10-phenanthroline) iron (III) and its derivatives in aqueous acidic perchlorate media. Inorg. Chem. 1976, 15, 2898–2900. [Google Scholar] [CrossRef]
- Pelizzetti, E.; Mentasti, E.; Pramauro, E. Outer-sphere oxidation of ascorbic acid. Inorg. Chem. 1978, 17, 1181–1186. [Google Scholar] [CrossRef]
- Macartiney, D.H.; Mc Auley, A. The outer-sphere oxidation of ascorbic acid by the thioureapentacyanoferrate(III) ion. Can. J. Chem. 1981, 59, 132. [Google Scholar] [CrossRef]
- Amjad, Z.; Brodovitch, J.C.; McAuley, A. Metal-ion oxidations in solution. Part XXI. Kinetics and mechanism of the reaction of ascorbic acid, hydroquinone, and catechol with 12- tungstocobaltoate (III). Can. J. Chem. 1977, 55, 3581–3586. [Google Scholar] [CrossRef]
- Tsukahara, K.; Yamamota, Y. A Kinetic Study of the Reactions of Several Cobalt(III) Complexes with Ascorbic Acid. Bull. Chem. Soc. Jpn. 1981, 54, 2642. [Google Scholar] [CrossRef]
- Williams, N.H.; Yamdell, J.K. Outer-sphere electrontransfer reactions of ascorbate anions. Aust. J. Chem. 1982, 35, 1133–1144. [Google Scholar] [CrossRef]
- Kimura, M.; Yamab, S. Kinetics and mechanism of the oxidation of L-ascorbic acid by tris(oxalato)cobaltate(III) and tris(1,10-phenanthroline)iron(III) complexes in aqueous solution. J. Chem. Soc. Dalton Trans. 1982, 2, 423–427. [Google Scholar] [CrossRef]
- Ou, B.; Hampsch-Woodill, M.; Flanagan, J.; Deemer, E.K.; Prior, R.L.; Huang, D. Novel fluorometric assay for hydroxyl radical prevention capacity using fluorescein as the probe. J. Agric. Food Chem. 2002, 50, 2772–2777. [Google Scholar] [CrossRef]
- Munteanu, I.G.; Apetrei, C. Analytical Methods Used in Determining Antioxidant Activity: A Review. Int. J. Mol. Sci. 2021, 22, 3380. [Google Scholar] [CrossRef] [PubMed]
- Benzie, I.F.; Strain, J.J. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: The FRAP assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef]
- Xiao, F. Guidelines for antioxidant assays for food components. Food Front. 2020, 1, 60–69. [Google Scholar] [CrossRef]
- Kim, E.; Winkler, T.E.; Kitchen, C.; Kang, M.; Banis, G.; Bentley, W.E.; Kelly, D.L.; Ghodssi, R.; Payne, G.F. Redox Probing for Chemical Information of Oxidative Stress. Anal. Chem. 2017, 89, 1583–1592. [Google Scholar] [CrossRef]
- Pietrzak, S.; Marciniak, W.; Derkacz, R.; Matuszczak, M.; Kiljańczyk, A.; Baszuk, P.; Bryśkiewicz, M.; Sikorski, A.; Gronwald, J.; Słojewski, M. Correlation between Selenium and Zinc Levels and Survival among Prostate Cancer Patients. Nutrients 2024, 16, 527. [Google Scholar] [CrossRef]
- Matuszczak, M.; Kiljańczyk, A.; Marciniak, W.; Derkacz, R.; Stempa, K.; Baszuk, P.; Bryśkiewicz, M.; Cybulski, C.; Dębniak, T.; Gronwald, J. Antioxidant Properties of Zinc and Copper—Blood Zinc-to Copper-Ratio as a Marker of Cancer Risk BRCA1 Mutation Carriers. Antioxidants 2024, 13, 841. [Google Scholar] [CrossRef] [PubMed]
- Matuszczak, M.; Kiljańczyk, A.; Marciniak, W.; Derkacz, R.; Stempa, K.; Baszuk, P.; Bryśkiewicz, M.; Sun, P.; Cheriyan, A.; Cybulski, C. Zinc and Its Antioxidant Properties: The Potential Use of Blood Zinc Levels as a Marker of Cancer Risk in BRCA1 Mutation Carriers. Antioxidants 2024, 13, 609. [Google Scholar] [CrossRef] [PubMed]
- Farrel, N.; Ugo, R.; James, B.R. Catalysis By Metal Complexes; Kluwer: Dordrecht, The Netherlands, 1989; Volume 11, p. 50. [Google Scholar]
- Weiss, R.B.; Christian, M.C. New cisplatin analogues in development: A review. Drugs 1993, 46, 360–377. [Google Scholar] [CrossRef]
- Keage, M.C.; Kelland, M.J.; Neidles, L.R.; Warning, M.J. Molecular Aspects of Anticover Drug DNA Interactions; CRC Press: New York, NY, USA, 1993; Volume 1, pp. 243–269. [Google Scholar]
- Dasari, S.; Bernard Tchounwou, P. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef]
- Zhong, Y.; Jia, C.; Zhang, X.; Liao, X.; Yang, B.; Cong, Y.; Pu, S.; Gao, C. Targeting drug delivery system for platinum(IV)-Based antitumor complexes. Eur. J. Med. Chem. 2020, 194, 112229. [Google Scholar] [CrossRef]
- Sadhana Senapati, S.P.; Das, A.K. Patnaik, Kinetics and Mechanism of Oxidation of L-Ascorbic Acid by Pt(IV)(aq) in Aqueous Hydrochloric Acid Medium. Adv. Chem. Phys. 2012, 2012, 143734. [Google Scholar]
- Lemma, K.; Berglund, J.; Farrell, N.; Elding, L.I. Kinetics and mechanism for reduction of anticancer-active tetrachloroam(m)ine platinum(IV) compounds by glutathione. J. Biol. Inorg. Chem. 2000, 5, 300–306. [Google Scholar] [CrossRef]
- Zorov, D.; Bannikova, S.Y.; Belousov, V.; Vyssokikh, M.Y.; Zorova, L.; Isaev, N.; Krasnikov, B.; Plotnikov, E.Y. Reactive oxygen and nitrogen species: Friends or foes? Biochemistry 2005, 70, 215–221. [Google Scholar] [CrossRef]
- Zorov, D.; Plotnikov, E.Y.; Jankauskas, S.; Isaev, N.; Silachev, D.; Zorova, L.; Pevzner, I.; Pulkova, N.; Zorov, S.; Morosanova, M. The phenoptosis problem: What is causing the death of an organism? Lessons from acute kidney injury. Biochemistry 2012, 77, 742–753. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- McQuade, R.M.; Stojanovska, V.; Bornstein, J.C.; Nurgali, K. PARP inhibition in platinum-based chemotherapy: Chemopotentiation and neuroprotection. Pharmacol. Res. 2018, 137, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhou, Z.; Li, K.; Liu, S. Nanomaterials-Induced Redox Imbalance: Challenged and Opportunities for Nanomaterials in Cancer Therapy. Adv. Sci. 2024, 16, e2308632. [Google Scholar] [CrossRef]
- Verma, P.; Rishi, B.; George, N.G.; Kushwaha, N.; Dhandha, H.; Kaur, M.; Jain, A.; Jain, A.; Chaudhry, S.; Singh, A.; et al. Recent advances and future directions in etiopathogenesis and mechanisms of reactive oxygen species in cancer treatment. Pathol. Oncol. Res. 2023, 29, 1611415. [Google Scholar] [CrossRef]
- Juan, C.A.; Pérez de la Lastra, J.M.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef] [PubMed]
- Iqubal, A.; Haque, S.E. Molecular Mechanism of Oxidative Stress in Cancer and Its Therapeutics. In Handbook of Oxidative Stress in Cancer: Therapeutic Aspects, 1st ed.; Chakraborti, S., Ed.; Springer: Singapore, 2022; pp. 3401–3415. [Google Scholar]
- Bredesen, D.E. Apoptosis: Overview and signal transduction pathways. J. Neurotrauma 2000, 17, 801–810. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.; Huang, Z.; Lin, Z.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef]
- Liu, J.; Cao, Y.; Hu, B.; Li, T.; Zhang, W.; Zhang, Z.; Gao, J.; Niu, H.; Ding, T.; Wu, J.; et al. Older but Stronger: Development of Platinum-Based Antitumor Agents and Research Advances in Tumor Immunity. Inorganics 2023, 11, 145. [Google Scholar] [CrossRef]
- Florea, A.M.; Büsselberg, D. Cisplatin as an Anti-Tumor Drug: Cellular Mechanisms of Activity, Drug Resistance and Induced Side Effects. Cancers 2011, 3, 1351–1371. [Google Scholar] [CrossRef]
- Spiro, T.G. Nucleic Acid-Metal Ion Interactions, 1st ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1980; Volume 1, pp. 1–29. [Google Scholar]
- Jia, C.; Deacon, G.B.; Zhang, Y.; Gao, C. Platinum(IV) antitumor complexes and their nano-drug delivery. Coord. Chem. Rev. 2021, 429, 213640. [Google Scholar] [CrossRef]
- Sánchez-Camacho, J.; Infante-Tadeo, S.; Carrasco, A.C.; Scoditti, S.; Martínez, Á.; Barroso-Bujans, F.; Sicilia, E.; Pizarro, A.M.; Salassa, L. Flavin-Conjugated Pt(IV) Anticancer Agents. Inorg. Chem. 2023, 62, 5644–5651. [Google Scholar] [CrossRef] [PubMed]
- Carr, J.L.; Tingle, M.D.; McKeage, M.J. Satraplatin activation by haemoglobin, cytochrome C and liver microsomes in vitro. Cancer Chemother. Pharmacol. 2006, 57, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Theiner, S.; Varbanov, H.P.; Galanski, M.S.; Egger, A.E.; Berger, W.; Heffeter, P.; Keppler, B.K. Comparative in vitro and in vivo pharmacological investigation of platinum(IV) complexes as novel anticancer drug candidates for oral application. J. Biol. Inorg. Chem. 2015, 20, 89–99. [Google Scholar] [CrossRef]
- Canil, G.; Gurruchaga-Pereda, J.; Braccini, S.; Marchetti, L.; Funaioli, T.; Marchetti, F.; Pratesi, A.; Salassa, L.; Gabbiani, C. Synthesis, Characterization and Photoactivation Studies on the Novel Pt(IV)-Based [Pt(OCOCH3)3(phterpy)] Complex. Int. J. Mol. Sci. 2023, 24, 1106. [Google Scholar] [CrossRef]
- Xu, L.; Kong, X.; Li, X.; Zhang, B.; Deng, Y.; Wang, J.; Duan, C.; Zhang, D.; Liu, W. Current Status of Novel Multifunctional Targeted Pt(IV) Compounds and Their Reductive Release Properties. Molecules 2024, 29, 746. [Google Scholar] [CrossRef]
- Afifah, N.N.; Diantini, A.; Intania, R.; Abdulah, R.; Barliana, M.I. Genetic Polymorphisms and the Efficacy of Platinum-Based Chemotherapy: Review. Pharmgenomics Pers. Med. 2020, 8, 427–444. [Google Scholar] [CrossRef]
- Rahman, A.; Roh, J.K.; Wolpert-DeFilippes, M.K.; Goldin, A.; Venditti, J.M.; Woolley, P.V. Therapeutic and Pharmacological Studies of Tetrachloro(d,l-trans)1,2-diaminocyclohexane Platinum (IV) (Tetraplatin), a New Platinum Analogue. Cancer Res. 1988, 48, 1745–1752. [Google Scholar]
- Gibbons, G.R.; Wyrick, S.D.; Chaney, S.G. Rapid Reduction of Tetrachloro(d,l-trans)1,2-diaminocyclohexaneplatinum(IV) (Tetraplatin) in RPMI 1640 Tissue Culture Medium. Cancer Res. 1989, 49, 1402–1407. [Google Scholar]
- Christian, M.C. Phase I Trials with Ormaplatin (Tetraplatin). In Platinum and Other Metal Coordination Compounds in Cancer Chemotherapy, 1st ed.; Howell, S.B., Ed.; Springer: Boston, MA, USA, 1991; pp. 453–458. [Google Scholar]
- Pendyala, L.; Cowens, J.W.; Chheda, G.B.; Dutta, S.P.; Creaven, P.J. Identification of cis-dichloro-bis-isopropylamine platinum(II) as a major metabolite of iproplatin in humans. Cancer Res. 1988, 48, 3533–3536. [Google Scholar]
- Kim, S.D.; Vrana, O.; Kleinwächter, V.; Niki, K.; Brabec, V. Polarographic Determination of Subnanogram Quantities of Free Platinum in Reaction Mixture with Dna. Anal. Lett. 1990, 23, 1505–1518. [Google Scholar] [CrossRef]
- Blatter, E.E.; Vollano, J.F.; Krishnan, B.S.; Dabrowiak, J.C. Interaction of the antitumor agents cis, cis, trans-Pt (IV)(NH3) 2Cl2 (OH)2 and cis, cis, trans-Pt (IV)[(CH3) 2CHNH2] 2Cl2 (OH)2 and their reduction products with PM2 DNA. Biochemistry 1984, 23, 4817–4820. [Google Scholar] [CrossRef]
- Zhiqin, D.; Houzong, Y.; Zhigang, W.; Guangyu, Z. Platinum anticancer drugs: Targeting and delivery. In Comprehensive Inorganic Chemistry III, 3rd ed.; Reedijk, J., Poeppelmeier, K.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2023; pp. 808–846. [Google Scholar]
- Novakova, O.; Vrana, O.; Kiseleva, V.I.; Brabec, V. DNA Interactions of Antitumor Platinum(IV) Complexes. Eur. J. Biochem. 1995, 228, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhong, X.; Yuan, H.; Guo, Y.; Song, D.; Qi, F.; Guo, Z. Interfering in apoptosis and DNA repair of cancer cells to conquer cisplatin resistance by platinum (IV) prodrugs. Chem. Sci. 2020, 11, 3829–3835. [Google Scholar] [CrossRef]
- Elding, L.I.; Gustafson, L. Bromide anation kinetics for some platinum(IV) bromo aqua complexes. Inorg. Chim. Acta 1977, 22, 201–207. [Google Scholar] [CrossRef]
- Laverick, M.; Nias, A.H.W.; Sadler, P.J.; Ismail, I.M. Transition from laboratory to clinic in cancer treatment. Abstracts of symposium papers. Br. J. Cancer 1981, 43, 732. [Google Scholar]
- van der Veer, J.L.; Peters, A.R.; Reedijk, J. Reaction products from platinum (IV) amine compounds and 5′-GMP are mainly bis (5′-GMP) platinum (II) amine adducts. J. Inorg. Biochem. 1986, 26, 137–142. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Q.; Ng, K.Y.; Xu, Z.; Xu, W.; Zhu, G. Advances in technical strategies for monitoring the reduction of platinum (iv) complexes. Inorg. Chem. Front. 2024, 11, 3085–3118. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, Z.; Deng, Z.; Zhu, G. Recent advances in the synthesis, stability, and activation of platinum(iv) anticancer prodrugs. Coord. Chem. Rev. 2021, 442, 213991. [Google Scholar] [CrossRef]
- Johnstone, T.C.; Alexander, S.M.; Wilson, J.J.; Lippard, S.J. Oxidative halogenation of cisplatin and carboplatin: Synthesis, spectroscopy, and crystal and molecular structures of Pt(iv) prodrugs. Dalton Trans. 2015, 44, 119–129. [Google Scholar] [CrossRef]
- Hu, X.; Li, F.; Noor, N.; Ling, D. Platinum drugs: From Pt(ii) compounds, Pt(iv) prodrugs, to Pt nanocrystals/nanoclusters. Sci. Bull. 2017, 62, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Imberti, C.; Sadler, P.J. Diazido platinum(IV) complexes for photoactivated anticancer chemotherapy. Inorg. Chem. Front. 2019, 6, 1623–1638. [Google Scholar] [CrossRef]
- Lemma, K.; Shi, T.; Elding, L.I. Kinetics and mechanism for reduction of the anticancer prodrug trans,trans,trans-[PtCl2(OH)2(c,-C6H11NH2)(NH3)] (JM335) by thiols. Inorg. Chem. 2000, 39, 1728–1734. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Filotto, C.; Bisanzo, M.; Delaney, S.; Lagasee, D.; Whitworth, J.L.; Jusko, A.; Li, C.; Wood, N.A.; Willingham, J.; et al. Reduction and anticancer activity of platinum(iv) complexes. Inorg. Chem. 1998, 37, 2500–2504. [Google Scholar] [CrossRef]
- Still, B.M.; Kumar, P.G.A.; Aldrich-Wright, J.R.; Price, W.S. 195Pt NMR—Theory and application. Chem. Soc. Rev. 2007, 36, 665–686. [Google Scholar] [CrossRef]
- Pregosin, P.S.; Kretschmer, M.; Preetz, W.; Rimkus, G. 195Pt NMR studies on stereoisomeric chloro bromo platinates(IV). Z. Naturforsch. 1982, 37, 1422–1424. [Google Scholar] [CrossRef]
- Wexselblatt, E.; Gibson, D. What do we know about the reduction of Pt(iv) pro-drugs? J. Inorg. Biochem. 2012, 117, 220–229. [Google Scholar] [CrossRef]
- Berners-Price, S.J.; Ronconi, L.; Sadler, P.J. Insights into the mechanism of action of platinum anticancer drugs from multinuclear NMR spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc. 2006, 49, 65–98. [Google Scholar] [CrossRef]
- Sahoo, D.; Deb, P.; Basu, T.; Bardhan, S.; Patra, S.; Sukul, P.K. Advancements in platinum-based anticancer drug development: A comprehensive review of strategies, discoveries, and future perspectives. Bioorg. Med. Chem. 2024, 112, 117894. [Google Scholar] [CrossRef]
- Parveen, S. Platinum-based cancer chemotherapeutics: Recent trends and future perspectives. Curr. Chin. Sci. 2022, 2, 275–293. [Google Scholar] [CrossRef]
- Marotta, C.; Giorgi, E.; Binacchi, F.; Cirri, D.; Gabbiani, C.; Pratesi, A. An overview of recent advancements in anticancer Pt (IV) prodrugs: New smart drug combinations, activation and delivery strategies. Inorganica Chim. Acta 2023, 548, 121388. [Google Scholar] [CrossRef]
- Canil, G.; Braccini, S.; Marzo, T.; Marchetti, L.; Pratesi, A.; Biver, T.; Gabbiani, C. Photocytotoxic Pt (iv) complexes as prospective anticancer agents. Dalton Trans. 2019, 48, 10933–10944. [Google Scholar] [CrossRef]
- Ozols, R.F.; Bundy, B.N.; Greer, B.E.; Fowler, J.M.; Clarke-Pearson, D.; Burger, R.A.; Mannel, R.S.; DeGeest, K.; Hartenbach, E.M.; Baergen, R.; et al. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: A Gynecologic Oncology Group study. J. Clin. Oncol. 2003, 21, 3194–3200. [Google Scholar] [CrossRef] [PubMed]
- Kondagunta, G.V.; Bacik, J.; Donadio, A.; Bajorin, D.; Marion, S.; Sheinfeld, J.; Bosl, G.J.; Motzer, R.J. Combination of paclitaxel, ifosfamide, and cisplatin is an effective second-line therapy for patients with relapsed testicular germ cell tumors. J. Clin. Oncol. 2005, 23, 6549–6555. [Google Scholar] [CrossRef] [PubMed]
- Gold, J.M.; Raja, A. Cisplatin. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Silver, D.P.; Richardson, A.L.; Eklund, A.C.; Wang, Z.C.; Szallasi, Z.; Li, Q.; Juul, N.; Leong, C.O.; Calogrias, D.; Buraimoh, A.; et al. Efficacy of neoadjuvant Cisplatin in triple-negative breast cancer. J. Clin. Oncol. 2010, 28, 1145–1153. [Google Scholar] [CrossRef]
- Whitney, C.W.; Sause, W.; Bundy, B.N.; Malfetano, J.H.; Hannigan, E.V.; Fowler, W.C.; Clarke-Pearson, D.L.; Liao, S.Y. Randomized comparison of fluorouracil plus cisplatin versus hydroxyurea as an adjunct to radiation therapy in stage IIB-IVA carcinoma of the cervix with negative para-aortic lymph nodes: A Gynecologic Oncology Group and Southwest Oncology Group study. J. Clin. Oncol. 1999, 17, 1339–1348. [Google Scholar] [CrossRef]
- Fleming, G.F.; Brunetto, V.L.; Cella, D.; Look, K.Y.; Reid, G.C.; Munkarah, A.R.; Kline, R.; Burger, R.A.; Goodman, A.; Burks, R.T. Phase III trial of doxorubicin plus cisplatin with or without paclitaxel plus filgrastim in advanced endometrial carcinoma: A Gynecologic Oncology Group Study. J. Clin. Oncol. 2004, 22, 2159–2166. [Google Scholar] [CrossRef]
- Fornasiero, A.; Daniele, O.; Ghiotto, C.; Piazza, M.; Fiore-Donati, L.; Calabró, F.; Rea, F.; Fiorentino, M.V. Chemotherapy for invasive thymoma. A 13-year experience. Cancer 1991, 68, 30–33. [Google Scholar] [CrossRef]
- Velasquez, W.S.; Cabanillas, F.; Salvador, P.; McLaughlin, P.; Fridrik, M.; Tucker, S.; Jagannath, S.; Hagemeister, F.B.; Redman, J.R.; Swan, F. Effective salvage therapy for lymphoma with cisplatin in combination with high-dose Ara-C and dexamethasone (DHAP). Blood 1988, 71, 117–122. [Google Scholar] [CrossRef]
- Velasquez, W.S.; McLaughlin, P.; Tucker, S.; Hagemeister, F.B.; Swan, F.; Rodriguez, M.A.; Romaguera, J.; Rubenstein, E.; Cabanillas, F. ESHAP--an effective chemotherapy regimen in refractory and relapsing lymphoma: A 4-year follow-up study. J. Clin. Oncol. 1994, 12, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S. Cisplatin: The first metal based anticancer drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer. 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Rabik, C.A.; Dolan, M.E. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat. Rev. 2007, 33, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gou, S.; Zhao, J.; Chen, F.; Xu, G.; Liu, X. Cytotoxicity profile of novel sterically hindered platinum(II) complexes with (1R,2R)-N(1),N(2)-dibutyl-1,2-diaminocyclohexane. Eur. J. Med. Chem. 2015, 96, 187–195. [Google Scholar] [CrossRef]
- Yu, H.; Gou, S.; Wang, Z.; Chen, F.; Fang, L. Toward overcoming cisplatin resistance via sterically hindered platinum(II) complexes. Eur. J. Med. Chem. 2016, 114, 141–152. [Google Scholar] [CrossRef]
- Wilson, J.J.; Lippard, S.J. In Vitro Anticancer Activity of cis -Diammineplatinum(II) Complexes with β-Diketonate Leaving Group Ligands. J. Med. Chem. 2012, 55, 5326–5336. [Google Scholar] [CrossRef]
- Piccinonna, S.; Margiotta, N.; Pacifico, C.; Lopalco, A.; Denora, N.; Fedi, S.; Corsini, M.; Natile, G. Dinuclear Pt(ii)-bisphosphonate complexes: A scaffold for multinuclear or different oxidation state platinum drugs. Dalt. Trans. 2012, 41, 9689–9699. [Google Scholar] [CrossRef]
- Hall, M.D.; Mellor, H.R.; Callaghan, R.; Hambley, T.W. Basis for design and development of platinum(IV) anticancer complexes. J. Med. Chem. 2007, 50, 3403–3411. [Google Scholar] [CrossRef]
- Galanski, M.S.; Jakupec, M.A.; Keppler, B.K. Update of the Preclinical Situation of Anticancer Platinum Complexes: Novel Design Strategies and Innovative Analytical Approaches. Curr. Med. Chem. 2005, 12, 2075–2094. [Google Scholar] [CrossRef]
- Schmidt, C.; Babu, T.; Kostrhunova, H.; Timm, A.; Basu, U.; Ott, I.; Gandin, V.; Brabec, V.; Gibson, D. Are Pt(IV) Prodrugs That Release Combretastatin A4 True Multi-action Prodrugs? J. Med. Chem. 2021, 64, 11364–11378. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D. Multi-action Pt(IV) anticancer agents; do we understand how they work? J. Inorg. Biochem. 2019, 191, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, V.; Sadler, P.J. Platinum(IV) Prodrugs. Met. Ions Life Sci. 2018, 18, 9783110470734-009. [Google Scholar]
- Kenny, R.G.; Marmion, C.J. Toward multi-targeted platinum and ruthenium drugsa new paradigm in cancer drug treatment regimens? Chem. Rev. 2019, 119, 1058. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Z.; Bonnitcha, P.; Wexselblatt, E.; Klein, A.V.; Najajreh, Y.; Gibson, D.; Hambley, T.W. Facile preparation of mono-, Di- and mixed-carboxylato platinum (IV) complexes for versatile anticancer prodrug design. Chem. Eur. J. 2013, 19, 1672–1676. [Google Scholar] [CrossRef]
- Song, Y.; Suntharalingam, K.; Yeung, J.S.; Royzen, M.; Lippard, S.J. Synthesis and characterization of Pt(IV) fluorescein conjugates to investigate Pt(IV) intracellular transformations. Bioconjug. Chem. 2013, 24, 1733–1740. [Google Scholar] [CrossRef]
- Nemirovski, A.; Vinograd, I.; Takrouri, K.; Mijovilovich, A.; Rompel, A.; Gibson, D. New reduction pathways for ctc-[PtCl2(CH3CO2)2(NH3)(Am)] anticancer prodrugs. Chem. Commun. 2010, 46, 1842–1844. [Google Scholar] [CrossRef]
- Jungwirth, U.; Kowol, C.R.; Keppler, B.K.; Hartinger, C.G.; Berger, W.; Heffeter, P. Anticancer activity of metal complexes: Involvement of redox processes. Antioxid. Redox Signal. 2011, 15, 1085–1127. [Google Scholar] [CrossRef]
- Han, X.; Sun, J.; Wang, Y.; He, Z. Recent advances in platinum (IV) complex-based delivery systems to improve platinum (II) anticancer therapy. Med. Res. Rev. 2015, 35, 1268–1299. [Google Scholar] [CrossRef]
- Grek, C.L.; Tew, K.D. Redox metabolism and malignancy. Curr. Opin. Pharmacol. 2010, 10, 362–368. [Google Scholar] [CrossRef]
- Shi, Y.; Liu, S.A.; Kerwood, D.J.; Goodisman, J.; Dabrowiak, J.C. Pt(IV) complexes as prodrugs for cisplatin. J. Inorg. Biochem. 2012, 107, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Washko, P.; Rotrosen, D.; Levine, M. Ascorbic acid in human neutrophils, Am. J. Clin. Nutr. 1991, 54, 1221S–1227S. [Google Scholar] [CrossRef] [PubMed]
- Michelet, F.; Gueguen, R.; Leroy, P.; Wellman, M.; Nicolas, A.; Siest, G. Blood and plasma glutathione measured in healthy subjects by HPLC: Relation to sex, aging, biological variables, and life habits. Clin. Chem. 1995, 41, 1509–1517. [Google Scholar] [CrossRef]
- Reiber, H.; Ruff, M.; Uhr, M. Ascorbate concentration in human cerebrospinal fluid (CSF) and serum. Intrathecal accumulation and CSF flow rate. Clin. Chim. Acta. 1993, 217, 163–173. [Google Scholar] [CrossRef]
- Hall, M.D.; Hambley, T.W. Platinum(IV) antitumour compounds: Their bioinorganic chemistry. Coord. Chem. Rev. 2002, 232, 49–67. [Google Scholar] [CrossRef]
- Ellis, L.T.; Er, H.M.; Hambley, T.W. The influence of the axial ligands of a series of platinum(iv) anti-Cancer complexes on their reduction to platinum(ii) and reaction with DNA. Aust. J. Chem. 1995, 48, 793–806. [Google Scholar] [CrossRef]
- Thomson, A.J.; Williams, R.J.P.; Reslova, S. The chemistry of complexes related to cis-Pt(NH3)2Cl2. An anti-tumour drug. In Biochemistry, 1st ed.; Thomson, A.J., Williams, R.J.P., Reslova, S., Wood, J.M., Brown, D.G., Bray, R.C., Swann, J.C., Neilands, J.B., Eds.; Springer: Berlin/Heidelberg, Germany, 1972; Volume 11, pp. 1–46. [Google Scholar]
- Platts, J.A.; Hibbs, D.E.; Hambley, T.W.; Hall, M.D. Calculation of the hydrophobicity of platinum drugs. J. Med. Chem. 2001, 44, 472–474. [Google Scholar] [CrossRef]
- Ponte, F.; Scoditti, S.; Mazzone, G.; Sicilia, E. The current status in computational exploration of Pt(iv) pro-drug activation by reduction. Phys. Chem. Chem. Phys. 2023, 25, 15586–15599. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, Y.; Tian, H. Current developments in Pt(IV) prodrugs conjugated with bioactive ligands. Bioinorg. Chem. Appl. 2018, 2018, 8276139. [Google Scholar] [CrossRef]
- Spector, D.; Krasnovskaya, O.; Pavlov, K.; Erofeev, A.; Gorelkin, P.; Beloglazkina, E.; Majouga, A. Pt(IV) Prodrugs with NSAIDs as Axial Ligands. Int. J. Mol. Sci. 2021, 22, 3817. [Google Scholar] [CrossRef]
- Chaney, S.G.; Till, G.K.; Wyrick, S. In vitro biotransformations of tetrachloro(d,1-trans)-1,2-diaminocyclohexaneplatinum(IV) (tetraplatin) in rat plasma. Cancer Res. 1990, 50, 4539–4545. [Google Scholar]
- Nandi, A.; Yan, L.J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxid. Med. Cell Longev. 2019, 11, 9613090. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Valko, M. Several lines of antioxidant defense against oxidative stress: Antioxidant enzymes, nanomaterials with multiple enzyme-mimicking activities, and low-molecular-weight antioxidants. Arch. Toxicol. 2024, 98, 1323–1367. [Google Scholar] [CrossRef]
- Rosa, A.C.; Corsi, D.; Cavi, N.; Bruni, N.; Dosio, F. Superoxide Dismutase Administration: A Review of Proposed Human Uses. Molecules 2021, 26, 1844. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gao, W.; Ma, Y.; Cheng, L.; Zhang, L.; Liu, Q.; Liu, C. Integrating Pt nanoparticles with carbon nanodots to achieve robust cascade superoxide dismutase-catalase nanozyme for antioxidant therapy. Nano Today 2023, 49, 101768. [Google Scholar] [CrossRef]
- Al-Fahdawi, M.Q.; Al-Doghachi, F.A.J.; Abdullah, Q.K.; Hammad, R.T.; Rasedee, A.; Ibrahim, W.N.; Alshwyeh, H.A.; Alosaimi, A.A.; Aldosary, S.K.; Eid, E.E.M.; et al. Oxidative stress cytotoxicity induced by platinum-doped magnesia nanoparticles in cancer cells. Biomed. Pharmacother. 2021, 138, 111483. [Google Scholar] [CrossRef]
- Catala, A. Lipid peroxidation of membrane phospholipids generates hydroxy-alkenals and oxidized phospholipids active in physiological and/or pathological conditions. Chem. Phys. Lipids 2009, 157, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, R.M. Reactive oxygen species at phospholipid bilayers: Distribution, mobility and permeation. Biochim. Biophys. Acta (BBA)-Biomembr. 2014, 1838, 438–444. [Google Scholar] [CrossRef]
- Aioub, M.; Panikkanvalappil, S.R.; El-Sayed, M.A. Platinum-coated gold nanorods: Efficient reactive oxygen scavengers that prevent oxidative damage toward healthy, untreated cells during plasmonic photothermal therapy. ACS Nano. 2017, 11, 579–586. [Google Scholar] [CrossRef]
- Nejdl, L.; Kudr, J.; Moulick, A.; Hegerova, D.; Ruttkay-Nedecky, B.; Gumulec, J.; Cihalova, K.; Smerkova, K.; Dostalova, S.; Krizkova, S. Platinum nanoparticles induce damage to DNA and inhibit DNA replication. PLoS ONE 2017, 12, e0180798. [Google Scholar] [CrossRef]
- Al-Qubaisi, M.; Rozita, R.; Yeap, S.K.; Omar, A.R.; Ali, A.M.; Alitheen, N.B. Selective cytotoxicity of goniothalamin against hepatoblastoma HepG2 cells. Molecules 2011, 16, 2944–2959. [Google Scholar] [CrossRef] [PubMed]
- Al-Qubaisi, M.; Rosli, R.; Subramani, T.; Omar, A.R.; Yeap, S.K.; Ali, A.M.; Alitheen, N.B. Goniothalamin selectively induces apoptosis on human hepatoblastoma cells through caspase-3 activation. Nat. Prod. Res. 2013, 27, 2216–2218. [Google Scholar] [CrossRef] [PubMed]
- Slee, E.A.; Adrain, C.; Martin, S.J. Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J. Biol. Chem. 2001, 276, 7320–7326. [Google Scholar] [CrossRef]
- Karlsson, J.O.G.; Jynge, P. Manganese- and Platinum-Driven Oxidative and Nitrosative Stress in Oxaliplatin-Associated CIPN with Special Reference to Ca4Mn(DPDP)5, MnDPDP and DPDP. Int. J. Mol. Sci. 2024, 25, 4347. [Google Scholar] [CrossRef]
- Zajączkowska, R.; Kocot-Kępska, M.; Leppert, W.; Wrzosek, A.; Mika, J.; Wordliczek, J. Mechanisms of Chemotherapy-Induced Peripheral Neuropathy. Int. J. Mol. Sci. 2019, 20, 1451. [Google Scholar] [CrossRef] [PubMed]
- Canta, A.; Pozzi, E.; Carozzi, V.A. Mitochondrial Dysfunction in Chemotherapy-Induced Peripheral Neuropathy (CIPN). Toxics 2015, 3, 198–223. [Google Scholar] [CrossRef]
- Kober, K.M.; Olshen, A.; Conley, Y.P.; Schumacher, M.; Topp, K.; Smoot, B.; Mazor, M.; Chesney, M.; Hammer, M.; Paul, S.M. Expression of mitochondrial dysfunction-related genes and pathways in paclitaxel-induced peripheral neuropathy in breast cancer survivors. Mol. Pain. 2018, 14, 1744806918816462. [Google Scholar] [CrossRef]
- Podratz, J.L.; Knight, A.M.; Ta, L.E.; Staff, N.P.; Gass, J.M.; Genelin, K.; Schlattau, A.; Lathroum, L.; Windebank, A.J. Cisplatin induced mitochondrial DNA damage in dorsal root ganglion neurons. Neurobiol. Dis. 2011, 41, 661–668. [Google Scholar] [CrossRef]
- Yang, Y.; Luo, L.; Cai, X.; Fang, Y.; Wang, J.; Chen, G.; Yang, J.; Zhou, Q.; Sun, X.; Cheng, X. Nrf2 inhibits oxaliplatin-induced peripheral neuropathy via protection of mitochondrial function. Free Radic. Biol. Med. 2018, 120, 13–24. [Google Scholar] [CrossRef]
- Joseph, E.K.; Chen, X.; Bogen, O.; Levine, J.D. Oxaliplatin acts on IB4-positive nociceptors to induce an oxidative stress-dependent acute painful peripheral neuropathy. J. Pain 2008, 9, 463–472. [Google Scholar] [CrossRef]
- Valko, M.; Morris, H.; Cronin, M.T. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Xiao, W.H.; Bennett, G.J. Functional deficits in peripheral nerve mitochondria in rats with paclitaxel- and oxaliplatin-evoked painful peripheral neuropathy. Exp. Neurol. 2011, 232, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Di Cesare Mannelli, L.; Zanardelli, M.; Failli, P.; Ghelardini, C. Oxaliplatin-induced neuropathy: Oxidative stress as pathological mechanism. Protective effect of silibinin. J. Pain 2012, 13, 276–284. [Google Scholar] [CrossRef]
- Di Cesare Mannelli, L.; Zanardelli, M.; Failli, P.; Ghelardini, C. Oxaliplatin-induced oxidative stress in nervous system-derived cellular models: Could it correlate with in vivo neuropathy? Free Radic. Biol. Med. 2013, 61, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Waseem, M.; Kaushik, P.; Tabassum, H.; Parvez, S. Role of Mitochondrial Mechanism in Chemotherapy-Induced Peripheral Neuropathy. Curr. Drug Metab. 2018, 19, 47–54. [Google Scholar] [CrossRef]
- Pan, L.; Song, K.; Hu, F.; Sun, W.; Lee, I. Nitric oxide induces apoptosis associated with TRPV1 channel-mediated Ca(2+) entryvia S-nitrosylation in osteoblasts. Eur. J. Pharmacol. 2013, 715, 280–285. [Google Scholar] [CrossRef]
- Jamieson, S.M.; Liu, J.; Connor, B.; McKeage, M.J. Oxaliplatin causes selective atrophy of a subpopulation of dorsal root ganglion neurons without inducing cell loss. Cancer Chemother. Pharmacol. 2005, 56, 391–399. [Google Scholar] [CrossRef]
- Apostolidis, L.; Schwarz, D.; Xia, A.; Weiler, M.; Heckel, A.; Godel, T.; Heiland, S.; Schlemmer, H.P.; Jäger, D.; Bendszus, M. Dorsal root ganglia hypertrophy as in vivo correlate of oxaliplatin-induced polyneuropathy. PLoS ONE 2017, 12, e0183845. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.S.; Fox, E.; Dennie, C.; Morgan, L.B.; McCully, C.L.; Balis, F.M. Plasma and cerebrospinal fluid pharmacokinetics of intravenous oxaliplatin, cisplatin, and carboplatin in nonhuman primates. Clin. Cancer Res. 2005, 11, 1669–1674. [Google Scholar] [CrossRef]
- Rochfort, K.D.; Collins, L.E.; Murphy, R.P.; Cummins, P.M. Downregulation of blood–brain barrier phenotype by proinflammatory cytokines involves NADPH oxidase-dependent ROS generation: Consequences for interendothelial adherens and tight junctions. PLoS ONE 2014, 9, e101815. [Google Scholar] [CrossRef]
- Argaw, A.T.; Zhang, Y.; Snyder, B.J.; Zhao, M.L.; Kopp, N.; Lee, S.C.; Raine, C.S.; Brosnan, C.F.; John, G.R. IL-1beta regulates blood–brain barrier permeability via reactivation of the hypoxia angiogenesis program. J. Immunol. 2006, 177, 5574–5584. [Google Scholar] [CrossRef] [PubMed]
- Branca, J.J.V.; Maresca, M.; Morucci, G.; Becatti, M.; Paternostro, F.; Gulisano, M.; Ghelardini, C.; Salvemini, D.; Di Cesare Mannelli, L.; Pacini, A. Oxaliplatin-induced blood brain barrier loosening: A new point of view on chemotherapy-induced neurotoxicity. Oncotarget 2018, 9, 23426–23438. [Google Scholar] [CrossRef]
- Sanna, M.D.; Ghelardini, C.; Galeotti, N. Altered Expression of Cytoskeletal and Axonal Proteins in Oxaliplatin-Induced Neuropathy. Pharmacology 2016, 97, 146–150. [Google Scholar] [CrossRef]
- Chandrasekharan, B.; Anitha, M.; Blatt, R.; Shahnavaz, N.; Kooby, D.; Staley, C.; Mwangi, S.; Jones, D.P.; Sitaraman, S.V.; Srinivasan, S. Colonic motor dysfunction in human diabetes is associated with enteric neuronal loss and increased oxidative stress. Neurogastroenterol. Motil. 2011, 23, 131-e26. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.M.; Russell, J.W.; Low, P.; Feldman, E.L. Oxidative stress in the pathogenesis of diabetic neuropathy. Endocr. Rev. 2004, 25, 612–628. [Google Scholar] [CrossRef] [PubMed]
- Areti, A.; Yerra, V.G.; Naidu, V.; Kumar, A. Oxidative stress and nerve damage: Role in chemotherapy induced peripheral neuropathy. Redox Biol. 2014, 2, 289–295. [Google Scholar] [CrossRef]
- Saifi, G.M.; Szigeti, K.; Snipes, G.J.; Garcia, C.A.; Lupski, J.R. Molecular mechanisms, diagnosis, and rational approaches to management of and therapy for Charcot-Marie-Tooth disease and related peripheral neuropathies. JIM 2003, 51, 261–283. [Google Scholar]
- Preston, T.J.; Henderson, J.T.; McCallum, G.P.; Wells, P.G. Base excision repair of reactive oxygen species-initiated 7,8-dihydro-8-oxo-2′-deoxyguanosine inhibits the cytotoxicity of platinum anticancer drugs. Mol. Cancer Ther. 2009, 8, 2015–2026. [Google Scholar] [CrossRef]
- Harrison, J.F.; Hollensworth, S.B.; Spitz, D.R.; Copeland, W.C.; Wilson, G.L.; LeDoux, S.P. Oxidative stress-induced apoptosis in neurons correlates with mitochondrial DNA base excision repair pathway imbalance. Nucleic Acids Res. 2005, 33, 4660–4671. [Google Scholar] [CrossRef]
- Jihoon, K. Polymeric biomaterials for the delivery of platinum-based anticancer drugs. Biomater. Sci. 2015, 3, 1002–1017. [Google Scholar]
- Zheng, S.; Li, G.; Shi, J.; Liu, X.; Li, M.; He, Z.; Tian, C.; Kamei, K.I. Emerging platinum(IV) prodrug nanotherapeutics: A new epoch for platinum-based cancer therapy. J. Control. Release 2023, 361, 819–846. [Google Scholar] [CrossRef]
- Bansal, A.; Simon, M.C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Meierhofer, D. Glutathione Metabolism in Renal Cell Carcinoma Progression and Implications for Therapies. Int. J. Mol. Sci. 2019, 20, 3672. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Fang, Y.Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Aspects Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [PubMed]
- Carretero, J.; Obrador, E.; Anasagasti, M.J.; Martin, J.J.; Vidal-Vanaclocha, F.; Estrela, J.M. Growth-associated changes in glutathione content correlate with liver metastatic activity of B16 melanoma cells. Clin. Exp. Metastasis. 1999, 17, 567–574. [Google Scholar] [CrossRef]
- Huang, Z.Z.; Chen, C.; Zeng, Z.; Yang, H.; Oh, J.; Chen, L.; Lu, S.C. Mechanism and significance of increased glutathione level in human hepatocellular carcinoma and liver regeneration. FASEB J. 2001, 15, 19–21. [Google Scholar] [CrossRef]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef]
- Li, B.; Qiu, B.; Lee, D.S.; Walton, Z.E.; Ochocki, J.D.; Mathew, L.K.; Mancuso, A.; Gade, T.P.; Keith, B.; Nissim, I.; et al. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature 2014, 513, 251–255. [Google Scholar] [CrossRef]
- Zhang, H.; Forman, H.J. Redox regulation of gamma-glutamyl transpeptidase. Am. J. Respir. Cell Mol. Biol. 2009, 41, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Gamcsik, M.P.; Kasibhatla, M.S.; Teeter, S.D.; Colvin, O.M. Glutathione levels in human tumors. Biomarkers 2012, 17, 671–691. [Google Scholar] [CrossRef] [PubMed]
- Hakimi, A.A.; Reznik, E.; Lee, C.H.; Creighton, C.J.; Brannon, A.R.; Luna, A.; Aksoy, B.A.; Liu, E.M.; Shen, R.; Lee, W.; et al. An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma. Cancer Cell 2016, 29, 104–116. [Google Scholar] [CrossRef]
- Desideri, E.; Ciccarone, F.; Ciriolo, M.R. Targeting Glutathione Metabolism: Partner in Crime in Anticancer Therapy. Nutrients 2019, 11, 1926. [Google Scholar] [CrossRef]
- Mi, S.; Gong, L.; Sui, Z. Friend or Foe? An Unrecognized Role of Uric Acid in Cancer Development and the Potential Anticancer Effects of Uric Acid-lowering Drugs. J. Cancer 2020, 11, 5236–5244. [Google Scholar] [CrossRef]
- Kennedy, L.; Sandhu, J.K.; Harper, M.-E.; Cuperlovic-Culf, M. Role of Glutathione in Cancer: From Mechanisms to Therapies. Biomolecules 2020, 10, 1429. [Google Scholar] [CrossRef] [PubMed]
- Gęgotek, A.; Skrzydlewska, E. Antioxidative and Anti-Inflammatory Activity of Ascorbic Acid. Antioxidants 2022, 11, 1993. [Google Scholar] [CrossRef]
- Nishikimi, M.; Yagi, K. Molecular basis for the deficiency in humans of gulonolactone oxidase, a key enzyme for ascorbic acid biosynthesis. Am. J. Clin. Nutr. 1991, 54, 1203S–1208S. [Google Scholar] [CrossRef] [PubMed]
- Reang, J.; Sharma, P.C.; Thakur, V.K.; Majeed, J. Understanding the Therapeutic Potential of Ascorbic Acid in the Battle to Overcome Cancer. Biomolecules 2021, 11, 1130. [Google Scholar] [CrossRef]
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.H.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.T.; Mitchell-Flack, M.; et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 2017, 170, 1079–1095. [Google Scholar] [CrossRef]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Darwiche, W.; Gomila, C.; Ouled-Haddou, H.; Naudot, M.; Doualle, C.; Morel, P.; Nguyen-Khac, F.; Garçon, L.; Marolleau, J.P.; Ghamlouch, H. Ascorbic acid (vitamin C) synergistically enhances the therapeutic effect of targeted therapy in chronic lymphocytic leukemia. J. Exp. Clin. Cancer Res. 2020, 39, 228. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.C.; Cook, J. Intravenous vitamin C for cancer therapy—Identifying the current gaps in our knowledge. Front. Physiol. 2018, 9, 1182. [Google Scholar] [CrossRef]
- Andrews, G.C.; Crawford, T. Recent Advances in the Derivatization of L-Ascorbic Acid. In Ascorbic Acid: Chemistry, Metabolism, and Uses; Seib, P.A., Tolbert, B.M., Eds.; American Chemical Society: Washington, DC, USA, 1982; Volume 200, pp. 59–79. [Google Scholar]
- Tsao, C.S. Vitamin C in health and disease. Antioxids. Health Dis. 1997, 5, 25–58. [Google Scholar]
- Macan, A.M.; Harej, A.; Cazin, I.; Klobučar, M.; Stepanić, V.; Pavelić, K.; Pavelić, S.K.; Schols, D.; Snoeck, R.; Andrei, G.; et al. Antitumor and antiviral activities of 4-substituted 1,2,3-triazolyl-2,3-dibenzyl-L-ascorbic acid derivatives. Eur. J. Med. Chem. 2019, 184, 111739. [Google Scholar] [CrossRef] [PubMed]
- Harej, A.; Meščić Macan, A.; Stepanić, V.; Klobučar, M.; Pavelić, K.; Pavelić, S.K.; Raić-Malić, S. The antioxidant and antiproliferative activities of 1,2,3-triazolyl-L-ascorbic acid derivatives. Int. J. Mol. Sci. 2019, 20, 4735. [Google Scholar] [CrossRef]
- Miura, K.; Haraguchi, M.; Ito, H.; Tai, A. Potential antitumor activity of 2-O-α-D-glucopyranosyl-6-O-(2-pentylheptanoyl)-L-ascorbic acid. Int. J. Mol. Sci. 2018, 19, 535. [Google Scholar] [CrossRef]
- Bordignon, B.; Chiron, J.; Fontés, M. Ascorbic acid derivatives as a new class of antiproliferative molecules. Cancer Lett. 2013, 338, 317–327. [Google Scholar] [CrossRef]
- Rosenberg, B. Some Biological Effects of Platinum Compounds New Agents for the Control of Tumours. Platin. Met. Rev. 1971, 15, 42–51. [Google Scholar] [CrossRef]
- Mehrotra, U.S.; Agarwal, M.C.; Mushran, S.P. Reduction of hexachloroplatinate by ascorbic acid. J. Inorg. Nucl. Chem. 1970, 32, 2325–2329. [Google Scholar] [CrossRef]
- Liu, Y.; Tian, H.; Xu, L.; Zhou, L.; Wang, J.; Xu, B.; Liu, C.; Elding, L.I.; Shi, T. Investigations of the Kinetics and Mechanism of Reduction of a Carboplatin Pt(IV) Prodrug by the Major Small-Molecule Reductants in Human Plasma. Int. J. Mol. Sci. 2019, 20, 5660. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iova, V.; Tincu, R.C.; Scrobota, I.; Tudosie, M.S. Pt(IV) Complexes as Anticancer Drugs and Their Relationship with Oxidative Stress. Biomedicines 2025, 13, 981. https://doi.org/10.3390/biomedicines13040981
Iova V, Tincu RC, Scrobota I, Tudosie MS. Pt(IV) Complexes as Anticancer Drugs and Their Relationship with Oxidative Stress. Biomedicines. 2025; 13(4):981. https://doi.org/10.3390/biomedicines13040981
Chicago/Turabian StyleIova, Vlad, Radu Ciprian Tincu, Ioana Scrobota, and Mihail Silviu Tudosie. 2025. "Pt(IV) Complexes as Anticancer Drugs and Their Relationship with Oxidative Stress" Biomedicines 13, no. 4: 981. https://doi.org/10.3390/biomedicines13040981
APA StyleIova, V., Tincu, R. C., Scrobota, I., & Tudosie, M. S. (2025). Pt(IV) Complexes as Anticancer Drugs and Their Relationship with Oxidative Stress. Biomedicines, 13(4), 981. https://doi.org/10.3390/biomedicines13040981