

Protective Action of 3,5-Diiodo-L-Thyronine on Cigarette Smoke-Induced Mitochondrial Dysfunction in Human Alveolar Epithelial Cells
 , , ,
, , ,  ,
,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
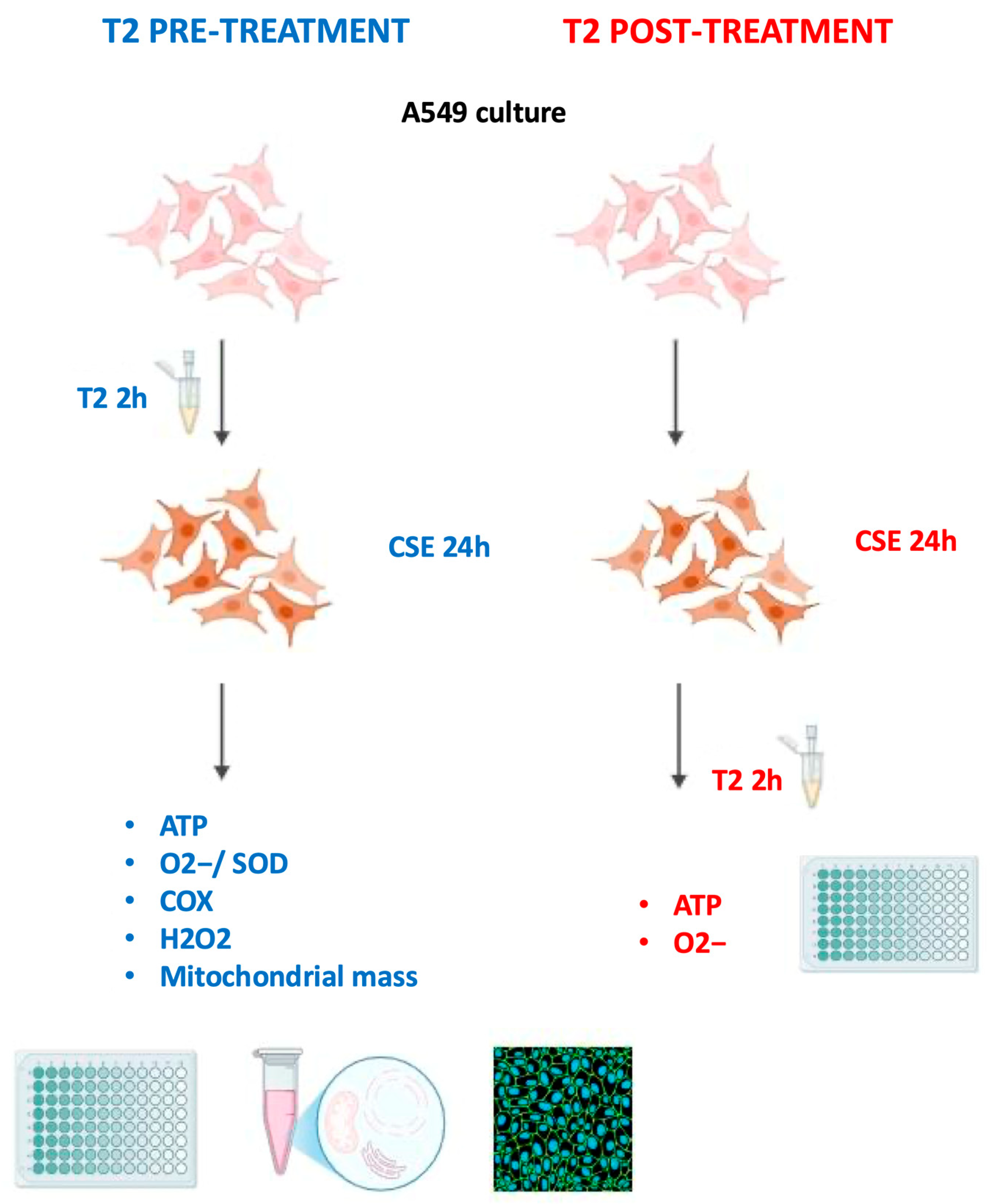
2.1. Cell Culture and Treatment
2.2. Preparation of Cigarette Smoke Extract
2.3. Thiazolyl Blue Tetrazolium Bromide Assay
2.4. ATP Assay
2.5. Mitochondrial Superoxide Radical Assay
2.6. Superoxide Dismutase Activity Assay
2.7. Measurement of Hydrogen Peroxide in Cell Lysate Samples
2.8. Mitochondria Isolation and Determination of Cytochrome Oxidase Activity
2.9. MitoTracker Green Flow Cytometry Analysis and Cell Staining
2.10. Statistical Analysis
3. Results
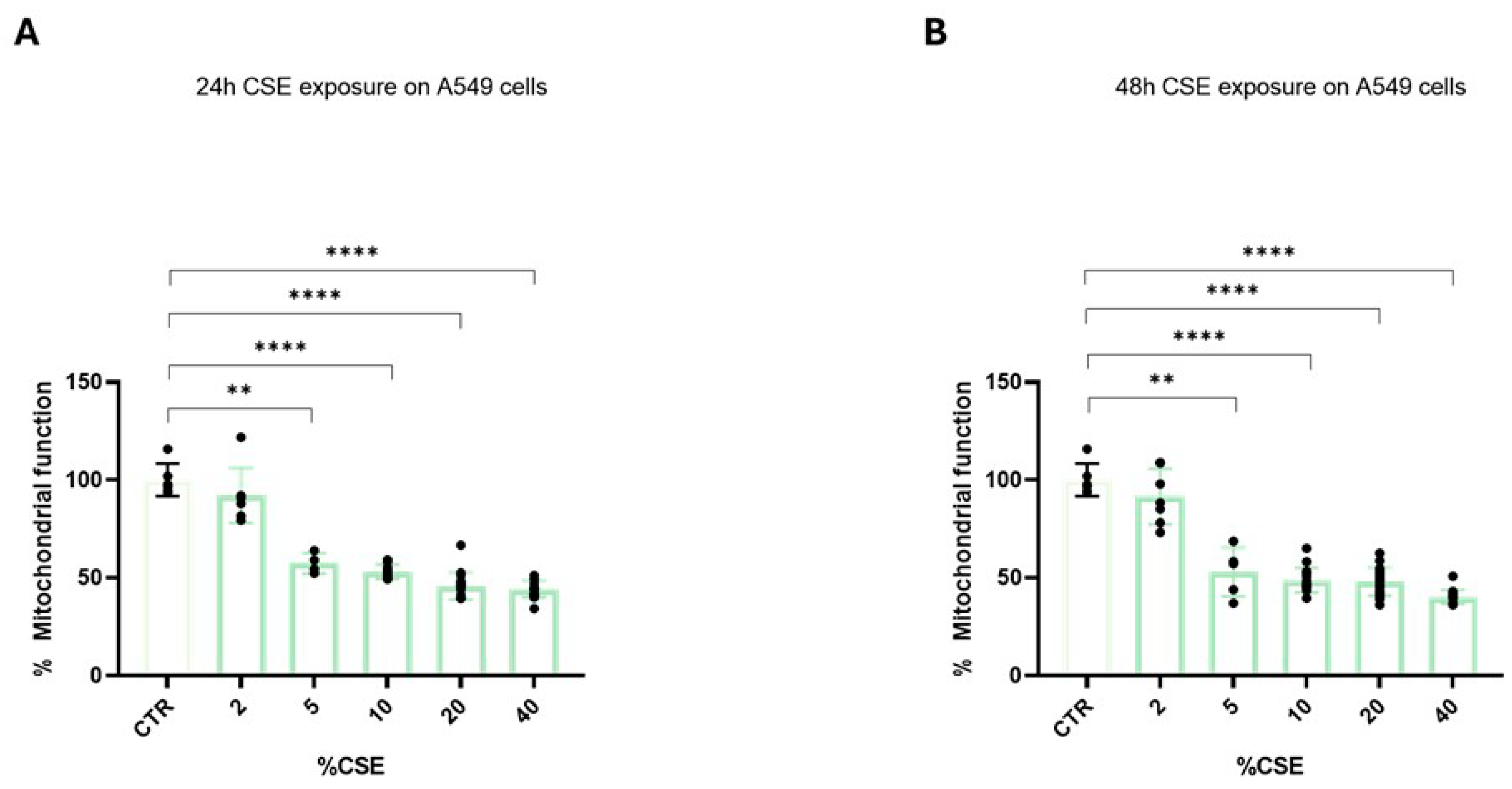
3.1. Cigarette Smoke Extract Significantly Affects Mitochondrial Reduction Properties
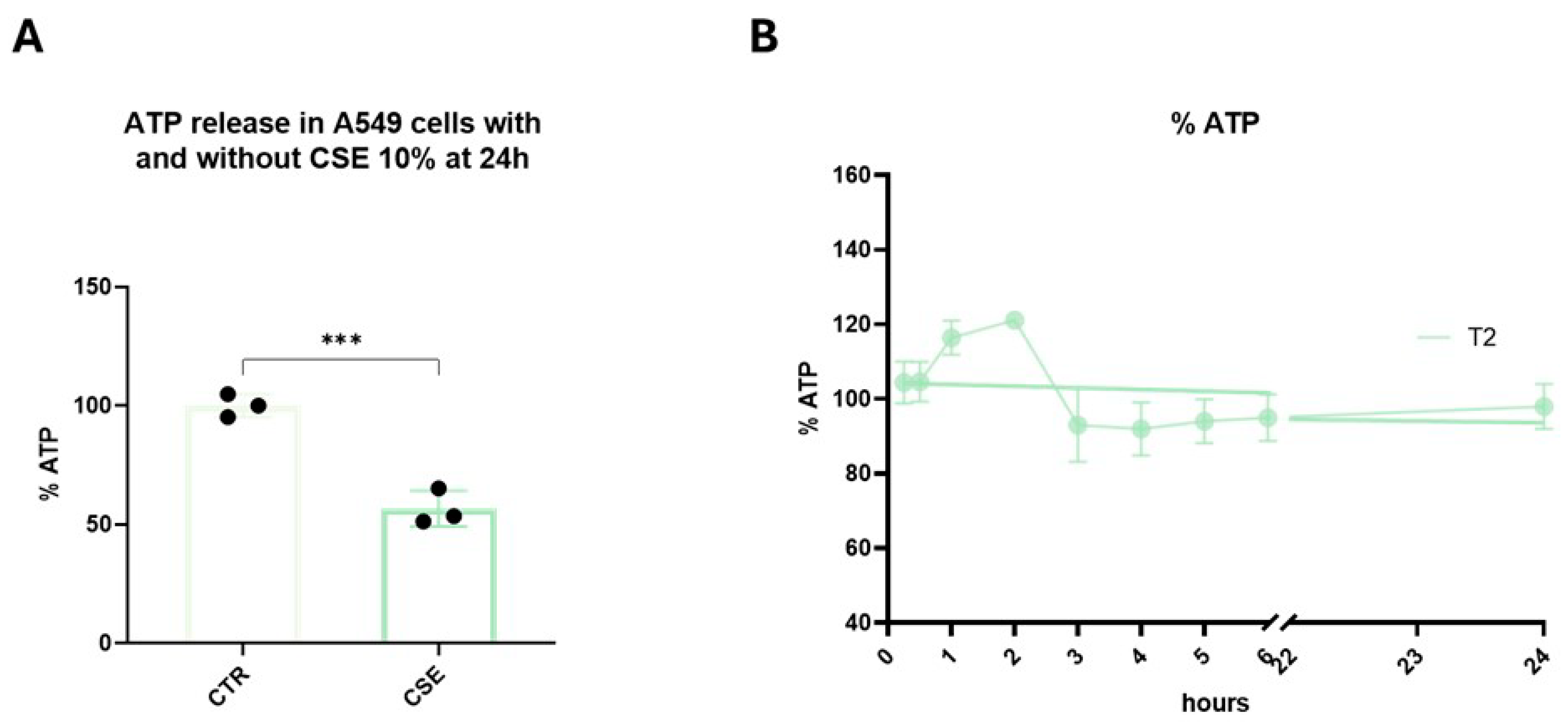
3.2. Mitochondrial Function and ATP Synthesis
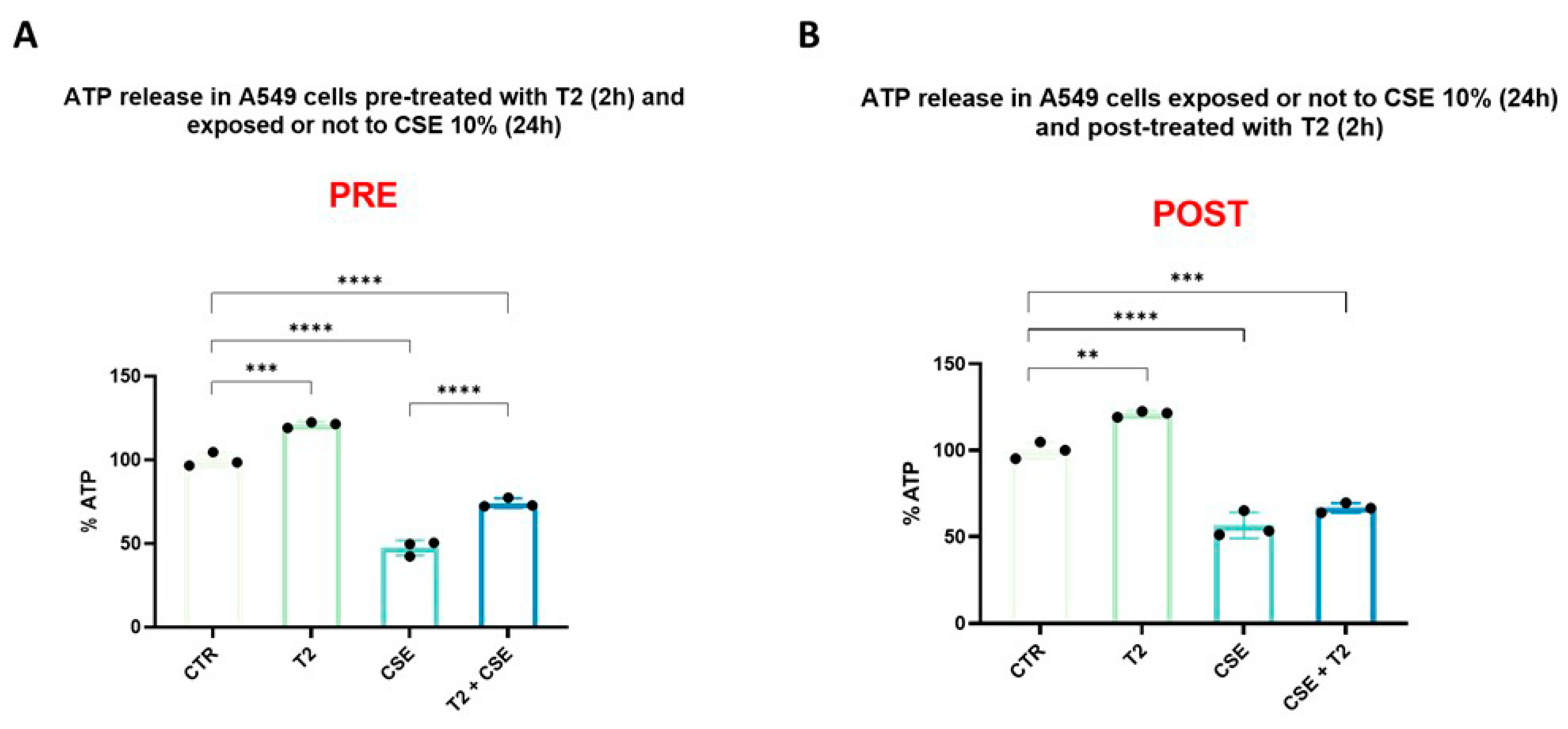
3.3. T2 Pretreatment Prevents Cigarette Smoke-Induced ATP Impairment
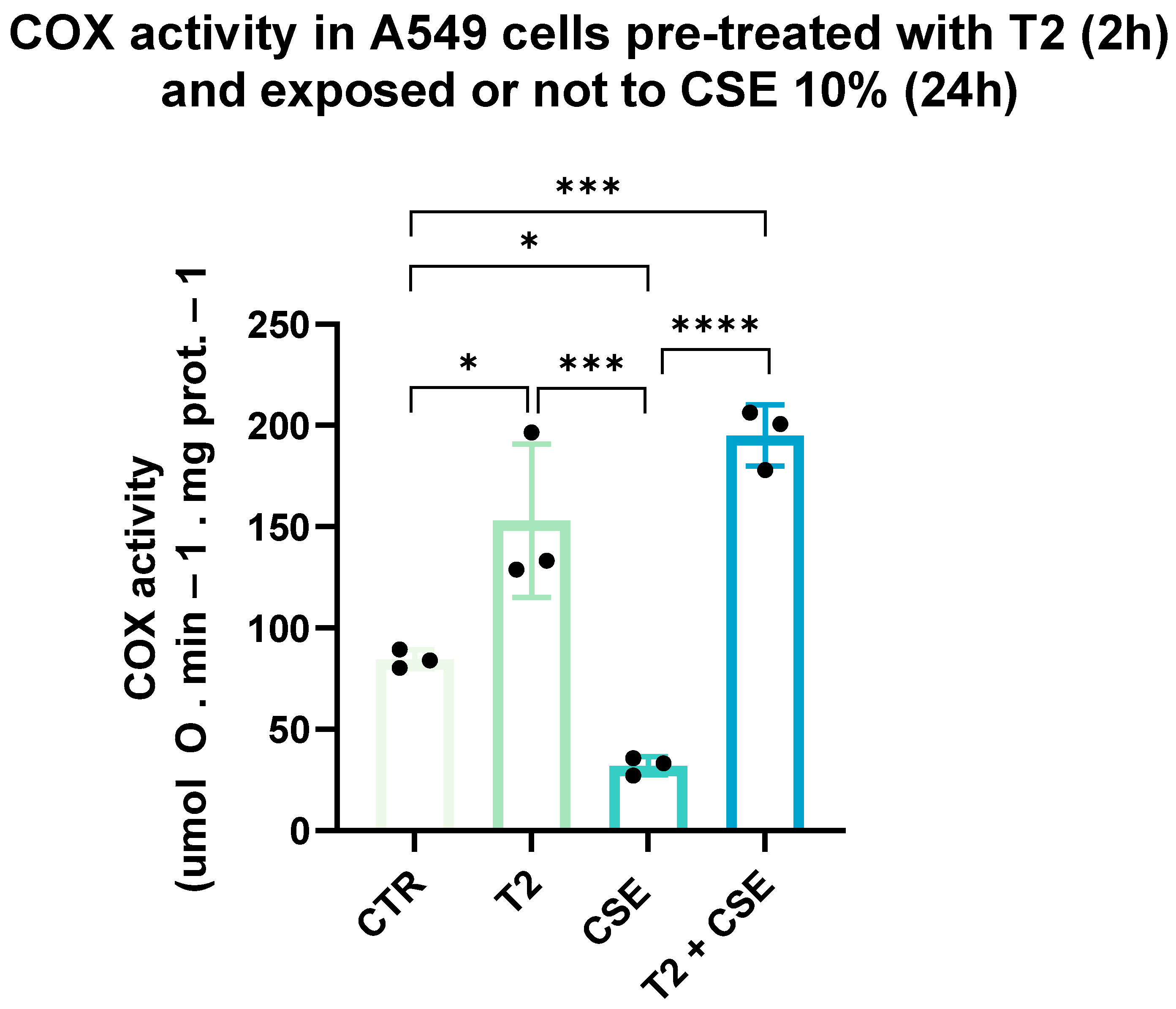
3.4. T2 Pretreatment Mitigates Cigarette Smoke-Induced Effects on Cellular Oxidative Capacity
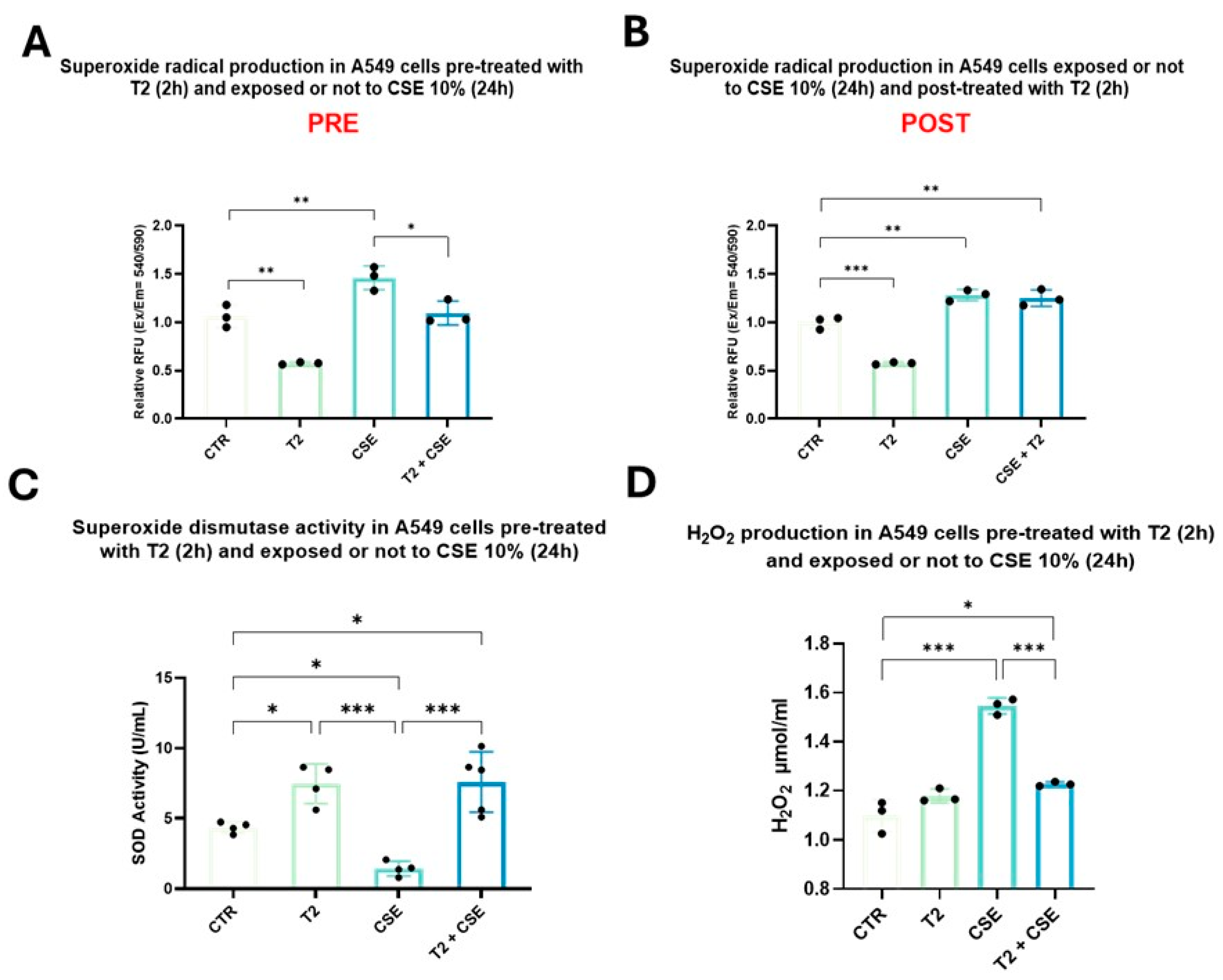
3.5. T2 Pretreatment Prevents Cigarette Smoke-Induced Oxidative Imbalance and Antioxidant Defense Impairment
3.6. T2 Effects on Cigarette Smoke-Induced Increment in Mitochondrial Mass
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bellou, V.; Gogali, A.; Kostikas, K. Asthma and Tobacco Smoking. J. Pers. Med. 2022, 12, 1231. [Google Scholar] [CrossRef]
- Wheaton, A.G.; Liu, Y.; Croft, J.B.; VanFrank, B.; Croxton, T.L.; Punturieri, A.; Postow, L.; Greenlund, K.J. Chronic Obstructive Pulmonary Disease and Smoking Status—United States, 2017. MMWR Morb. Mortal. Wkly. Rep. 2019, 68, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Mirra, D.; Cione, E.; Spaziano, G.; Esposito, R.; Sorgenti, M.; Granato, E.; Cerqua, I.; Muraca, L.; Iovino, P.; Gallelli, L.; et al. Circulating MicroRNAs Expression Profile in Lung Inflammation: A Preliminary Study. J. Clin. Med. 2022, 11, 5446. [Google Scholar] [CrossRef] [PubMed]
- Walser, T.; Cui, X.; Yanagawa, J.; Lee, J.M.; Heinrich, E.; Lee, G.; Sharma, S.; Dubinett, S.M. Smoking and Lung Cancer: The Role of Inflammation. Proc. Am. Thorac. Soc. 2008, 5, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.R.; Jang, J.; Park, S.M.; Ryu, S.M.; Cho, S.J.; Yang, S.R. Cigarette Smoke-Induced Respiratory Response: Insights into Cellular Processes and Biomarkers. Antioxidants 2023, 12, 1210. [Google Scholar] [CrossRef]
- Zhang, L.; Xu, J.; Li, Y.; Meng, F.; Wang, W. Smoking on the risk of acute respiratory distress syndrome: A systematic review and meta-analysis. Crit. Care 2024, 28, 122. [Google Scholar] [CrossRef]
- Esposito, R.; Mirra, D.; Sportiello, L.; Spaziano, G.; D’Agostino, B. Overview of Antiviral Drug Therapy for COVID-19: Where Do We Stand? Biomedicines 2022, 10, 2815. [Google Scholar] [CrossRef]
- Esposito, R.; Mirra, D.; Spaziano, G.; Panico, F.; Gallelli, L.; D’Agostino, B. The Role of MMPs in the Era of CFTR Modulators: An Additional Target for Cystic Fibrosis Patients? Biomolecules 2023, 13, 350. [Google Scholar] [CrossRef]
- Mirra, D.; Esposito, R.; Spaziano, G.; La Torre, C.; Vocca, C.; Tallarico, M.; Cione, E.; Gallelli, L.; D’Agostino, B. Lung microRNAs Expression in Lung Cancer and COPD: A Preliminary Study. Biomedicines 2023, 11, 736. [Google Scholar] [CrossRef]
- Wang, Q.; Ji, X.; Rahman, I. Dysregulated Metabolites Serve as Novel Biomarkers for Metabolic Diseases Caused by E-Cigarette Vaping and Cigarette Smoking. Metabolites 2021, 11, 345. [Google Scholar] [CrossRef]
- Schaible, A.M.; Filosa, R.; Krauth, V.; Temml, V.; Pace, S.; Garscha, U.; Liening, S.; Weinigel, C.; Rummler, S.; Schieferdecker, S.; et al. The 5-lipoxygenase inhibitor RF-22c potently suppresses leukotriene biosynthesis in cellulo and blocks bronchoconstriction and inflammation in vivo. Biochem. Pharmacol. 2016, 112, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Mirra, D.; Esposito, R.; Spaziano, G.; Sportiello, L.; Panico, F.; Squillante, A.; Falciani, M.; Cerqua, I.; Gallelli, L.; Cione, E.; et al. MicroRNA Monitoring in Human Alveolar Macrophages from Patients with Smoking-Related Lung Diseases: A Preliminary Study. Biomedicines 2024, 12, 1050. [Google Scholar] [CrossRef] [PubMed]
- Gallelli, L.; D’Agostino, B.; Marrocco, G.; De Rosa, G.; Filippelli, W.; Rossi, F.; Advenier, C. Role of tachykinins in the bronchoconstriction induced by HCl intraesophageal instillation in the rabbit. Life Sci. 2003, 72, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Nyunoya, T.; Mebratu, Y.; Contreras, A.; Delgado, M.; Chand, H.S.; Tesfaigzi, Y. Molecular processes that drive cigarette smoke-induced epithelial cell fate of the lung. Am. J. Respir. Cell Mol. Biol. 2014, 50, 471–482. [Google Scholar] [CrossRef]
- Madan, S.; Uttekar, B.; Chowdhary, S.; Rikhy, R. Mitochondria lead the way: Mitochondrial dynamics and function in cellular movements in development and disease. Front. Cell Dev. Biol. 2022, 9, 781933. [Google Scholar] [CrossRef]
- Fetterman, J.L.; Sammy, M.I.; Ballinger, S.W. Mitochondrial toxicity of tobacco smoke and air pollution. Toxicology 2017, 391, 18–33. [Google Scholar] [CrossRef]
- Tulen, C.B.; Opperhuizen, A.; van Schooten, F.J.; Remels, A.H. Disruption of the Molecular Regulation of Mitochondrial Metabolism in Airway and Lung Epithelial Cells by Cigarette Smoke: Are Aldehydes the Culprit? Cells 2023, 12, 299. [Google Scholar] [CrossRef]
- Yildiz, L.; Kayaoğlu, N.; Aksoy, H. The changes of superoxide dismutase, catalase, and glutathione peroxidase activities in erythrocytes of active and passive smokers. Clin. Chem. Lab. Med. 2002, 40, 612–615. [Google Scholar] [CrossRef]
- Kondo, T.; Tagami, S.; Yoshioka, A.; Nishimura, M.; Kawakami, Y. Current smoking of elderly men reduces antioxidants in alveolar macrophages. Am. J. Respir. Crit. Care Med. 1994, 149, 178–182. [Google Scholar] [CrossRef]
- Mandraffino, G.; Sardo, M.A.; Riggio, S.; D’Ascola, A.; Loddo, S.; Alibrandi, A.; Saitta, C.; Imbalzano, E.; Mandraffino, R.; Venza, M.; et al. Smoke exposure and circulating progenitor cells: Evidence for modulation of antioxidant enzymes and cell count, Clin. Biochem. 2010, 43, 1436–1442. [Google Scholar] [CrossRef]
- Joshi, B.; Singh, S.; Sharma, P.; Mohapatra, T.; Kumar, P. Effect of cigarette smoking on selected antioxidant enzymes and oxidative stress biomarkers. J. Clin. Diagn. Res. 2020, 14, BC19–BC23. [Google Scholar] [CrossRef]
- Sauleda, J.; García-Palmer, F.; Wiesner, R.J.; Tarraga, S.; Harting, I.; Tomás, P.; Gómez, C.; Saus, C.; Palou, A.; Agustí, A.G. Cytochrome oxidase activity and mitochondrial gene expression in skeletal muscle of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1998, 157, 1413–1417. [Google Scholar] [CrossRef]
- Global Initiative for Chronic Obstructive Lung Disease. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease—2025 Update. Available online: https://www.goldcopd.com (accessed on 28 November 2024).
- Rosenwasser, Y.; Berger, I.; Loewy, Z.G. Therapeutic Approaches for Chronic Obstructive Pulmonary Disease (COPD) Exacerbations. Pathogens. 2022, 11, 1513. [Google Scholar] [CrossRef]
- Damiano, F.; Rochira, A.; Gnoni, A.; Siculella, L. Action of thyroid hormones, T3 and T2, on hepatic fatty acids: Differences in metabolic effects and molecular mechanisms. Int. J. Mol. Sci. 2017, 18, 744. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, G.; Kaminski, N.; Lee, P.J. PINK1 mediates the protective effects of thyroid hormone T3 in hyperoxia-induced lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2021, 320, L1118–L1125. [Google Scholar] [CrossRef] [PubMed]
- Folkesson, H.G.; Norlin, A.; Wang, Y.; Abedinpour, P.; Matthay, M.A. Dexamethasone and thyroid hormone pretreatment upregulate alveolar epithelial fluid clearance in adult rats. J. Appl. Physiol. 2000, 88, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Mullur, R.; Liu, Y.Y.; Brent, G.A. Thyroid hormone regulation of metabolism. Physiol. Rev. 2014, 94, 355–382. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.; Giacco, A.; Di Munno, C.; Goglia, F. Direct and rapid effects of 3,5-diiodo-L-thyronine (T2). Mol. Cell. Endocrinol. 2017, 458, 121–126. [Google Scholar] [CrossRef]
- Senese, R.; de Lange, P.; Petito, G.; Moreno, M.; Goglia, F.; Lanni, A. 3,5-Diiodothyronine: A novel thyroid hormone metabolite and potent modulator of energy metabolism. Front. Endocrinol. 2018, 9, 427. [Google Scholar] [CrossRef]
- De Lange, P.; Cioffi, F.; Senese, R.; Moreno, M.; Lombardi, A.; Silvestri, E.; De Matteis, R.; Lionetti, L.; Mollica, M.P.; Goglia, F.; et al. Nonthyrotoxic prevention of diet-induced insulin resistance by 3,5-diiodo-L-thyronine in rats. Diabetes 2011, 60, 2730–2739. [Google Scholar] [CrossRef]
- Polverino, F.; Mirra, D.; Yang, C.X.; Esposito, R.; Spaziano, G.; Rojas-Quintero, J.; Sgambato, M.; Piegari, E.; Cozzolino, A.; Cione, E.; et al. Similar programmed death ligand 1 (PD-L1) expression profile in patients with mild COPD and lung cancer. Sci. Rep. 2022, 12, 22402. [Google Scholar] [CrossRef] [PubMed]
- Sacripanti, G.; Nguyen, N.M.; Lorenzini, L.; Frascarelli, S.; Saba, A.; Zucchi, R.; Ghelardoni, S. 3,5-Diiodo-l-Thyronine Increases Glucose Consumption in Cardiomyoblasts Without Affecting the Contractile Performance in Rat Heart. Front. Endocrinol. 2018, 9, 282. [Google Scholar] [CrossRef]
- Silvestri, E.; Lombardi, A.; Coppola, M.; Gentile, A.; Cioffi, F.; Senese, R.; Goglia, F.; Lanni, A.; Moreno, M.; De Lange, P. Differential Effects of 3,5-Diiodo-L-Thyronine and 3,5,3′-Triiodo-L-Thyronine On Mitochondrial Respiratory Pathways in Liver from Hypothyroid Rats. Cell Physiol Biochem. 2018, 47, 2471–2483. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.S.H.; Li, Z.L.; Huang, T.Y.; Su, K.W.; Lin, C.Y.; Huang, C.H.; Chen, H.Y.; Lu, M.C.; Huang, H.M.; Lee, S.Y.; et al. Effect of Estrogen on Heteronemin-Induced Anti-proliferative Effect in Breast Cancer Cells With Different Estrogen Receptor Status. Front. Cell Dev. Biol. 2021, 9, 688607. [Google Scholar] [CrossRef]
- Remigante, A.; Spinelli, S.; Straface, E.; Gambardella, L.; Russo, M.; Cafeo, G.; Caruso, D.; Falliti, G.; Dugo, P.; Dossena, S.; et al. Mechanisms underlying the anti-aging activity of bergamot (Citrus bergamia) extract in human red blood cells. Front. Physiol. 2023, 14, 1225552. [Google Scholar] [CrossRef]
- Li, T.; Gao, S.J. KSHV hijacks FoxO1 to promote cell proliferation and cellular transformation by antagonizing oxidative stress. J. Med. Virol. 2023, 95, e28676. [Google Scholar] [CrossRef]
- Petito, G.; Giacco, A.; Cioffi, F.; Mazzoli, A.; Magnacca, N.; Iossa, S.; Goglia, F.; Senese, R.; Lanni, A. Short-term fructose feeding alters tissue metabolic pathways by modulating microRNAs expression both in young and adult rats. Front. Cell Dev. Biol. 2023, 11, 1101844. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Stringer, J.M.; Liu, J.; Hutt, K.J. Evaluation of mitochondria in oocytes following γ-irradiation. Sci. Rep. 2019, 9, 19941. [Google Scholar] [CrossRef]
- Doherty, E.; Perl, A. Measurement of Mitochondrial Mass by Flow Cytometry during Oxidative Stress. React. Oxyg. Species 2017, 4, 275–283. [Google Scholar] [CrossRef]
- Long, G.; Gong, R.; Wang, Q.; Zhang, D.; Huang, C. Role of released mitochondrial DNA in acute lung injury. Front. Immunol. 2022, 13, 973089. [Google Scholar] [CrossRef]
- Vamesu, B.M.; Nicola, T.; Li, R.; Hazra, S.; Matalon, S.; Kaminski, N.; Ambalavanan, N.; Kandasamy, J. Thyroid hormone modulates hyperoxic neonatal lung injury and mitochondrial function. JCI Insight 2023, 8, e160697. [Google Scholar] [CrossRef] [PubMed]
- Neupane, P.; Bhuju, S.; Thapa, N.; Bhattarai, H.K. ATP synthase: Structure, function and inhibition. Biomol. Concepts 2019, 10, 1–10. [Google Scholar] [CrossRef]
- van der Toorn, M.; Slebos, D.J.; de Bruin, H.G.; Leuvenink, H.G.; Bakker, S.J.; Gans, R.O.; Koëter, G.H.; van Oosterhout, A.J.; Kauffman, H.F. Cigarette smoke-induced blockade of the mitochondrial respiratory chain switches lung epithelial cell apoptosis into necrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L1211–L1218. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Luan, G.; Xu, Y.; Shen, S.; Qian, S.; Zhu, Z.; Zhang, X.; Yin, S.; Ye, J. Cigarette smoke extract increases mitochondrial membrane permeability through activation of adenine nucleotide translocator (ANT) in lung epithelial cells. Biochem. Biophys. Res. Commun. 2020, 525, 733–739. [Google Scholar] [CrossRef]
- Cioffi, F.; Giacco, A.; Goglia, F.; Silvestri, E. Bioenergetic aspects of mitochondrial actions of thyroid hormones. Cells 2022, 11, 997. [Google Scholar] [CrossRef]
- Fontanesi, F.; Soto, I.C.; Barrientos, A. Cytochrome c oxidase biogenesis: New levels of regulation. IUBMB Life 2008, 60, 557–568. [Google Scholar] [CrossRef]
- Arnold, S.; Goglia, F.; Kadenbach, B. 3,5-Diiodothyronine binds to subunit Va of cytochrome-c oxidase and abolishes the allosteric inhibition of respiration by ATP. Eur. J. Biochem. 1998, 252, 325–330. [Google Scholar] [CrossRef]
- Alonso, J.R.; Cardellach, F.; Casademont, J.; Miró, O. Reversible inhibition of mitochondrial complex IV activity in PBMC following acute smoking. Eur. Respir. J. 2004, 23, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Chen, P.; Liu, X.M. The role of cigarette smoke-induced pulmonary vascular endothelial cell apoptosis in COPD. Respir Res. 2021, 22, 39. [Google Scholar] [CrossRef]
- Collin, F. Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 2407. [Google Scholar] [CrossRef]
- Juan, C.A.; de la Lastra, J.M.P.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef]
- Garmendia, J.; Morey, P.; Bengoechea, J.A. Impact of cigarette smoke exposure on host-bacterial pathogen interactions. Eur. Respir. J. 2012, 39, 467–477. [Google Scholar] [CrossRef]
- Martemucci, G.; Costagliola, C.; Mariano, M.; D’andrea, L.; Napolitano, P.; D’Alessandro, A.G. Free Radical Properties, Source and Targets, Antioxidant Consumption and Health. Oxygen 2022, 2, 48–78. [Google Scholar] [CrossRef]
- Padmavathi, P.; Raghu, P.S.; Reddy, V.D. Chronic cigarette smoking-induced oxidative/nitrosative stress in human erythrocytes and platelets. Mol. Cell. Toxicol. 2018, 14, 27–34. [Google Scholar] [CrossRef]
- Cioffi, F.; Senese, R.; Petito, G.; Lasala, P.; de Lange, P.; Silvestri, E.; Lombardi, A.; Moreno, M.; Goglia, F.; Lanni, A. Both 3,3′,5-triiodothyronine and 3,5-diodo-L-thyronine Are Able to Repair Mitochondrial DNA Damage but by Different Mechanisms. Front. Endocrinol. 2019, 10, 216. [Google Scholar] [CrossRef] [PubMed]
- Guerra, L.N.; Moiguer, S.; Karner, M.; de Molina, M.C.; Sreider, C.M.; Burdman, J.A. Antioxidants in the treatment of Graves Disease. IUBMB Life 2001, 51, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Cantin, A.M. Cellular response to cigarette smoke and oxidants: Adapting to survive. Proc. Am. Thorac. Soc. 2010, 7, 368–375. [Google Scholar] [CrossRef]
- Jomova, K.; Raptova, R.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Valko, M. Reactive oxygen species, toxicity, oxidative stress, and antioxidants: Chronic diseases and aging. Arch. Toxicol. 2023, 97, 2499–2574. [Google Scholar] [CrossRef]
- Tanni, S.E.; Correa, C.R.; Angeleli, A.Y.; Vale, S.A.; Coelho, L.S.; Godoy, I. Increased production of hydrogen peroxide by peripheral blood monocytes associated with smoking exposure intensity in smokers. J. Inflamm. 2012, 9, 45. [Google Scholar] [CrossRef]
- Aspera-Werz, R.H.; Ehnert, S.; Heid, D.; Zhu, S.; Chen, T.; Braun, B.; Sreekumar, V.; Arnscheidt, C.; Nussler, A.K. Nicotine and Cotinine Inhibit Catalase and Glutathione Reductase Activity Contributing to the Impaired Osteogenesis of SCP-1 Cells Exposed to Cigarette Smoke. Oxidative Med. Cell. Longev. 2018, 2018, 3172480. [Google Scholar] [CrossRef]
- Pereira, B.; Rosa, L.F.; Safi, D.A.; Bechara, E.J.; Curi, R. Control of superoxide dismutase, catalase and glutathione peroxidase activities in rat lymphoid organs by thyroid hormones. J. Endocrinol. 1994, 140, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Morini, P.; Casalino, E.; Sblano, C.; Landriscina, C. The response of rat liver lipid peroxidation, antioxidant enzyme activities and glutathione concentration to the thyroid hormone. Int. J. Biochem. 1991, 23, 1025–1030. [Google Scholar] [CrossRef]
- Miwa, S.; Kashyap, S.; Chini, E.; von Zglinicki, T. Mitochondrial dysfunction in cell senescence and aging. J. Clin. Investig. 2022, 132, e158447. [Google Scholar] [CrossRef]
- Korolchuk, V.I.; Miwa, S.; Carroll, B.; von Zglinicki, T. Mitochondria in Cell Senescence: Is Mitophagy the Weakest Link? EBioMedicine 2017, 21, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Menzies, K.J.; Robinson, B.H.; Hood, D.A. Effect of thyroid hormone on mitochondrial properties and oxidative stress in cells from patients with mtDNA defects. Am. J. Physiol. Cell Physiol. 2009, 296, C355–C362. [Google Scholar] [CrossRef] [PubMed]
- Jonas, W.; LietzowJ, J.; Wohlgemuth, F.; Hoefig, C.S.; Wiedmer, P.; Schweizer, U.; Köhrle, J.; Schürmann, A. 3,5-Diiodo-L-thyronine (3,5-t2) exerts thyromimetic effects on hypothalamus-pituitary-thyroid axis, body composition, and energy metabolism in male diet-induced obese mice. Endocrinology 2015, 156, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Louzada, R.A.; Padron, A.S.; Marques-Neto, S.R.; Maciel, L.; Werneck-de-Castro, J.P.; Ferreira, A.C.F.; Nascimento, J.M.H.; Carvalho, D.P. 3,5-Diiodothyronine protects against cardiac ischaemia-reperfusion injury in male rats. Exp. Physiol. 2021, 106, 2185–2197. [Google Scholar] [CrossRef]
- Antonelli, A.; Fallahi, P.; Ferrari, S.M.; Di Domenicantonio, A.; Moreno, M.; Lanni, A.; Goglia, F. 3,5-diiodo-L-thyronine increases resting metabolic rate and reduces body weight without undesirable side effects. J. Biol. Regul. Homeost. Agents. 2011, 25, 655–660. [Google Scholar] [PubMed]
- Horst, C.; Harneit, A.; Seitz, H.J.; Rokos, H. 3,5-Di-iodo-L-thyronine suppresses TSH in rats in vivo and in rat pituitary fragments in vitro. J. Endocrinol. 1995, 145, 291–297. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panico, F.; Mirra, D.; Petito, G.; Spaziano, G.; Del Vecchio, V.; Esposito, R.; Senese, R.; Desiderio, V.; Lanni, A.; D’Agostino, B. Protective Action of 3,5-Diiodo-L-Thyronine on Cigarette Smoke-Induced Mitochondrial Dysfunction in Human Alveolar Epithelial Cells. Biomedicines 2025, 13, 1014. https://doi.org/10.3390/biomedicines13051014
Panico F, Mirra D, Petito G, Spaziano G, Del Vecchio V, Esposito R, Senese R, Desiderio V, Lanni A, D’Agostino B. Protective Action of 3,5-Diiodo-L-Thyronine on Cigarette Smoke-Induced Mitochondrial Dysfunction in Human Alveolar Epithelial Cells. Biomedicines. 2025; 13(5):1014. https://doi.org/10.3390/biomedicines13051014
Chicago/Turabian StylePanico, Francesca, Davida Mirra, Giuseppe Petito, Giuseppe Spaziano, Vitale Del Vecchio, Renata Esposito, Rosalba Senese, Vincenzo Desiderio, Antonia Lanni, and Bruno D’Agostino. 2025. "Protective Action of 3,5-Diiodo-L-Thyronine on Cigarette Smoke-Induced Mitochondrial Dysfunction in Human Alveolar Epithelial Cells" Biomedicines 13, no. 5: 1014. https://doi.org/10.3390/biomedicines13051014
APA StylePanico, F., Mirra, D., Petito, G., Spaziano, G., Del Vecchio, V., Esposito, R., Senese, R., Desiderio, V., Lanni, A., & D’Agostino, B. (2025). Protective Action of 3,5-Diiodo-L-Thyronine on Cigarette Smoke-Induced Mitochondrial Dysfunction in Human Alveolar Epithelial Cells. Biomedicines, 13(5), 1014. https://doi.org/10.3390/biomedicines13051014