
Machine Learning-Driven Transcriptome Analysis of Keratoconus for Predictive Biomarker Identification

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset Collection
2.2. Data Processing and Identifying Differentially Expressed Genes (DEGs)
2.3. Gene Function Enrichment Analysis
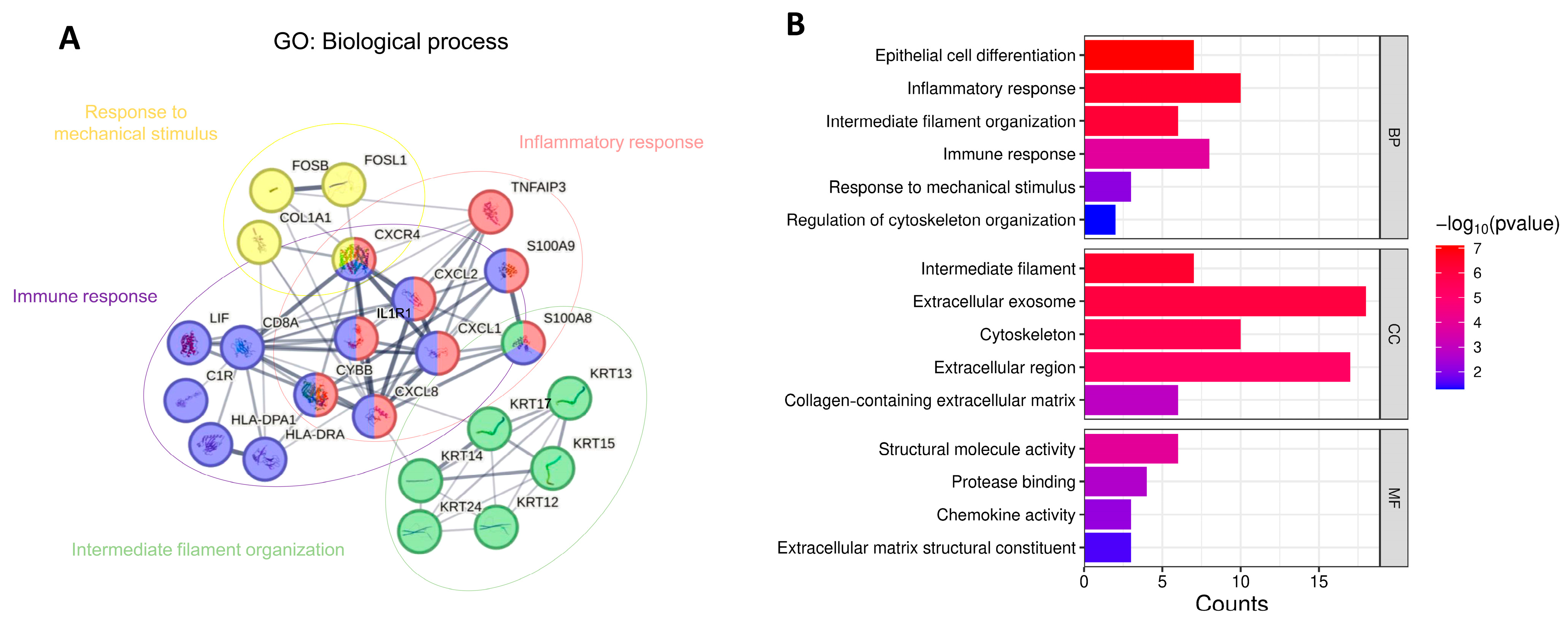
2.4. Protein–Protein Interaction (PPI) Network Construction and Gene Ontology (GO) Pathway Analysis
2.5. Machine Learning Models
2.6. SHAP Feature Selection
2.7. Statistical Analysis
3. Results
4. Discussion
Limitation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| KTCN | Keratoconus |
| DEGs | Differentially expressed genes |
| DGE | Differential gene expression |
| CECs | Corneal epithelial cells |
| CSCs | Corneal stromal cells |
| ImCs | Immune cells |
| GSEA | Gene set enrichment analysis |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| GO | Gene Ontology |
| BPs | Biological processes |
| CCs | Cellular components |
| MFs | Molecular functions |
| SHAP | SHapley Additive exPlanations |
| MMPs | Matrix metalloproteinases |
| ECM | Extracellular matrix |
| RNA-Seq | RNA sequencing |
| scRNA-seq | Single-cell RNA sequencing |
| ROC Curves | Receiver operating characteristic curves |
| AUC | Area under the curve |
| AI | Artificial Intelligence |
| ML | Machine learning |
| MSigDB | Molecular Signatures Database |
| FDR | False Discovery Rate |
| GEO | Gene Expression Omnibus |
| TP | True Positive |
| TN | True Negative |
| FP | False Positive |
| FN | False Negative |
| MCC | Matthews Correlation Coefficient |
| GAGs | Glycosaminoglycans |
| SVM | Support Vector Machine |
| XGBoost | eXtreme Gradient Boosting |
| RBF | Radial basis function |
References
- Krachmer, J.H.; Feder, R.S.; Belin, M.W. Keratoconus and related noninflammatory corneal thinning disorders. Surv. Ophthalmol. 1984, 28, 293–322. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, H.; Heydarian, S.; Hooshmand, E.; Saatchi, M.; Yekta, A.; Aghamirsalim, M.; Valadkhan, M.; Mortazavi, M.; Hashemi, A.; Khabazkhoob, M. The Prevalence and Risk Factors for Keratoconus: A Systematic Review and Meta-Analysis. Cornea 2020, 39, 263–270. [Google Scholar] [CrossRef]
- Ferrari, G.; Rama, P. The keratoconus enigma: A review with emphasis on pathogenesis. Ocul. Surf. 2020, 18, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Lasagni Vitar, R.M.; Bonelli, F.; Rama, P.; Ferrari, G. Nutritional and Metabolic Imbalance in Keratoconus. Nutrients 2022, 14, 913. [Google Scholar] [CrossRef]
- Balasubramanian, S.A.; Pye, D.C.; Willcox, M.D. Effects of eye rubbing on the levels of protease, protease activity and cytokines in tears: Relevance in keratoconus. Clin. Exp. Optom. 2013, 96, 214–218. [Google Scholar] [CrossRef]
- Hung, K.H.; Tan, H.Y.; Chen, H.C.; Yeh, L.K. Clinical characteristics and topographic findings of corneal ectasia in patients with symptomatic Demodex blepharitis. Taiwan. J. Ophthalmol. 2021, 11, 146–155. [Google Scholar] [CrossRef]
- Chang, S.H.; Mohammadvali, A.; Chen, K.J.; Ji, Y.R.; Young, T.H.; Wang, T.J.; Willoughby, C.E.; Hamill, K.J.; Elsheikh, A. The Relationship Between Mechanical Properties, Ultrastructural Changes, and Intrafibrillar Bond Formation in Corneal UVA/Riboflavin Cross-linking Treatment for Keratoconus. J. Refract. Surg. 2018, 34, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.H.; Zhou, D.; Eliasy, A.; Li, Y.C.; Elsheikh, A. Experimental evaluation of stiffening effect induced by UVA/Riboflavin corneal cross-linking using intact porcine eye globes. PLoS ONE 2020, 15, e0240724. [Google Scholar] [CrossRef]
- Blackburn, B.J.; Jenkins, M.W.; Rollins, A.M.; Dupps, W.J. A Review of Structural and Biomechanical Changes in the Cornea in Aging, Disease, and Photochemical Crosslinking. Front. Bioeng. Biotechnol. 2019, 7, 66. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, H.; Shan, M.; Dong, Y.; Zhang, L.; Chen, L.; Wang, Y. Comprehensive Transcriptome Analysis of Patients With Keratoconus Highlights the Regulation of Immune Responses and Inflammatory Processes. Front. Genet. 2022, 13, 782709. [Google Scholar] [CrossRef]
- Shinde, V.; Hu, N.; Mahale, A.; Maiti, G.; Daoud, Y.; Eberhart, C.G.; Maktabi, A.; Jun, A.S.; Al-Swailem, S.A.; Chakravarti, S. RNA sequencing of corneas from two keratoconus patient groups identifies potential biomarkers and decreased NRF2-antioxidant responses. Sci. Rep. 2020, 10, 9907. [Google Scholar] [CrossRef]
- Dou, S.; Wang, Q.; Zhang, B.; Wei, C.; Wang, H.; Liu, T.; Duan, H.; Jiang, H.; Liu, M.; Qi, X.; et al. Single-cell atlas of keratoconus corneas revealed aberrant transcriptional signatures and implicated mechanical stretch as a trigger for keratoconus pathogenesis. Cell Discov. 2022, 8, 66. [Google Scholar] [CrossRef]
- Zhou, H.Y.; Wang, L.Y.; Xu, Y.; Zhang, Y.L.; Liu, Q. Role of Corneal Collagen Fibrils in Corneal Disorders and Related Pathological Conditions. Int. J. Ophthalmol. 2017, 10, 803–811. [Google Scholar]
- Shetty, R.; Nuijts, R.M.M.A.; Vazirani, J.; Jayadev, C.; Richoz, O.; Gomes, J.A.P.; Padmanabhan, P. Biochemical Markers and Alterations in Keratoconus. Asia Pac. J. Ophthalmol. 2020, 9, 533–540. [Google Scholar] [CrossRef]
- Cheung, I.M.Y.; Angelo, L.; Gokul, A.; Ziaei, M. Non-Genetic Risk Factors for Keratoconus and Its Progression. Clin. Exp. Optom. 2025. [Google Scholar] [CrossRef]
- Wentz-Hunter, K.; Cheng, E.L.; Ueda, J.; Sugar, J.; Yue, B.Y. Keratocan expression is increased in the stroma of keratoconus corneas. Mol. Med. 2001, 7, 470–477. [Google Scholar] [CrossRef]
- Zhou, L.; Yue, B.Y.; Twining, S.S.; Sugar, J.; Feder, R.S. Expression of wound healing and stress-related proteins in keratoconus corneas. Curr. Eye Res. 1996, 15, 1124–1131. [Google Scholar] [CrossRef]
- Garcia, B.; Garcia-Suarez, O.; Merayo-Lloves, J.; Alcalde, I.; Alfonso, J.F.; Fernandez-Vega Cueto, L.; Meana, A.; Vazquez, F.; Quiros, L.M. Differential Expression of Proteoglycans by Corneal Stromal Cells in Keratoconus. Investig. Ophthalmol. Vis. Sci. 2016, 57, 2618–2628. [Google Scholar] [CrossRef]
- Balasubramanian, S.A.; Mohan, S.; Pye, D.C.; Willcox, M.D. Proteases, proteolysis and inflammatory molecules in the tears of people with keratoconus. Acta Ophthalmol. 2012, 90, e303–e309. [Google Scholar] [CrossRef]
- Mazzotta, C.; Traversi, C.; Mellace, P.; Bagaglia, S.A.; Zuccarini, S.; Mencucci, R.; Jacob, S. Keratoconus Progression in Patients with Allergy and Elevated Surface Matrix Metalloproteinase 9 Point-of-Care Test. Eye Contact Lens 2018, 44 (Suppl. 2), S48–S53. [Google Scholar] [CrossRef]
- Shi, Y. Strategies for improving the early diagnosis of keratoconus. Clin. Optom. 2016, 8, 13–21. [Google Scholar] [CrossRef]
- Chan, H.C.S.; Shan, H.; Dahoun, T.; Vogel, H.; Yuan, S. Advancing Drug Discovery via Artificial Intelligence. Trends Pharmacol. Sci. 2019, 40, 801. [Google Scholar] [CrossRef]
- Gore, J.C. Artificial intelligence in medical imaging. Magn. Reson. Imaging 2020, 68, A1–A4. [Google Scholar] [CrossRef]
- Xu, Y.; Verma, D.; Sheridan, R.P.; Liaw, A.; Ma, J.; Marshall, N.M.; McIntosh, J.; Sherer, E.C.; Svetnik, V.; Johnston, J.M. Deep Dive into Machine Learning Models for Protein Engineering. J. Chem. Inf. Model. 2020, 60, 2773–2790. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.; Masarone, S.; Watson, D.; Barnes, M.R. The benefits and pitfalls of machine learning for biomarker discovery. Cell Tissue Res. 2023, 394, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Prelaj, A.; Miskovic, V.; Zanitti, M.; Trovo, F.; Genova, C.; Viscardi, G.; Rebuzzi, S.E.; Mazzeo, L.; Provenzano, L.; Kosta, S.; et al. Artificial intelligence for predictive biomarker discovery in immuno-oncology: A systematic review. Ann. Oncol. 2024, 35, 29–65. [Google Scholar] [CrossRef]
- Li, H.; Sun, X.; Li, Z.; Zhao, R.; Li, M.; Hu, T. Machine learning-based integration develops biomarkers initial the crosstalk between inflammation and immune in acute myocardial infarction patients. Front. Cardiovasc. Med. 2022, 9, 1059543. [Google Scholar] [CrossRef] [PubMed]
- Mohd Faizal, A.S.; Hon, W.Y.; Thevarajah, T.M.; Khor, S.M.; Chang, S.W. A biomarker discovery of acute myocardial infarction using feature selection and machine learning. Med. Biol. Eng. Comput. 2023, 61, 2527–2541. [Google Scholar] [CrossRef]
- Morabito, F.; Adornetto, C.; Monti, P.; Amaro, A.; Reggiani, F.; Colombo, M.; Rodriguez-Aldana, Y.; Tripepi, G.; D’Arrigo, G.; Vener, C.; et al. Genes selection using deep learning and explainable artificial intelligence for chronic lymphocytic leukemia predicting the need and time to therapy. Front. Oncol. 2023, 13, 1198992. [Google Scholar] [CrossRef]
- Cohen, E.; Bank, D.; Sorkin, N.; Giryes, R.; Varssano, D. Use of machine learning to achieve keratoconus detection skills of a corneal expert. Int. Ophthalmol. 2022, 42, 3837–3847. [Google Scholar] [CrossRef]
- Cao, K.; Verspoor, K.; Sahebjada, S.; Baird, P.N. Accuracy of Machine Learning Assisted Detection of Keratoconus: A Systematic Review and Meta-Analysis. J. Clin. Med. 2022, 11, 478. [Google Scholar] [CrossRef]
- Kabza, M.; Karolak, J.A.; Rydzanicz, M.; Szczesniak, M.W.; Nowak, D.M.; Ginter-Matuszewska, B.; Polakowski, P.; Ploski, R.; Szaflik, J.P.; Gajecka, M. Collagen Synthesis Disruption and Downregulation of Core Elements of TGF-beta, Hippo, and Wnt Pathways in Keratoconus Corneas. Eur. J. Hum. Genet. 2017, 25, 582–590. [Google Scholar] [CrossRef]
- Reimand, J.; Isserlin, R.; Voisin, V.; Kucera, M.; Tannus-Lopes, C.; Rostamianfar, A.; Wadi, L.; Meyer, M.; Wong, J.; Xu, C.; et al. Pathway Enrichment Analysis and Visualization of Omics Data Using g:Profiler, GSEA, Cytoscape, and EnrichmentMap. Nat. Protoc. 2019, 14, 482–517. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Adler, P.; Vilo, J.; Peterson, H. g:Profiler-Interoperable Web Service for Functional Enrichment Analysis and Gene Identifier Mapping (2023 Update). Nucleic Acids Res. 2023, 51, W207–W212. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Dong, L.; Xiao, Z.; He, W.; Zhao, J.; Pan, H.; Chu, B.; Cheng, J.; Wang, H. Integrated Analysis of the Proteome and Transcriptome in a MCAO Mouse Model Revealed the Molecular Landscape During Stroke Progression. J. Adv. Res. 2020, 24, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.I.; Mitchell, T.M. Machine Learning: Trends, Perspectives, and Prospects. Science 2015, 349, 255–260. [Google Scholar] [CrossRef]
- DeGroat, W.; Abdelhalim, H.; Patel, K.; Mendhe, D.; Zeeshan, S.; Ahmed, Z. Discovering Biomarkers Associated and Predicting Cardiovascular Disease with High Accuracy Using a Novel Nexus of Machine Learning Techniques for Precision Medicine. Sci. Rep. 2024, 14, 1. [Google Scholar] [CrossRef]
- Sepulveda, J.L. Using R and Bioconductor in Clinical Genomics and Transcriptomics. J. Mol. Diagn. 2020, 22, 3–20. [Google Scholar] [CrossRef]
- Liu, B.; Lin, H.; Chen, Y.; Yang, C. Prediction of Rock Unloading Strength Based on PSO-XGBoost Hybrid Models. Materials 2024, 17, 17. [Google Scholar] [CrossRef]
- Noble, W.S. What is a Support Vector Machine? Nat. Biotechnol. 2006, 24, 1565–1567. [Google Scholar] [CrossRef] [PubMed]
- Bennett-Lenane, H.; Griffin, B.T.; O’Shea, J.P. Machine Learning Methods for Prediction of Food Effects on Bioavailability: A Comparison of Support Vector Machines and Artificial Neural Networks. Eur. J. Pharm. Sci. 2022, 168, 106018. [Google Scholar] [CrossRef]
- Brown, S.S.G.; Mak, E.; Clare, I.; Grigorova, M.; Beresford-Webb, J.; Walpert, M.; Jones, E.; Hong, Y.T.; Fryer, T.D.; Coles, J.P.; et al. Support Vector Machine Learning and Diffusion-Derived Structural Networks Predict Amyloid Quantity and Cognition in Adults with Down’s Syndrome. Neurobiol. Aging 2022, 115, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Pontil, M.; Verri, A. Properties of Support Vector Machines. Neural Comput. 1998, 10, 955–974. [Google Scholar] [CrossRef] [PubMed]
- Raza, A.; Uddin, J.; Almuhaimeed, A.; Akbar, S.; Zou, Q.; Ahmad, A. AIPs-SnTCN: Predicting Anti-Inflammatory Peptides Using fastText and Transformer Encoder-Based Hybrid Word Embedding with Self-Normalized Temporal Convolutional Networks. J. Chem. Inf. Model. 2023, 63, 6537–6554. [Google Scholar] [CrossRef]
- Xu, H.; Peng, X.; Peng, Z.; Wang, R.; Zhou, R.; Fu, L. Construction and SHAP Interpretability Analysis of a Risk Prediction Model for Feeding Intolerance in Preterm Newborns Based on Machine Learning. BMC Med. Inform. Decis. Mak. 2024, 24, 342. [Google Scholar] [CrossRef]
- Lupasco, T.; Notarnicola, C.; Grieve, K.; Ayello-Scheer, S.; Gabison, E.E.; Baudouin, C.; Labbe, A. Corneal Epithelium in Keratoconus Underexpresses Active NRF2 and a Subset of Oxidative Stress-Related Genes. PLoS ONE 2022, 17, e0273807. [Google Scholar] [CrossRef]
- Li, S.; Zhang, J.; Qian, S.; Wu, X.; Sun, L.; Ling, T.; Jin, Y.; Li, W.; Sun, L.; Lai, M.; et al. S100A8 Promotes Epithelial-Mesenchymal Transition and Metastasis Under TGF-beta/USF2 Axis in Colorectal Cancer. Cancer Commun. 2021, 41, 154–170. [Google Scholar] [CrossRef]
- Zheng, J.; Wang, J.; Liu, H.; Chen, F.; Wang, H.; Chen, S.; Xie, J.; Zheng, Z.; Li, Z. Alarmins S100A8/A9 Promote Intervertebral Disc Degeneration and Inflammation-Related Pain in a Rat Model Through Toll-Like Receptor-4 and Activation of the NF-kappaB Signaling Pathway. Osteoarthr. Cartil. 2022, 30, 998–1011. [Google Scholar] [CrossRef]
- Stachon, T.; Nastaranpour, M.; Seitz, B.; Meese, E.; Latta, L.; Taneri, S.; Ardjomand, N.; Szentmary, N.; Ludwig, N. Altered Regulation of mRNA and miRNA Expression in Epithelial and Stromal Tissue of Keratoconus Corneas. Investig. Ophthalmol. Vis. Sci. 2022, 63, 7. [Google Scholar] [CrossRef]
- Rabinowitz, Y.S. Keratoconus. Surv. Ophthalmol. 1998, 42, 297–319. [Google Scholar] [CrossRef] [PubMed]
- Fini, M.E.; Yue, B.Y.; Sugar, J. Collagenolytic/Gelatinolytic Metalloproteinases in Normal and Keratoconus Corneas. Curr. Eye Res. 1992, 11, 849–862. [Google Scholar] [CrossRef]
- Khaled, M.L.; Bykhovskaya, Y.; Yablonski, S.E.R.; Li, H.; Drewry, M.D.; Aboobakar, I.F.; Estes, A.; Gao, X.R.; Stamer, W.D.; Xu, H.; et al. Differential Expression of Coding and Long Noncoding RNAs in Keratoconus-Affected Corneas. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2717–2728. [Google Scholar] [CrossRef]
- Lema, I.; Brea, D.; Rodriguez-Gonzalez, R.; Diez-Feijoo, E.; Sobrino, T. Proteomic Analysis of the Tear Film in Patients with Keratoconus. Mol. Vis. 2010, 16, 2055–2061. [Google Scholar]
- Balasubramanian, S.A.; Wasinger, V.C.; Pye, D.C.; Willcox, M.D. Preliminary Identification of Differentially Expressed Tear Proteins in Keratoconus. Mol. Vis. 2013, 19, 2124–2134. [Google Scholar] [PubMed]
- Nielsen, K.; Birkenkamp-Demtroder, K.; Ehlers, N.; Orntoft, T.F. Identification of Differentially Expressed Genes in Keratoconus Epithelium Analyzed on Microarrays. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2466–2476. [Google Scholar] [CrossRef]
- Maruri, D.P.; Miron-Mendoza, M.; Kivanany, P.B.; Hack, J.M.; Schmidtke, D.W.; Petroll, W.M.; Varner, V.D. ECM Stiffness Controls the Activation and Contractility of Corneal Keratocytes in Response to TGF-β1. Biophys. J. 2020, 119, 1865–1877. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.; Thompson, B.; Fisk, J.N.; Nebert, D.W.; Bruford, E.A.; Vasiliou, V.; Bunick, C.G. Update of the Keratin Gene Family: Evolution, Tissue-Specific Expression Patterns, and Relevance to Clinical Disorders. Hum. Genom. 2022, 16, 1. [Google Scholar] [CrossRef]
- Joseph, R.J.; Srivastava, O.P.; Pfister, R.R. Differential Epithelial and Stromal Protein Profiles in Keratoconus and Normal Human Corneas. Exp. Eye Res. 2011, 92, 282–298. [Google Scholar] [CrossRef]
- Noorunnahar, M.; Chowdhury, A.H.; Mila, F.A. A Tree-Based eXtreme Gradient Boosting (XGBoost) Machine Learning Model to Forecast the Annual Rice Production in Bangladesh. PLoS ONE 2023, 18, e0283452. [Google Scholar] [CrossRef]
- Sorenson, B.S.; Lantz, T.L.; Handa, J.T.; Salyer, A.C.; Wessells, R.J.; Ramesh, R.; McCormick, T.S.; Ward, N.L. IL-1 Receptor Regulates S100A8/A9-Dependent Keratinocyte Resistance to Bacterial Invasion. Mucosal Immunol. 2012, 5, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Zhang, X.; Li, Y.; Wang, J.; Liu, H. A Novel Combined Oxidative Stress and Extracellular Matrix Related Predictive Gene Signature for Keratoconus. Biochem. Biophys. Res. Commun. 2025, 742, 151144. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, S.; Zhou, Y.; Chen, L.; Li, Y.; Liu, C. Comprehensive Bioinformatics Analysis to Reveal Key RNA Targets and Hub Competitive Endogenous RNA Network of Keratoconus. Front. Genet. 2022, 13, 896780. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
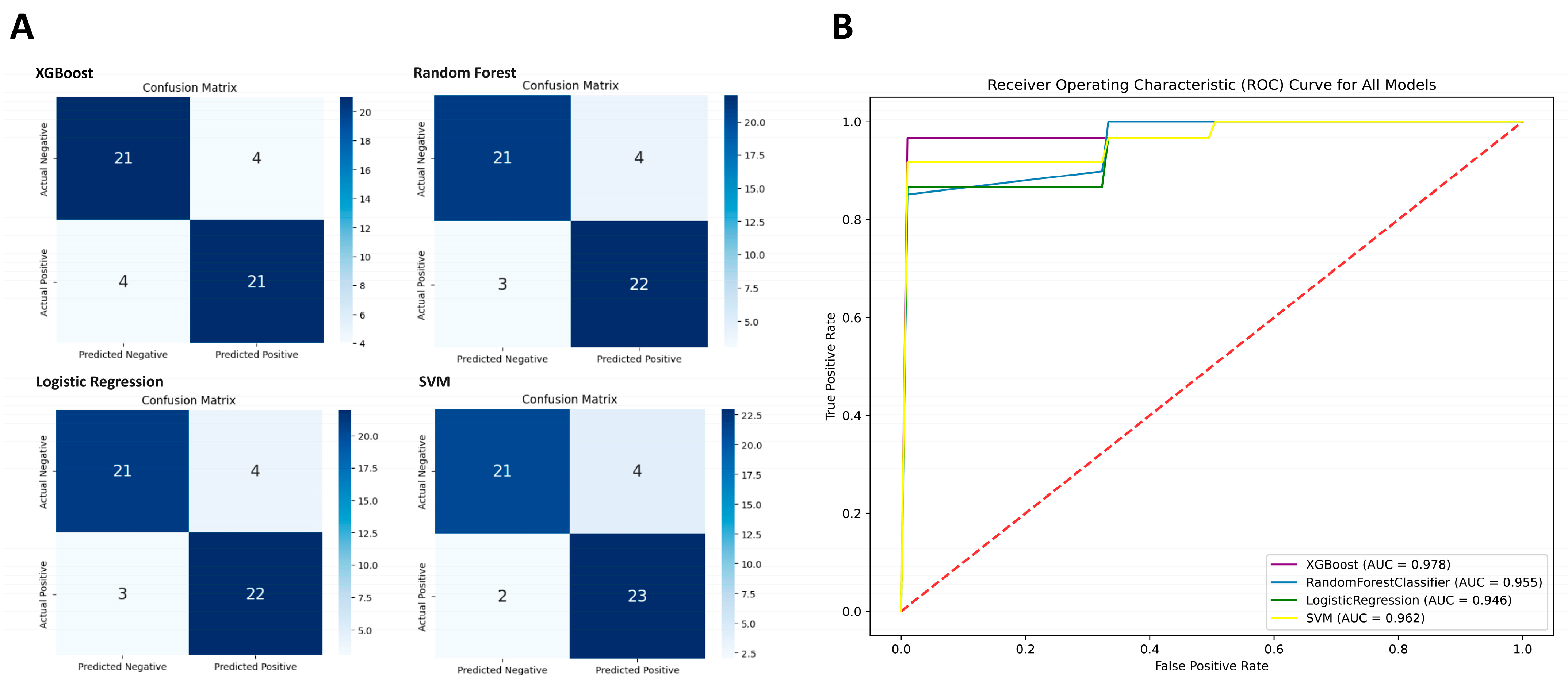
| XGBoost | Random Forest | Logistic Regression | SVM | |
|---|---|---|---|---|
| True Positive | 21 | 22 | 22 | 23 |
| True Negative | 21 | 21 | 21 | 21 |
| False Positive | 4 | 4 | 4 | 4 |
| False Negative | 4 | 3 | 3 | 2 |
| Sensitivity | 84% | 88% | 88% | 92% |
| Specificity | 84% | 84% | 84% | 84% |
| Accuracy | 84% | 86% | 86% | 88% |
| F1-score | 0.84 | 0.863 | 0.863 | 0.885 |
| MCC | 0.86 | 0.72 | 0.72 | 0.76 |
| AUC | 0.97 | 0.95 | 0.94 | 0.96 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, S.-H.; Yeh, L.-K.; Hung, K.-H.; Chiu, Y.-J.; Hsieh, C.-H.; Ma, C.-P. Machine Learning-Driven Transcriptome Analysis of Keratoconus for Predictive Biomarker Identification. Biomedicines 2025, 13, 1032. https://doi.org/10.3390/biomedicines13051032
Chang S-H, Yeh L-K, Hung K-H, Chiu Y-J, Hsieh C-H, Ma C-P. Machine Learning-Driven Transcriptome Analysis of Keratoconus for Predictive Biomarker Identification. Biomedicines. 2025; 13(5):1032. https://doi.org/10.3390/biomedicines13051032
Chicago/Turabian StyleChang, Shao-Hsuan, Lung-Kun Yeh, Kuo-Hsuan Hung, Yen-Jung Chiu, Chia-Hsun Hsieh, and Chung-Pei Ma. 2025. "Machine Learning-Driven Transcriptome Analysis of Keratoconus for Predictive Biomarker Identification" Biomedicines 13, no. 5: 1032. https://doi.org/10.3390/biomedicines13051032
APA StyleChang, S.-H., Yeh, L.-K., Hung, K.-H., Chiu, Y.-J., Hsieh, C.-H., & Ma, C.-P. (2025). Machine Learning-Driven Transcriptome Analysis of Keratoconus for Predictive Biomarker Identification. Biomedicines, 13(5), 1032. https://doi.org/10.3390/biomedicines13051032