
Synergistic Anticancer Activity of HSP70 Inhibitor and Doxorubicin in Gain-of-Function Mutated p53 Breast Cancer Cells

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Drug Treatment
2.2. CCK8 Assay
2.3. Drug Interaction Analysis
2.4. Thioflavin T Staining
2.5. Real-Time RT-PCR
2.6. Flow Cytometry
2.7. Immunocytochemistry
2.8. Activated Caspase-3/7 Detection Staining
2.9. Statistical Analysis
3. Results
3.1. Doxorubicin Resistance Appears in Two High ThT-Staining Breast Cancer Cell Lines, MDA-MB-231 and T47D
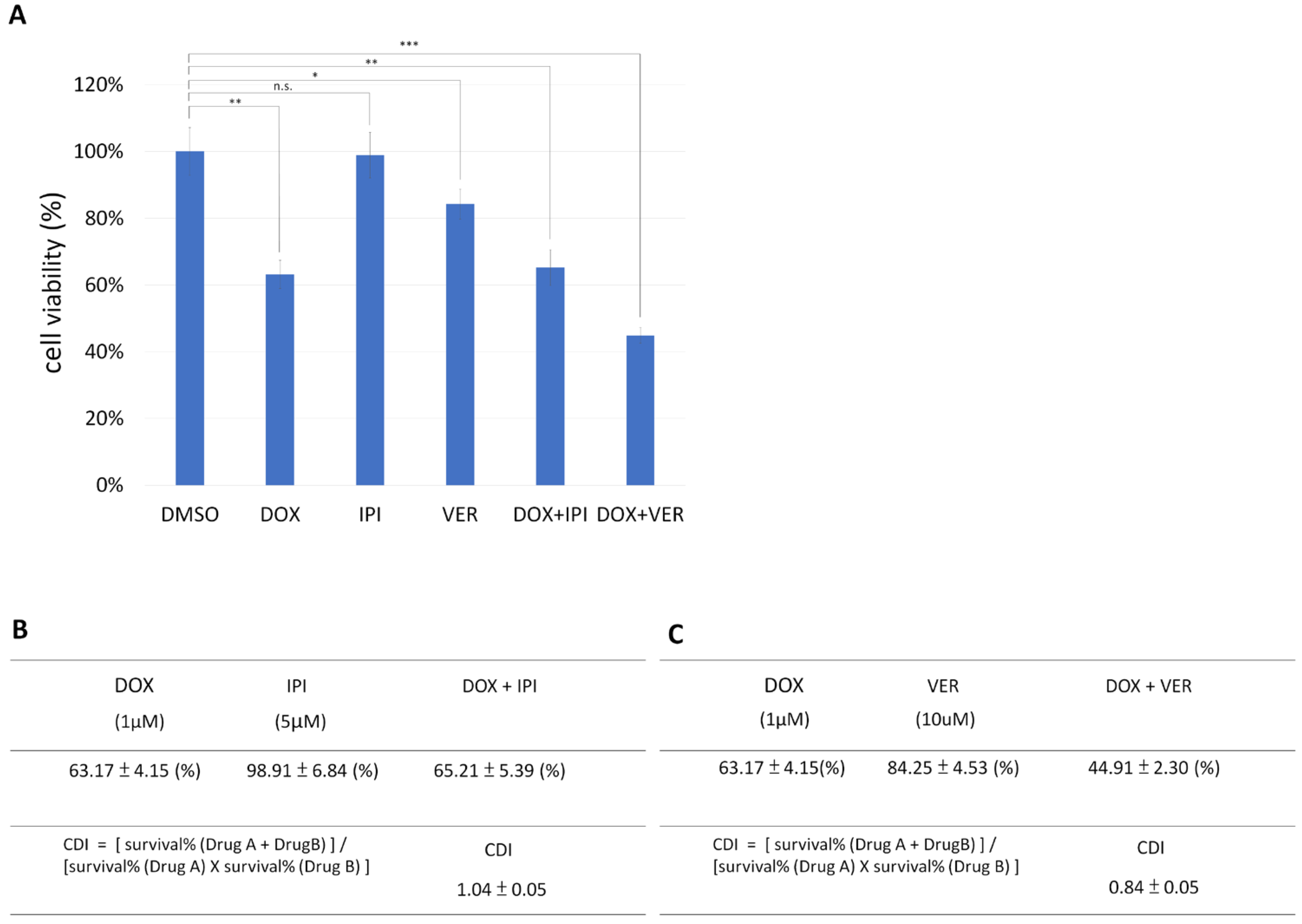
3.2. HSP70 Inhibitor and Doxorubicin Exhibit Synergistic Anticancer Activity in MDA-MB-231
3.3. HSP70 Inhibitor and Doxorubicin Have Synergistic Anticancer Activity in T47D, and HSP90 Inhibitor Has No Cytotoxic Reponse in T47D
3.4. Combination Therapy of HSP70 Inhibitor and Doxorubicin Induces Apoptosis in MDA-MB-231 Cells
3.5. The Expression of hsp70 Family Members in Breast Invasive Carcinoma (BRCA)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- de Andrade, K.C.; Lee, E.E.; Tookmanian, E.M.; Kesserwan, C.A.; Manfredi, J.J.; Hatton, J.N.; Loukissas, J.K.; Zavadil, J.; Zhou, L.; Olivier, M.; et al. The TP53 Database: Transition from the International Agency for Research on Cancer to the US National Cancer Institute. Cell Death Differ. 2022, 29, 1071–1073. [Google Scholar] [CrossRef] [PubMed]
- Macleod, K.F.; Sherry, N.; Hannon, G.; Beach, D.; Tokino, T.; Kinzler, K.; Vogelstein, B.; Jacks, T. p53-dependent and independent expression of p21 during cell growth, differentiation, and DNA damage. Genes Dev. 1995, 9, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Hermeking, H.; Lengauer, C.; Polyak, K.; He, T.C.; Zhang, L.; Thiagalingam, S.; Kinzler, K.W.; Vogelstein, B. 14-3-3 sigma is a p53-regulated inhibitor of G2/M progression. Mol. Cell 1997, 1, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.H.; Chen, J.Y.; Lu, M.H.; Chang, L.T.; Lin, H.C.; Chang, Y.M.; Chao, C.F. Functional four-base A/T gap core sequence CATTAG of P53 response elements specifically bound tetrameric P53 differently than two-base A/T gap core sequence CATG bound both dimeric and tetrameric P53. Nucleic Acids Res. 2009, 37, 1984–1990. [Google Scholar] [CrossRef]
- Rangel, L.P.; Costa, D.C.; Vieira, T.C.; Silva, J.L. The aggregation of mutant p53 produces prion-like properties in cancer. Prion 2014, 8, 75–84. [Google Scholar] [CrossRef]
- Kim, S.; An, S.S. Role of p53 isoforms and aggregations in cancer. Medicine 2016, 95, e3993. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Xu, D.; Zhang, T.; Hu, W.; Feng, Z. Gain-of-function mutant p53 in cancer progression and therapy. J. Mol. Cell Biol. 2020, 12, 674–687. [Google Scholar] [CrossRef]
- Zhou, G.; Wang, J.; Zhao, M.; Xie, T.X.; Tanaka, N.; Sano, D.; Patel, A.A.; Ward, A.M.; Sandulache, V.C.; Jasser, S.A.; et al. Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Mol. Cell 2014, 54, 960–974. [Google Scholar] [CrossRef]
- Ano Bom, A.P.; Rangel, L.P.; Costa, D.C.; de Oliveira, G.A.; Sanches, D.; Braga, C.A.; Gava, L.M.; Ramos, C.H.; Cepeda, A.O.; Stumbo, A.C.; et al. Mutant p53 aggregates into prion-like amyloid oligomers and fibrils: Implications for cancer. J. Biol. Chem. 2012, 287, 28152–28162. [Google Scholar] [CrossRef]
- Wilcken, R.; Wang, G.; Boeckler, F.M.; Fersht, A.R. Kinetic mechanism of p53 oncogenic mutant aggregation and its inhibition. Proc. Natl. Acad. Sci. USA 2012, 109, 13584–13589. [Google Scholar] [CrossRef]
- Cai, B.H.; Bai, Z.Y.; Lien, C.F.; Yu, S.J.; Lu, R.Y.; Wu, M.H.; Wu, W.C.; Chen, C.C.; Hsu, Y.C. NAMPT Inhibitor and P73 Activator Represses P53 R175H Mutated HNSCC Cell Proliferation in a Synergistic Manner. Biomolecules 2022, 12, 438. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.H.; Wang, Y.T.; Chen, C.C.; Yeh, F.Y.; Lin, Y.R.; Lin, Y.C.; Wu, T.Y.; Wu, K.Y.; Lien, C.F.; Shih, Y.C.; et al. Chlorophyllides repress gain-of-function p53 mutated HNSCC cell proliferation via activation of p73 and repression of p53 aggregation in vitro and in vivo. Biochim. Biophys. Acta Mol. Basis Dis. 2025, 1871, 167662. [Google Scholar] [CrossRef]
- Levy, C.B.; Stumbo, A.C.; Ano Bom, A.P.; Portari, E.A.; Cordeiro, Y.; Silva, J.L.; De Moura-Gallo, C.V. Co-localization of mutant p53 and amyloid-like protein aggregates in breast tumors. Int. J. Biochem. Cell Biol. 2011, 43, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Gao, J.L.; Ji, J.W.; Gao, M.; Yin, Q.S.; Qiu, Q.L.; Wang, C.; Chen, S.Z.; Xu, J.; Liang, R.S.; et al. Cytotoxicity enhancement in MDA-MB-231 cells by the combination treatment of tetrahydropalmatine and berberine derived from Corydalis yanhusuo W. T. Wang. J. Intercult. Ethnopharmacol. 2014, 3, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Luchtel, R.A.; Bhagat, T.; Pradhan, K.; Jacobs, W.R.; Levine, M.; Verma, A.; Shenoy, N. High-dose ascorbic acid synergizes with anti-PD1 in a lymphoma mouse model. Proc. Natl. Acad. Sci. USA 2020, 117, 1666–1677. [Google Scholar] [CrossRef]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; DeZonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef]
- Huang, X.; Shi, D.; Zou, X.; Wu, X.; Huang, S.; Kong, L.; Yang, M.; Xiao, Y.; Chen, B.; Chen, X.; et al. BAG2 drives chemoresistance of breast cancer by exacerbating mutant p53 aggregate. Theranostics 2023, 13, 339–354. [Google Scholar] [CrossRef]
- Yu, H.; Ding, J.; Zhu, H.; Jing, Y.; Zhou, H.; Tian, H.; Tang, K.; Wang, G.; Wang, X. LOXL1 confers antiapoptosis and promotes gliomagenesis through stabilizing BAG2. Cell Death Differ. 2020, 27, 3021–3036. [Google Scholar] [CrossRef]
- Liburkin-Dan, T.; Toledano, S.; Neufeld, G. Lysyl Oxidase Family Enzymes and Their Role in Tumor Progression. Int. J. Mol. Sci. 2022, 23, 6249. [Google Scholar] [CrossRef]
- Ramos, S.; Ferreira, S.; Fernandes, A.S.; Saraiva, N. Lysyl Oxidases Expression and Breast Cancer Progression: A Bioinformatic Analysis. Front. Pharmacol. 2022, 13, 883998. [Google Scholar] [CrossRef]
- Shi, Y. Mechanisms of caspase activation and inhibition during apoptosis. Mol. Cell 2002, 9, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S. Caspase function in programmed cell death. Cell Death Differ. 2007, 14, 32–43. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, S.H.; Clark, A.C. Targeting cell death in tumors by activating caspases. Curr. Cancer Drug Targets 2008, 8, 98–109. [Google Scholar] [CrossRef]
- Tanaka, N.; Okada, H.; Yamaguchi, K.; Seki, M.; Matsubara, D.; Gotoh, N.; Suzuki, Y.; Furukawa, Y.; Yamashita, T.; Inoue, J.I.; et al. Mint3-depletion-induced energy stress sensitizes triple-negative breast cancer to chemotherapy via HSF1 inactivation. Cell Death Dis. 2023, 14, 815. [Google Scholar] [CrossRef]
- Shen, M.; Cao, S.; Long, X.; Xiao, L.; Yang, L.; Zhang, P.; Li, L.; Chen, F.; Lei, T.; Gao, H.; et al. DNAJC12 causes breast cancer chemotherapy resistance by repressing doxorubicin-induced ferroptosis and apoptosis via activation of AKT. Redox Biol. 2024, 70, 103035. [Google Scholar] [CrossRef]
- Radons, J. The human HSP70 family of chaperones: Where do we stand? Cell Stress Chaperones 2016, 21, 379–404. [Google Scholar] [CrossRef]
- Jiang, W.; Pan, X.; Yan, H.; Wang, G. Prognostic Significance of the Hsp70 Gene Family in Colorectal Cancer. Med. Sci. Monit. 2021, 27, e928352. [Google Scholar] [CrossRef]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Karthikeyan, S.K.; Korla, P.K.; Patel, H.; Shovon, A.R.; Athar, M.; Netto, G.J.; Qin, Z.S.; Kumar, S.; Manne, U.; et al. UALCAN: An update to the integrated cancer data analysis platform. Neoplasia 2022, 25, 18–27. [Google Scholar] [CrossRef]
- Westerheide, S.D.; Anckar, J.; Stevens, S.M.; Sistonen, L.; Morimoto, R.I. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science 2009, 323, 1063–1066. [Google Scholar] [CrossRef]
- Zhang, T.; Berrocal, J.G.; Frizzell, K.M.; Gamble, M.J.; DuMond, M.E.; Krishnakumar, R.; Yang, T.; Sauve, A.A.; Kraus, W.L. Enzymes in the NAD+ salvage pathway regulate SIRT1 activity at target gene promoters. J. Biol. Chem. 2009, 284, 20408–20417. [Google Scholar] [CrossRef] [PubMed]
- Ongkeko, W.M.; An, Y.; Chu, T.S.; Aguilera, J.; Dang, C.L.; Wang-Rodriguez, J. Gleevec suppresses p63 expression in head and neck squamous cell carcinoma despite p63 activation by DNA-damaging agents. Laryngoscope 2006, 116, 1390–1396. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Zhang, C.; Zwahlen, M.; von Feilitzen, K.; Karlsson, M.; Shi, M.; Yuan, M.; Song, X.; Li, X.; Yang, H.; et al. Systematic transcriptional analysis of human cell lines for gene expression landscape and tumor representation. Nat. Commun. 2023, 14, 5417. [Google Scholar] [CrossRef]
- Xu, J.; Reumers, J.; Couceiro, J.R.; De Smet, F.; Gallardo, R.; Rudyak, S.; Cornelis, A.; Rozenski, J.; Zwolinska, A.; Marine, J.C.; et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 2011, 7, 285–295. [Google Scholar] [CrossRef]
- Kehrloesser, S.; Osterburg, C.; Tuppi, M.; Schäfer, B.; Vousden, K.H.; Dötsch, V. Intrinsic aggregation propensity of the p63 and p73 TI domains correlates with p53R175H interaction and suggests further significance of aggregation events in the p53 family. Cell Death Differ. 2016, 23, 1952–1960. [Google Scholar] [CrossRef]
- Cai, B.H.; Hsu, Y.C.; Yeh, F.Y.; Lin, Y.R.; Lu, R.Y.; Yu, S.J.; Shaw, J.F.; Wu, M.H.; Tsai, Y.Z.; Lin, Y.C.; et al. P63 and P73 Activation in Cancers with p53 Mutation. Biomedicines 2022, 10, 1490. [Google Scholar] [CrossRef]
- Qin, Z.Q.; Li, Q.G.; Yi, H.; Lu, S.S.; Huang, W.; Rong, Z.X.; Tang, Y.Y.; Xiao, Z.Q. Heterozygous p53-R280T Mutation Enhances the Oncogenicity of NPC Cells Through Activating PI3K-Akt Signaling Pathway. Front. Oncol. 2020, 10, 104. [Google Scholar] [CrossRef]
- Norris, B.; Pritchard, K.I.; James, K.; Myles, J.; Bennett, K.; Marlin, S.; Skillings, J.; Findlay, B.; Vandenberg, T.; Goss, P.; et al. Phase III comparative study of vinorelbine combined with doxorubicin versus doxorubicin alone in disseminated metastatic/recurrent breast cancer: National Cancer Institute of Canada Clinical Trials Group Study MA8. J. Clin. Oncol. 2000, 18, 2385–2394. [Google Scholar] [CrossRef]
- Bisht, A.; Avinash, D.; Sahu, K.K.; Patel, P.; Das Gupta, G.; Kurmi, B.D. A comprehensive review on doxorubicin: Mechanisms, toxicity, clinical trials, combination therapies and nanoformulations in breast cancer. Drug Deliv. Transl. Res. 2025, 15, 102–133. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, B. Doxorubicin induces cardiotoxicity through upregulation of death receptors mediated apoptosis in cardiomyocytes. Sci. Rep. 2017, 7, 44735. [Google Scholar] [CrossRef]
- Christidi, E.; Brunham, L.R. Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell Death Dis. 2021, 12, 339. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, K.-Y.; Crucho, A.; Su, M.; Chen, S.-T.; Hung, C.-H.; Kou, Y.-L.; Liu, Y.-J.; Hsu, T.-C.; Yeh, F.-Y.; Lien, C.-F.; et al. Synergistic Anticancer Activity of HSP70 Inhibitor and Doxorubicin in Gain-of-Function Mutated p53 Breast Cancer Cells. Biomedicines 2025, 13, 1034. https://doi.org/10.3390/biomedicines13051034
Wu K-Y, Crucho A, Su M, Chen S-T, Hung C-H, Kou Y-L, Liu Y-J, Hsu T-C, Yeh F-Y, Lien C-F, et al. Synergistic Anticancer Activity of HSP70 Inhibitor and Doxorubicin in Gain-of-Function Mutated p53 Breast Cancer Cells. Biomedicines. 2025; 13(5):1034. https://doi.org/10.3390/biomedicines13051034
Chicago/Turabian StyleWu, Kuan-Yo, Ana Crucho, Mia Su, Sih-Tong Chen, Chen-Hsiu Hung, Yu-Ling Kou, Yu-Jie Liu, Tzu-Chi Hsu, Fang-Yu Yeh, Ching-Feng Lien, and et al. 2025. "Synergistic Anticancer Activity of HSP70 Inhibitor and Doxorubicin in Gain-of-Function Mutated p53 Breast Cancer Cells" Biomedicines 13, no. 5: 1034. https://doi.org/10.3390/biomedicines13051034
APA StyleWu, K.-Y., Crucho, A., Su, M., Chen, S.-T., Hung, C.-H., Kou, Y.-L., Liu, Y.-J., Hsu, T.-C., Yeh, F.-Y., Lien, C.-F., Chen, C.-C., & Cai, B.-H. (2025). Synergistic Anticancer Activity of HSP70 Inhibitor and Doxorubicin in Gain-of-Function Mutated p53 Breast Cancer Cells. Biomedicines, 13(5), 1034. https://doi.org/10.3390/biomedicines13051034