
Recent Progress and Advances in HGF/MET-Targeted Therapeutic Agents for Cancer Treatment

Abstract
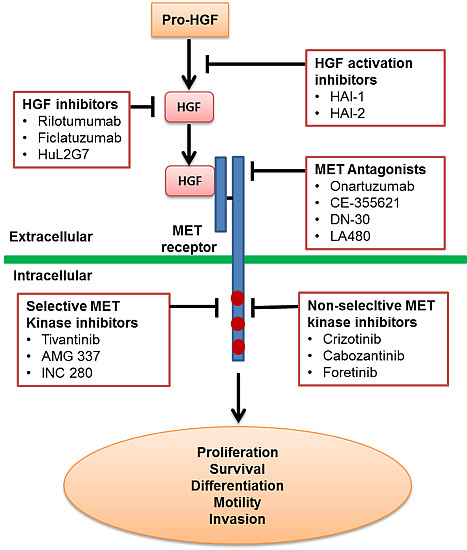
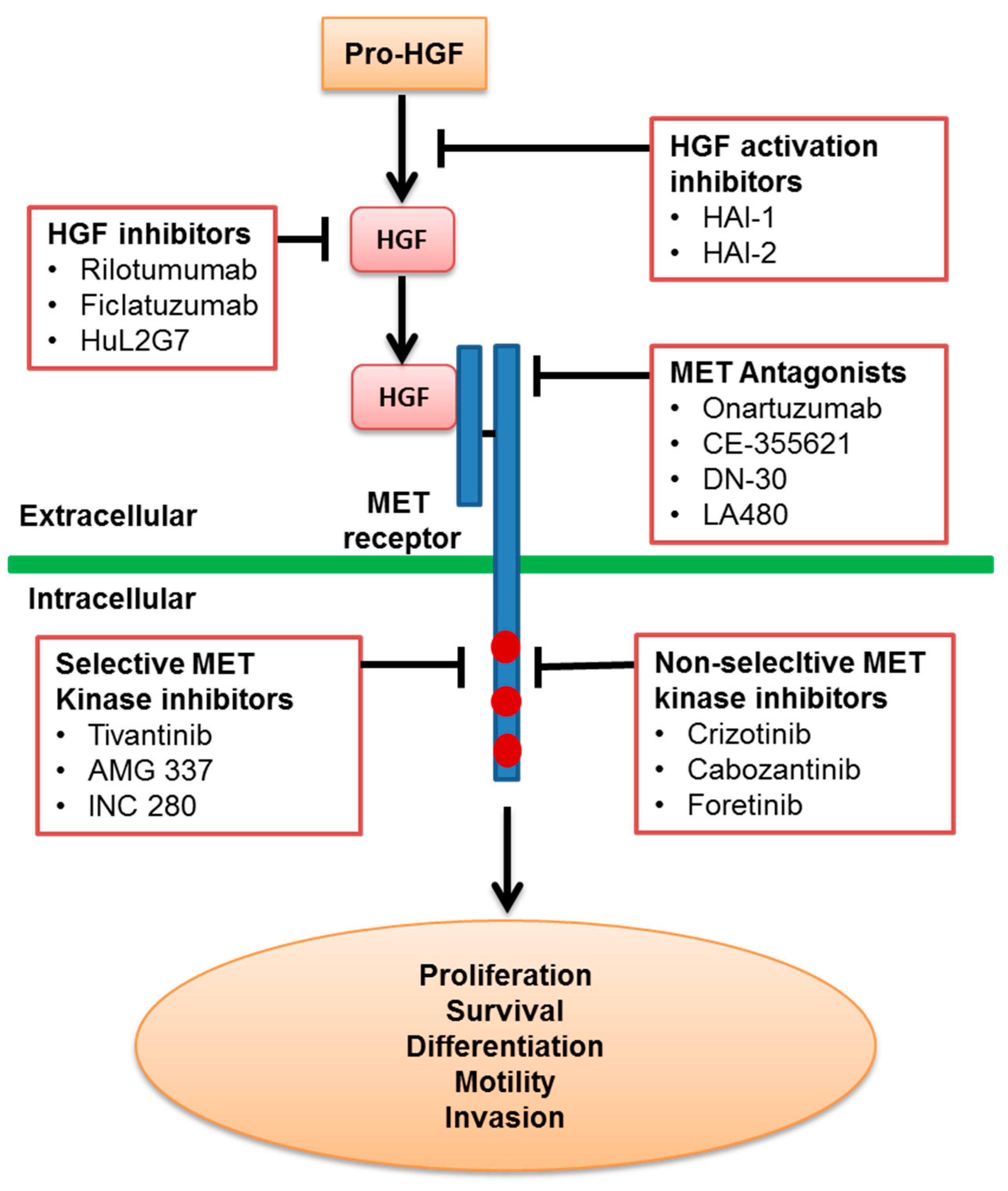
:
1. Introduction

{kind=link}
{kind=link}
{kind=link}
| Compound | Modality | Target(s) | Company | Cancer Type | Development Phase | |
|---|---|---|---|---|---|---|
| Rilotumumab (AMG 102) | Antibody | HGF | Amgen | Gastric, lung, colon, brain, ovary, renal | 2/3 | |
| Ficlatuzumab (AV-299) | Antibody | HGF | AVEO Pharmaceuticals | Lung | 1/2 | |
| HuL2G7 (TAK701) | Antibody | HGF | Galaxy Biotech | Solid tumors | 1 | |
| Onartuzumab (MetMab) | Antibody | MET | Genentech/Roche | Lung, colon, breast | 2/3 | |
| AMG 337 | Small molecule | MET | Amgen | Solid tumors | 1/2 | |
| INC 280 | Small molecule | MET | Novartis/Incyte | Renal, brain, liver, lung, melanoma, head and neck | 2 | |
| Tivantinib (ARQ 197) | Small molecule | MET | ArQule/Daiichi–Sankyo/Kyowa Hakko Kirin | Lung, colon, breast, liver, prostate, myeloma | 2/3 | |
| Crizotinib (PF-2341066) | Small molecule | MET, ALK | Pfizer | Lung, lymphoma | 2/3 | |
| Cabozantinib (XL 184) | Small molecule | MET, VEGFR2, RET, KIT, AXL, FLT3 | Exelixis/Bristol–Myers Squibb | Lung | 2/3 | |
| Foretinib (XL 880) | Small molecule | MET, VEGFR2, PDGFR, RON, FLT-2, FLT-3, AXL, TIE2 | Exelixis/GlaxoSmithKline | Lung, breast, liver, renal, stomach, head and neck | 1/2 | |
| Golvatinib (E7050) | Small molecule | MET, VEGFR2 | Eisai Inc. | Liver, head and neck, stomach | 1 | |
| MGCD265 | Small molecule | MET, VEGFR2, RON, TIE2 | MethylGene | Lung | 1/2 | |
| BMS-777607 | Small molecule | MET, RON | Bristol-Myers Squibb | Solid tumors | 1/2 |
- (1)
- Mechanism of action: targeting the ligand (i.e., HGF) vs. targeting the receptor (i.e., MET)
- (2)
- Modality of therapeutic agents: small molecules vs. therapeutic proteins
- (3)
- Treatment strategies: monotherapy and combination therapy
- (4)
- Value and clinical implications of biomarkers for the HGF/MET pathway
- (5)
- Challenges and perspectives on the development of HGF/MET therapeutics
2. Mechanism of Action: Targeting the HGF Ligand vs. Targeting the MET Receptor
2.1. Inhibition of HGF Activation

2.2. Inhibition of HGF Binding to the MET Receptor
2.3. MET Antagonists
2.4. MET Kinase Inhibitors
3. Modality of Therapeutic Agents: Clinical Features of Small Molecules and Therapeutic mAbs
| Compound | Top Five Grade ≥ 3 AE (%) | MTD or the Highest Tested Dose | CL | Vd | t1/2 |
|---|---|---|---|---|---|
| Rilotumumab (AMG 102) [44] | Hypoxia (3) | 20 mg/kg q2w | 0.104–0.176 mL/h/kg | 59 mL/kg [84] | 14.5–22.0 days |
| Dyspnea (3) | |||||
| Upper gastrointestinal hemorrhage (3) | |||||
| Colonic fistula (3) * | |||||
| Ficlatuzumab (AV-299) [41] | Hypokalemia (17) | 20 mg/kg q2w | 0.16 ± 0.06 mL/h/kg | 49.4 ± 11.0 mL/kg | 225 ± 74 h |
| Peripheral edema (8) | |||||
| Fatigue (4) | |||||
| Diarrhea (4) | |||||
| Vomiting (4) | |||||
| HuL2G7 (TAK701) [52,85] | Gastrointestinal ileus (2) | 20 mg/kg q2w | NR | NR | 8.6–14.1 days |
| Pleural effusion (2) | |||||
| Urinary tract infection (2) | |||||
| Dyspnea (1) * | |||||
| Onartuzumab (MetMab) [56] | Edema peripheral (9) | 15 mg/kg q3w | 6.85 ± 1.94 mL/day/kg | 96.8 ± 23.4 mL/kg | 11.5 ± 5.54 days |
| Abdominal pain (3) | |||||
| AST increased (3) | |||||
| Pyrexia (3) | |||||
| Hyponatremia (3) | |||||
| AMG 337 [70] | Headache (9) | 300 mg QD | NR | NR | 4.6–6.9 h |
| Fatigue (5) | |||||
| Dyspnea (5) | |||||
| Vomiting (3) | |||||
| Nausea (1) | |||||
| INC 280 [71] | Blood bilirubin increased (12) | 600 mg BID | NR | NR | 2.3–6.4 h |
| Anemia (9) | |||||
| Fatigue (9) | |||||
| Hyponatremia (9) | |||||
| Hypophagia (6) | |||||
| Tivantinib (ARQ 197) [59] | Anemia (4) | 360 mg BID | NR | NR | 1.5–2.7 h |
| Neutropenia (3) | |||||
| Leukopenia (1) | |||||
| Thrombocytopenia (1) | |||||
| Nausea (1) | |||||
| Crizotinib (PF-2341066) [75,86] | ALT elevation (17) | 250 mg BID | 100 L/h at single dose; 60 L/h at steady state | 1772 L | 42 h |
| Neutropenia (12) | |||||
| AST elevation (9) | |||||
| Lymphopenia (9) | |||||
| Hypophosphatemia (5) | |||||
| Cabozantinib (XL 184) [81] | Fatigue (10) | 175 mg QD | 4.4 L/h | 349 L | 91.3 ± 33.3 h |
| Palmar plantar erythrodysesthesia (10) | |||||
| Increased lipase (10) | |||||
| Diarrhea (7) | |||||
| Decreased weight (6) | |||||
| Foretinib (EXEL 2880) [82] | Hypertension (24.3) | 80 mg QD | 83 L/h | NR | NR |
| Fatigue (18.9) | |||||
| Nausea (10.8) | |||||
| Diarrhea (10.8) | |||||
| Dehydration (10.8) | |||||
| Golvatinib (E7050) [87] | Fatigue (14.7) | 400 mg QD | NR | 325–707 L | NR |
| Decreased appetite (8.8) | |||||
| Renal impairment (2.9) | |||||
| Elevated GGT (2.9) | |||||
| Elevated ALP (2.9) |
4. Treatment Strategies: Monotherapy and Drug Combination
4.1. Monotherapy
4.2. Combination of Chemotherapy with HGF/MET Inhibitors
4.3. Combination of Targeted Therapy with HGF/MET Inhibitors

4.4. Drug Combination against Drug Resistance
5. Value and Clinical Implications of HGF/MET Biomarkers
5.1. HGF/MET Biomarkers in Cancer Development
5.1.1. Circulating HGF
5.1.2. MET Protein Overexpression
5.1.3. MET Gene Amplification and Mutation
5.2. Predictive Values of HGF/MET Biomarkers for Clinical Outcome
5.2.1. Preclinical Evidence
5.2.2. Clinical Evidence
| Cancer Type | Biomarkers (Treatment) | Key Findings |
|---|---|---|
| Gastric cancer | MET polymorphism (surgery) | MET polymorphism of 161 Japanese, 101 US, and 63 Austrian patients with locoregional gastric cancer treated with surgery was examined. Patients with any G (A/G or G/G genotype) allele of MET rs40239 had significantly longer disease-free survival and OS compared with those with the AA genotype in male Japanese, but not in female Japanese as well as all patients in the US and Austrian cohorts [112]. |
| FGFR2, HER2 and MET, tissue from tumor body (TB), luminal surface (LS), and invasive edge (IE) (surgery) | High MET expression level was associated with worse OS after adjustment for other covariates (p = 0.006); HER2 (p = 0.004) and pERK (p = 0.001) expression level differed between tumor regions, with increased HER2 expression level in the LS compared with the TB and IE. The potential downstream markers pS6 and pERK were expressed across tumor regions, providing evidence that resections and biopsies would yield comparative results in clinical trials [113]. | |
| MET overexpression in tumor tissue | MET expression level was associated with improved trends in clinical outcomes [51]. | |
| Colorectal or rectal cancer | PTEN, PI3K p110a, MET, and CAIX in tumor specimens (bevacizumab) | In patients with metastatic colorectal cancer (CRC) treated with bevacizumab, expression level of CAIX, PI3K p110a, and MET in metastases did not predict objective response (OR). PTEN loss was associated with OR but not OS. Tumor heterogeneity should be taken into consideration [114]. |
| HGF, MET (hepatectomy) | High HGF is produced to promote liver regeneration post-hepatectomy, which was used for the treatment of liver metastasis of CRC. HGF produced after hepatectomy may stimulate the progression of CRC cells with MET in residual liver. Results from 94 CRC patients including 24 with liver metastasis showed that MET overexpression was closely associated with CRC liver metastases (87% of 24 patients), while in liver metastatic lesions, the MET expression level was reduced in comparison to primary lesions [115]. | |
| Gene expression profiles (chemo-radiotherapy) | To differentiate gene expression profiles based on tumor regression grading (TRG) in residual cancer cells after operative chemoradiotherapy (CRT), total RNA was obtained from 52 patients with locally advanced rectal cancer to examine the expression levels of 20 genes. Expressions of LGR5, PDRG1, GLUT, MK167, andBAX genes were significantly associated with clinical outcome in the grading systems, while other tested gene expressions, including HGF and MET, did not show any associations, indicating that TRG may reflect features of proliferation, stemness potency, and resistance to hypoxia of residual cancer cells following preoperative CRT [116]. | |
| Papillary renal cell carcinoma (PRCC) | MET pathway activation markers (foretinib) | PRCC patients were stratified by MET pathway activation (germline or somatic MET mutation, MET [7q31] amplification, or gain of chromosome 7). The presence of a germline MET mutation was highly predictive of a response (five of 10 vs. five of 57 patients with and without germline MET mutations, respectively) [117]. |
| Glioblastoma | HGF, EGFRvIII amplification (preclinical study with SGX523 and erlotinib) | HGF autocrine expression correlated with high phospho-MET levels in HGF autocrine cell lines, and these lines showed high sensitivity to MET inhibition in vivo. An HGF paracrine environment may enhance glioblastoma growth in vivo but may not indicate sensitivity to MET inhibition. EGFRvIII amplification predicted sensitivity to EGFR inhibition, but in the same tumor, increased copies of MET from gains of chromosome 7 did not increase MET activity or predict sensitivity to MET inhibitors. Thus, HGF autocrine glioblastoma bears an activated MET signaling pathway that may predict sensitivity to MET inhibitors. Serum HGF levels may be a biomarker for the presence of autocrine tumors and their responsiveness to MET therapeutics [118]. |
| HGF/MET (in vitro cell line assays) | HGF and MET are expressed in vitro in glioblastoma multiforme cell lines as well as in normal human astrocyte (NHA) cells. HGF stimulates tyrosine phosphorylation of MET in both glioma cell lines and NHA cells, but only the glioma cell lines proliferate and become motile and invasive in response to HGF, whereas the NHA cells are nonresponsive, implicating autocrine/paracrine MET/HGF signaling in glioma tumorigenesis; HGF signaling through MET is negatively regulated in NHA cells [119]. | |
| Breast cancer (BC) | MET tissue expression and serum level of HGF in female breast ductal carcinoma (surgery) | Significant increase in serum HGF levels was found in patients compared with healthy subjects. Serum level of HGF is an independent prognostic indicator of breast cancer. Fibrocystic disease of the breast showed weak HGFR expression, while in normal tissue, HGFR was scanty; breast invasive ductal carcinoma showed homogenous strong reaction to HGFR. Preoperative high serum HGF levels and malignancy occur together. MET overexpression in tumors was a poor prognostic factor for OS [120]. |
| Serum HGF level in patients and healthy subjects (surgery) | Higher serum soluble HGF was found in patients with invasive breast cancer compared with healthy subjects (p < 0.001). Multivariate analysis suggested that advanced tumor-node-metastasis (TNM) staging was an independent factor regarding the high level of HGF (p < 0.001). Preoperative serum HGF levels might reflect the severity of invasive breast cancer [121]. | |
| MET, p-MET (any available treatments for BC) | High protein levels of MET and p-MET were found in 257 patients and correlated with poor prognosis for PFS and OS in HER2-positive breast cancers and associated with a significantly higher risk of recurrence and death (p < 0.05) [122]. | |
| Breast cancer (BC) | MET or HGF gene copy number (trastuzumab based treatment) | High gene copy number of MET and HGF was found to significantly associate with an increased risk of trastuzumab-based therapy failure (e.g., shorter time to disease progression) in HER2-positive metastatic BC. MET and HGF FISH-positive status was highly correlated (p < 0.001) and combination of both biomarkers did not increase predictive value of either considered separately [123]. |
| Prostate cancer | HGF and PSA (surgery) | Combined PSA and HGF assessment increased accuracy in distinguishing patients with metastatic or localized disease [124]. |
| Multiple myeloma | HGF (melphalan plus prednisone with or without interferon-α) | HGF was elevated at diagnosis in 43% of myeloma patients compared with healthy subjects (p < 0.00001). In the group with elevated HGF levels, 46% of the patients reached a plateau phase, as compared with 60% of the patients with low HGF levels (p = 0.005), and the median survival time was 21 and 32 months, respectively (p = 0.002). HGF may be a useful follow-up parameter in myeloma patients. Measurement of HGF may identify a group of patients with poor response to melphalan-prednisone treatment and short survival. HGF was a prognostic factor in patients with high levels of beta 2-microglobulin [125]. |
| Squamous cell carcinoma of the oral tongue (SCCOT) | Expression level of MET and tumor invasion depth, lymph node metastasis (surgery) | The relationship between the expression level of MET and tumor invasion depth, lymph node metastasis, and patient survival in small (T(1-2)) SCCOT was assessed in 71 surgically treated patients. The presence of neck metastasis and >4 mm depth of tumor invasion significantly correlated with MET overexpression. The survival rates were significantly shorter in patients with MET overexpression. Constitutive activation of MET enhanced migration and invasion of tongue cancer cells in vitro through the expressions of matrix metalloproteinase-1, -2, and -9, and promoted tongue cancer cell growth in vitro and in vivo [126]. |
| Endometrial cancer (EC) | HGF, MET, and basic fibroblast growth factor (bFGF) (surgery) | The prognostic significance of HGF, MET, and bFGF that contributes to angiogenesis and proliferation in numerous cancers was assessed in the tumor and stroma of EC patients (n = 211). Tumor bFGF was significantly associated with high-grade endometrioid and clear cell histology (p < 0.001), advanced stage (p = 0.008), positive lymph-node involvement (p = 0.002), poor OS (log-rank test, p = 0.009), and poor RFS (p < 0.001). HGF-positive and stromal bFGF-positive tumors had a lower risk of death compared with HGF-positive and stromal bFGF-negative tumors (hazard ratio (HR) = 0.14; 95% CI: 0.03, 0.60). HGF-positive and bFGF-positive tumors had a higher risk of recurrence compared with cases with negative expression of both markers (HR = 9.88, 95% CI: 2.63, 37.16). These data show that tumor and stromal bFGF expression level have opposite associations with survival outcomes in EC patients, which deserves confirmation in larger studies [127]. |
6. Challenges and Perspectives on the Development of HGF/MET Therapeutics
6.1. Select Relevant Animal Species and Preclinical Models
6.2. Select Appropriate Methods to Analyze Data from Xenograft Models
6.3. Identifying Signal from Noise
6.4. Cancer Variety with a Given Cancer Type
6.5. Regional Differences in Cancer Treatment
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Scagliotti, G.V.; Novello, S.; von Pawel, J. The emerging role of MET/HGF inhibitors in oncology. Cancer Treat Rev. 2013, 39, 793–801. [Google Scholar] [CrossRef]
- Jung, K.H.; Park, B.H.; Hong, S.S. Progress in cancer therapy targeting c-Met signaling pathway. Arch. Pharm. Res. 2012, 35, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; vande Woude, G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003, 4, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Beppu, K.; Uchiyama, A.; Morisaki, T.; Nakamura, K.; Noshiro, H.; Matsumoto, K.; Nakamura, T.; Tanaka, M.; Katano, M. Elevation of serum hepatocyte growth factor concentration in patients with gastric cancer is mediated by production from tumor tissue. Anticancer Res. 2000, 20, 1263–1267. [Google Scholar] [PubMed]
- Drebber, U.; Baldus, S.E.; Nolden, B.; Grass, G.; Bollschweiler, E.; Dienes, H.P.; Holscher, A.H.; Monig, S.P. The overexpression of c-met as a prognostic indicator for gastric carcinoma compared to p53 and p21 nuclear accumulation. Oncol. Rep. 2008, 19, 1477–1483. [Google Scholar] [PubMed]
- Han, S.U.; Lee, J.H.; Kim, W.H.; Cho, Y.K.; Kim, M.W. Significant correlation between serum level of hepatocyte growth factor and progression of gastric carcinoma. World J. Surg. 1999, 23, 1176–1180. [Google Scholar] [CrossRef] [PubMed]
- Janjigian, Y.Y.; Tang, L.H.; Coit, D.G.; Kelsen, D.P.; Francone, T.D.; Weiser, M.R.; Jhanwar, S.C.; Shah, M.A. MET expression and amplification in patients with localized gastric cancer. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1021–1027. [Google Scholar] [CrossRef]
- Kubicka, S.; Claas, C.; Staab, S.; Kuhnel, F.; Zender, L.; Trautwein, C.; Wagner, S.; Rudolph, K.L.; Manns, M. p53 mutation pattern and expression of c-erbB2 and c-met in gastric cancer: Relation to histological subtypes, Helicobacter pylori infection, and prognosis. Dig. Dis. Sci. 2002, 47, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Lennerz, J.K.; Kwak, E.L.; Ackerman, A.; Michael, M.; Fox, S.B.; Bergethon, K.; Lauwers, G.Y.; Christensen, J.G.; Wilner, K.D.; Haber, D.A.; et al. MET amplification identifies a small and aggressive subgroup of esophagogastric adenocarcinoma with evidence of responsiveness to crizotinib. J. Clin. Oncol. 2011, 29, 4803–4810. [Google Scholar]
- Nakajima, M.; Sawada, H.; Yamada, Y.; Watanabe, A.; Tatsumi, M.; Yamashita, J.; Matsuda, M.; Sakaguchi, T.; Hirao, T.; Nakano, H. The prognostic significance of amplification and overexpression of c-met and c-erb B-2 in human gastric carcinomas. Cancer 1999, 85, 1894–1902. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Miki, C.; Wakuda, R.; Kobayashi, M.; Tonouchi, H.; Kusunoki, M. Circulating level of hepatocyte growth factor as a useful tumor marker in patients with early-stage gastric carcinoma. Scand. J. Gastroenterol. 2004, 39, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Yonemura, Y.; Nojima, N.; Hirono, Y.; Fushida, S.; Fujimura, T.; Miwa, K.; Endo, Y.; Yamamoto, H.; Watanabe, H. The relation between the growth patterns of gastric carcinoma and the expression of hepatocyte growth factor receptor (c-met), autocrine motility factor receptor, and urokinase-type plasminogen activator receptor. Cancer 1998, 82, 2112–2122. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Kitamura, M.; Arai, K.; Iwasaki, Y.; Yamamoto, Y.; Igari, A.; Toi, M. Increase in the circulating level of hepatocyte growth factor in gastric cancer patients. Br. J. Cancer 1997, 75, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Newton, R.C.; Scherle, P.A. Developing c-MET pathway inhibitors for cancer therapy: Progress and challenges. Trends Mol. Med. 2010, 16, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.C.; Tretiakova, M.S.; MacKinnon, A.C.; Ramnath, N.; Johnson, C.; Dietrich, S.; Seiwert, T.; Christensen, J.G.; Jagadeeswaran, R.; Krausz, T.; et al. Expression and mutational analysis of MET in human solid cancers. Genes Chromosom. Cancer 2008, 47, 1025–1037. [Google Scholar] [CrossRef]
- Jun, H.T.; Sun, J.; Rex, K.; Radinsky, R.; Kendall, R.; Coxon, A.; Burgess, T.L. AMG 102, a fully human anti-hepatocyte growth factor/scatter factor neutralizing antibody, enhances the efficacy of temozolomide or docetaxel in U-87 MG cells and xenografts. Clin. Cancer Res. 2007, 13, 6735–6742. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Wang, L.; Su, Y.C.; Gillespie, G.Y.; Salhotra, A.; Lal, B.; Laterra, J. Systemic anti-hepatocyte growth factor monoclonal antibody therapy induces the regression of intracranial glioma xenografts. Clin. Cancer Res. 2006, 12, 1292–1298. [Google Scholar] [CrossRef] [PubMed]
- Puri, N.; Khramtsov, A.; Ahmed, S.; Nallasura, V.; Hetzel, J.T.; Jagadeeswaran, R.; Karczmar, G.; Salgia, R. A selective small molecule inhibitor of c-Met, PHA665752, inhibits tumorigenicity and angiogenesis in mouse lung cancer xenografts. Cancer Res. 2007, 67, 3529–3534. [Google Scholar] [CrossRef] [PubMed]
- Previdi, S.; Scolari, F.; Chila, R.; Ricci, F.; Abbadessa, G.; Broggini, M. Combination of the c-Met inhibitor tivantinib and zoledronic acid prevents tumor bone engraftment and inhibits progression of established bone metastases in a breast xenograft model. PLoS One 2013, 8, e79101. [Google Scholar] [CrossRef] [PubMed]
- Mittra, E.S.; Fan-Minogue, H.; Lin, F.I.; Karamchandani, J.; Sriram, V.; Han, M.; Gambhir, S.S. Preclinical efficacy of the anti-hepatocyte growth factor antibody ficlatuzumab in a mouse brain orthotopic glioma model evaluated by bioluminescence, PET, and MRI. Clin. Cancer Res. 2013, 19, 5711–5721. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Yang, R.; Zheng, Z.; Romero, M.; Ross, J.; Bou-Reslan, H.; Carano, R.A.; Kasman, I.; Mai, E.; Young, J.; et al. MetMAb, the one-armed 5D5 anti-c-Met antibody, inhibits orthotopic pancreatic tumor growth and improves survival. Cancer Res. 2008, 68, 4360–4368. [Google Scholar]
- Zou, H.Y.; Li, Q.; Lee, J.H.; Arango, M.E.; Burgess, K.; Qiu, M.; Engstrom, L.D.; Yamazaki, S.; Parker, M.; Timofeevski, S.; et al. Sensitivity of selected human tumor models to PF-04217903, a novel selective c-Met kinase inhibitor. Mol. Cancer Ther. 2012, 11, 1036–1047. [Google Scholar]
- ClinicalTrails.gov. Available online: http://www.clinicaltrials.gov (accessed on 21 January 2015).
- Gak, E.; Taylor, W.G.; Chan, A.M.; Rubin, J.S. Processing of hepatocyte growth factor to the heterodimeric form is required for biological activity. FEBS Lett. 1992, 311, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Parr, C.; Sanders, A.J.; Jiang, W.G. Hepatocyte growth factor activation inhibitors—Therapeutic potential in cancer. Anticancer Agents Med. Chem. 2010, 10, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.D.; Lee, S.L. Matriptase is required for the active form of hepatocyte growth factor induced Met, focal adhesion kinase and protein kinase B activation on neural stem/progenitor cell motility. Biochim. Biophys. Acta 2014, 1843, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Owen, K.A.; Qiu, D.; Alves, J.; Schumacher, A.M.; Kilpatrick, L.M.; Li, J.; Harris, J.L.; Ellis, V. Pericellular activation of hepatocyte growth factor by the transmembrane serine proteases matriptase and hepsin, but not by the membrane-associated protease uPA. Biochem. J. 2010, 426, 219–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naldini, L.; Vigna, E.; Bardelli, A.; Follenzi, A.; Galimi, F.; Comoglio, P.M. Biological activation of pro-HGF (hepatocyte growth factor) by urokinase is controlled by a stoichiometric reaction. J. Biol. Chem. 1995, 270, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Nagakawa, O.; Yamagishi, T.; Fujiuchi, Y.; Junicho, A.; Akashi, T.; Nagaike, K.; Fuse, H. Serum hepatocyte growth factor activator (HGFA) in benign prostatic hyperplasia and prostate cancer. Eur. Urol. 2005, 48, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, H.; Hamasuna, R.; Itoh, H.; Kitamura, N.; Koono, M. Activation of hepatocyte growth factor/scatter factor in colorectal carcinoma. Cancer Res. 2000, 60, 6148–6159. [Google Scholar] [PubMed]
- Wader, K.F.; Fagerli, U.M.; Holt, R.U.; Stordal, B.; Borset, M.; Sundan, A.; Waage, A. Elevated serum concentrations of activated hepatocyte growth factor activator in patients with multiple myeloma. Eur. J. Haematol. 2008, 81, 380–383. [Google Scholar] [PubMed]
- Hu, C.; Jiang, N.; Wang, G.; Zheng, J.; Yang, W.; Yang, J. Expression of hepatocyte growth factor activator inhibitor-1 (HAI-1) gene in prostate cancer: Clinical and biological significance. J. BUON 2014, 19, 215–220. [Google Scholar] [PubMed]
- Yasuda, K.; Komiya, A.; Watanabe, A.; Morii, A.; Oya, T.; Nagakawa, O.; Fujiuchi, Y.; Fuse, H. Expression of Hepatocyte growth factor activator inhibitor type-1 (HAI-1) in prostate cancer. Anticancer Res. 2013, 33, 575–581. [Google Scholar] [PubMed]
- Nakamura, K.; Abarzua, F.; Hongo, A.; Kodama, J.; Nasu, Y.; Kumon, H.; Hiramatsu, Y. The role of hepatocyte growth factor activator inhibitor-1 (HAI-1) as a prognostic indicator in cervical cancer. Int. J. Oncol. 2009, 35, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Abarzua, F.; Hongo, A.; Kodama, J.; Nasu, Y.; Kumon, H.; Hiramatsu, Y. Hepatocyte growth factor activator inhibitor-2 (HAI-2) is a favorable prognosis marker and inhibits cell growth through the apoptotic pathway in cervical cancer. Ann. Oncol. 2009, 20, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Nagakawa, O.; Yamagishi, T.; Akashi, T.; Nagaike, K.; Fuse, H. Serum hepatocyte growth factor activator inhibitor type I (HAI-I) and type 2 (HAI-2) in prostate cancer. Prostate 2006, 66, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.R.; Gentle, D.; Abdulrahman, M.; Maina, E.N.; Gupta, K.; Banks, R.E.; Wiesener, M.S.; Kishida, T.; Yao, M.; Teh, B.; et al. Tumor suppressor activity and epigenetic inactivation of hepatocyte growth factor activator inhibitor type 2/SPINT2 in papillary and clear cell renal cell carcinoma. Cancer Res. 2005, 65, 4598–4606. [Google Scholar]
- Sanders, A.J.; Parr, C.; Mason, M.D.; Jiang, W.G. Suppression of hepatocyte growth factor activator inhibitor-1 leads to a more aggressive phenotype of prostate cancer cells in vitro. Int. J. Mol. Med. 2007, 20, 613–619. [Google Scholar] [PubMed]
- Parr, C.; Jiang, W.G. Hepatocyte growth factor activation inhibitors (HAI-1 and HAI-2) regulate HGF-induced invasion of human breast cancer cells. Int. J. Cancer 2006, 119, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Weiss, G.J.; Papadopoulos, K.P.; Hofmeister, C.C.; Tibes, R.; Tolcher, A.; Isaacs, R.; Jac, J.; Han, M.; Payumo, F.C.; et al. Phase I ficlatuzumab monotherapy or with erlotinib for refractory advanced solid tumours and multiple myeloma. Br. J. Cancer 2014, 111, 272–280. [Google Scholar] [PubMed]
- Tabernero, J.; Elez, M.E.; Herranz, M.; Rico, I.; Prudkin, L.; Andreu, J.; Mateos, J.; Carreras, M.J.; Han, M.; Gifford, J.; et al. A pharmacodynamic/pharmacokinetic study of ficlatuzumab in patients with advanced solid tumors and liver metastases. Clin. Cancer Res. 2014, 20, 2793–2804. [Google Scholar]
- Tan, E.; Park, K.; Lim, W.T.; Ahn, M.; Ng, Q.S.; Ahn, J.S.; Tan, D.S.; Sun, J.; Jac, J.; Han, M.; et al. Phase Ib study of ficlatuzumab (formerly AV-299), an anti-hepatocyte growth factor (HGF) monoclonal antibody (MAb) in combination with gefitinib (G) in Asian patients (pts) with NSCLC. ASCO Meet. Abstr. 2011, 29, 7571. [Google Scholar]
- Mok, T.S.K.; Park, K.; Geater, S.L.; Agarwal, S.; Han, M.; Komarnitsky, P.; Credi, M.; McKee, K.; Kuriyama, N.; Slichenmyer, W.; et al. A randomized phase 2 study with exploratory biomarker analysis of ficlatuzumab, a humanized hepatocyte growth factor (HGF) inhibitory monoclonal antibody, in combination with gefitinib versus gefitinib alone in Asian patients with lung adenocarcinoma. Ann. Oncol. 2012, 23, 1198P. [Google Scholar]
- Gordon, M.S.; Sweeney, C.S.; Mendelson, D.S.; Eckhardt, S.G.; Anderson, A.; Beaupre, D.M.; Branstetter, D.; Burgess, T.L.; Coxon, A.; Deng, H.; et al. Safety, pharmacokinetics, and pharmacodynamics of AMG 102, a fully human hepatocyte growth factor-neutralizing monoclonal antibody, in a first-in-human study of patients with advanced solid tumors. Clin. Cancer Res. 2010, 16, 699–710. [Google Scholar]
- Martin, L.P.; Sill, M.; Shahin, M.S.; Powell, M.; DiSilvestro, P.; Landrum, L.M.; Gaillard, S.L.; Goodheart, M.J.; Hoffman, J.; Schilder, R.J. A phase II evaluation of AMG 102 (rilotumumab) in the treatment of persistent or recurrent epithelial ovarian, fallopian tube or primary peritoneal carcinoma: A Gynecologic Oncology Group study. Gynecol. Oncol. 2014, 132, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Rosen, P.J.; Sweeney, C.J.; Park, D.J.; Beaupre, D.M.; Deng, H.; Leitch, I.M.; Shubhakar, P.; Zhu, M.; Oliner, K.S.; Anderson, A.; et al. A phase Ib study of AMG 102 in combination with bevacizumab or motesanib in patients with advanced solid tumors. Clin. Cancer Res. 2010, 16, 2677–2687. [Google Scholar] [PubMed]
- Ryan, C.J.; Rosenthal, M.; Ng, S.; Alumkal, J.; Picus, J.; Gravis, G.; Fizazi, K.; Forget, F.; Machiels, J.P.; Srinivas, S.; et al. Targeted MET inhibition in castration-resistant prostate cancer: A randomized phase II study and biomarker analysis with rilotumumab plus mitoxantrone and prednisone. Clin. Cancer Res. 2013, 19, 215–224. [Google Scholar]
- Schoffski, P.; Garcia, J.A.; Stadler, W.M.; Gil, T.; Jonasch, E.; Tagawa, S.T.; Smitt, M.; Yang, X.; Oliner, K.S.; Anderson, A.; et al. A phase II study of the efficacy and safety of AMG 102 in patients with metastatic renal cell carcinoma. BJU Int. 2011, 108, 679–686. [Google Scholar]
- Van Cutsem, E.; Eng, C.; Nowara, E.; Swieboda-Sadlej, A.; Tebbutt, N.C.; Mitchell, E.; Davidenko, I.; Stephenson, J.; Elez, E.; Prenen, H.; et al. Randomized Phase Ib/II Trial of Rilotumumab or Ganitumab with Panitumumab versus Panitumumab Alone in Patients with Wild-type KRAS Metastatic Colorectal Cancer. Clin. Cancer Res. 2014, 20, 4240–4250. [Google Scholar] [PubMed]
- Wen, P.Y.; Schiff, D.; Cloughesy, T.F.; Raizer, J.J.; Laterra, J.; Smitt, M.; Wolf, M.; Oliner, K.S.; Anderson, A.; Zhu, M.; et al. A phase II study evaluating the efficacy and safety of AMG 102 (rilotumumab) in patients with recurrent glioblastoma. Neuro Oncol. 2011, 13, 437–446. [Google Scholar]
- Iveson, T.; Donehower, R.C.; Davidenko, I.; Tjulandin, S.; Deptala, A.; Harrison, M.; Nirni, S.; Lakshmaiah, K.; Thomas, A.; Jiang, Y.; et al. Rilotumumab in combination with epirubicin, cisplatin, and capecitabine as first-line treatment for gastric or oesophagogastric junction adenocarcinoma: An open-label, dose de-escalation phase 1b study and a double-blind, randomised phase 2 study. Lancet Oncol. 2014, 15, 1007–1018. [Google Scholar] [PubMed]
- Jones, S.F.; Cohen, R.B.; Bendell, J.C.; Denlinger, C.S.; Harvey, R.D.; Parasuraman, S.; Chi, X.; Scholz, C.; Wyant, T.; Kauh, J. Safety, tolerability, and pharmacokinetics of TAK-701, a humanized anti-hepatocyte growth factor (HGF) monoclonal antibody, in patients with advanced nonhematologic malignancies: First-in-human phase I dose-escalation study. ASCO Meet. Abstr. 2010, 28, 3081. [Google Scholar]
- Tseng, J.R.; Kang, K.W.; Dandekar, M.; Yaghoubi, S.; Lee, J.H.; Christensen, J.G.; Muir, S.; Vincent, P.W.; Michaud, N.R.; Gambhir, S.S. Preclinical efficacy of the c-Met inhibitor CE-355621 in a U87 MG mouse xenograft model evaluated by 18F-FDG small-animal PET. J. Nucl. Med. 2008, 49, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Pacchiana, G.; Chiriaco, C.; Stella, M.C.; Petronzelli, F.; de Santis, R.; Galluzzo, M.; Carminati, P.; Comoglio, P.M.; Michieli, P.; Vigna, E. Monovalency unleashes the full therapeutic potential of the DN-30 anti-Met antibody. J. Biol. Chem. 2010, 285, 36149–36157. [Google Scholar] [CrossRef] [PubMed]
- Wortinger, M.A.; Peek, V.; Zeng, W.; Yan, L.; Tetreault, J.; Xia, J.; Jirong, L.; Chow, C.; Manro, J.R.; Stephens, J.R.; et al. c-Met antibody LY2875358 (LA480) has pre-clinical enhanced efficacy with gastric cancer standard-of-care in vitro and in vivo. Cancer Res. 2012, 72, 2738. [Google Scholar]
- Salgia, R.; Patel, P.; Bothos, J.; Yu, W.; Eppler, S.; Hegde, P.; Bai, S.; Kaur, S.; Nijem, I.; Catenacci, D.V.; et al. Phase I dose-escalation study of onartuzumab as a single agent and in combination with bevacizumab in patients with advanced solid malignancies. Clin. Cancer Res. 2014, 20, 1666–1675. [Google Scholar]
- Spigel, D.R.; Ervin, T.J.; Ramlau, R.A.; Daniel, D.B.; Goldschmidt, J.H., Jr.; Blumenschein, G.R., Jr.; Krzakowski, M.J.; Robinet, G.; Godbert, B.; Barlesi, F.; et al. Randomized phase II trial of Onartuzumab in combination with erlotinib in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2013, 31, 4105–4114. [Google Scholar]
- Koeppen, H.; Yu, W.; Zha, J.; Pandita, A.; Penuel, E.; Rangell, L.; Raja, R.; Mohan, S.; Patel, R.; Desai, R.; et al. Biomarker analyses from a placebo-controlled phase ii study evaluating erlotinib +/− onartuzumab in advanced non-small cell lung cancer: MET expression levels are predictive of patient benefit. Clin. Cancer Res. 2014, 20, 4488–4498. [Google Scholar]
- Pant, S.; Saleh, M.; Bendell, J.; Infante, J.R.; Jones, S.; Kurkjian, C.D.; Moore, K.M.; Kazakin, J.; Abbadessa, G.; Wang, Y.; et al. A phase I dose escalation study of oral c-MET inhibitor tivantinib (ARQ 197) in combination with gemcitabine in patients with solid tumors. Ann. Oncol. 2014, 25, 1416–1421. [Google Scholar]
- Goldberg, J.M.; Gavcovich, T.; Saigal, G.; Goldman, J.W.; Rosen, L.S. Extended progression-free survival in two patients with alveolar soft part sarcoma exposed to tivantinib. J. Clin. Oncol. 2014, 32, e114–e116. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.K.; Muro, K.; Ryu, M.H.; Yasui, H.; Nishina, T.; Ryoo, B.Y.; Kamiya, Y.; Akinaga, S.; Boku, N. A phase II trial of a selective c-Met inhibitor tivantinib (ARQ 197) monotherapy as a second- or third-line therapy in the patients with metastatic gastric cancer. Investig. New Drugs 2014, 32, 355–361. [Google Scholar] [CrossRef]
- Feldman, D.R.; Einhorn, L.H.; Quinn, D.I.; Loriot, Y.; Joffe, J.K.; Vaughn, D.J.; Flechon, A.; Hajdenberg, J.; Halim, A.B.; Zahir, H.; et al. A phase 2 multicenter study of tivantinib (ARQ 197) monotherapy in patients with relapsed or refractory germ cell tumors. Investig. New Drugs 2013, 31, 1016–1022. [Google Scholar] [PubMed]
- Broggini, M.; Garassino, M.C.; Damia, G. Evaluation of safety and efficacy of tivantinib in the treatment of inoperable or recurrent non-small-cell lung cancer. Cancer Manag. Res. 2013, 5, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Simonelli, M.; Rodriguez-Lope, C.; Zucali, P.; Camacho, L.H.; Granito, A.; Senzer, N.; Rimassa, L.; Abbadessa, G.; Schwartz, B.; et al. A Phase-1b study of tivantinib (ARQ 197) in adult patients with hepatocellular carcinoma and cirrhosis. Br. J. Cancer 2013, 108, 21–24. [Google Scholar]
- Trojan, J.; Zeuzem, S. Tivantinib in hepatocellular carcinoma. Expert Opin. Investig. Drugs 2013, 22, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.J.; Goldberg, J.M.; Dubois, S.G.; Choy, E.; Rosen, L.; Pappo, A.; Geller, J.; Judson, I.; Hogg, D.; Senzer, N.; et al. Tivantinib (ARQ 197), a selective inhibitor of MET, in patients with microphthalmia transcription factor-associated tumors: Results of a multicenter phase 2 trial. Cancer 2012, 118, 5894–5902. [Google Scholar]
- Goldman, J.W.; Laux, I.; Chai, F.; Savage, R.E.; Ferrari, D.; Garmey, E.G.; Just, R.G.; Rosen, L.S. Phase 1 dose-escalation trial evaluating the combination of the selective MET (mesenchymal-epithelial transition factor) inhibitor tivantinib (ARQ 197) plus erlotinib. Cancer 2012, 118, 5903–5911. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; von Pawel, J.; Garmey, E.G.; Akerley, W.L.; Brugger, W.; Ferrari, D.; Chen, Y.; Costa, D.B.; Gerber, D.E.; Orlov, S.; et al. Randomized phase II study of erlotinib plus tivantinib versus erlotinib plus placebo in previously treated non-small-cell lung cancer. J. Clin. Oncol. 2011, 29, 3307–3315. [Google Scholar]
- Azuma, K.; Yoshioka, H.; Yamamoto, N.; Takahashi, T.; Nishio, M.; Katakami, N.; Ahn, M.-J.; Hirashima, T.; Maemondo, M.; Kim, S.-W.; et al. Tivantinib plus erlotinib versus placebo plus erlotinib in Asian patients with previously treated nonsquamous NSCLC with wild-type EGFR: First report of a phase III ATTENTION trial. ASCO Meet. Abstr. 2014, 32, 8044. [Google Scholar]
- Hong, D.S.; LoRusso, P.; Hamid, O.; Beaupre, D.M.; Janku, F.; Khan, R.; Kittaneh, M.; Loberg, R.D.; Amore, B.; Caudillo, I.; et al. First-in-human study of AMG 337, a highly selective oral inhibitor of MET, in adult patients (pts) with advanced solid tumors. ASCO Meet. Abstr. 2014, 32, 2508. [Google Scholar]
- Bang, Y.-J.; Su, W.-C.; Nam, D.-H.; Lim, W.-T.; Bauer, T.M.; Brana, I.; Poon, R.T.-P.; Hong, D.S.; Lin, C.-C.; Peng, B.; et al. Phase I study of the safety and efficacy of INC280 in patients with advanced MET-dependent solid tumors. ASCO Meet. Abstr. 2014, 32, 2520. [Google Scholar]
- Rodig, S.J.; Shapiro, G.I. Crizotinib, a small-molecule dual inhibitor of the c-Met and ALK receptor tyrosine kinases. Curr. Opin. Investig. Drugs 2010, 11, 1477–1490. [Google Scholar] [PubMed]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar]
- Qian, F.; Engst, S.; Yamaguchi, K.; Yu, P.; Won, K.A.; Mock, L.; Lou, T.; Tan, J.; Li, C.; Tam, D.; et al. Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer Res. 2009, 69, 8009–8016. [Google Scholar]
- Kazandjian, D.; Blumenthal, G.M.; Chen, H.Y.; He, K.; Patel, M.; Justice, R.; Keegan, P.; Pazdur, R. FDA approval summary: Crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements. Oncologist 2014, 19, e5–e11. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Ou, S.-H.I.; Shapiro, G.; Otterson, G.A.; Villaruz, L.C.; Villalona-Calero, M.A.; Iafrate, A.J.; Varella-Garcia, M.; Dacic, S.; Cardarella, S.; et al. Efficacy and safety of crizotinib in patients with advanced c-MET-amplified non-small cell lung cancer (NSCLC). ASCO Meet. Abstr. 2014, 32, 8001. [Google Scholar]
- Liu, L.; Aleksandrowicz, E.; Fan, P.; Schonsiegel, F.; Zhang, Y.; Sahr, H.; Gladkich, J.; Mattern, J.; Depeweg, D.; Lehner, B.; et al. Enrichment of c-Met+ tumorigenic stromal cells of giant cell tumor of bone and targeting by cabozantinib. Cell Death Dis. 2014, 5, e1471. [Google Scholar]
- Viola, D.; Cappagli, V.; Elisei, R. Cabozantinib (XL184) for the treatment of locally advanced or metastatic progressive medullary thyroid cancer. Future Oncol. 2013, 9, 1083–1092. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, B.H.; Bendell, J.C.; Modiano, M.R.; Machiels, J.-P.H.; Versola, M.J.; Hodge, J.P.; Sawarna, K.; Tse, N. Phase I/II study of E7050 (golvantinib) in combination with sorafenib in patients (pts) with advanced hepatocellular carcinoma (HCC): Phase I results. ASCO Meet. Abstr. 2013, 31, 294. [Google Scholar]
- Beeram, M.; Patnaik, A.; Amaravadi, R.K.; Haas, N.B.; Papadopoulos, K.P.; Tolcher, A.W.; Smith, L.S.; Harlacker, K.; Espino, G.; Drouin, M.A.; et al. MGCD265, a multitargeted oral tyrosine kinase receptor inhibitor of Met and VEGFR, in combination with docetaxel. ASCO Meet. Abstr. 2012, 30, e13604. [Google Scholar]
- Kurzrock, R.; Sherman, S.I.; Ball, D.W.; Forastiere, A.A.; Cohen, R.B.; Mehra, R.; Pfister, D.G.; Cohen, E.E.; Janisch, L.; Nauling, F.; et al. Activity of XL184 (Cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J. Clin. Oncol. 2011, 29, 2660–2666. [Google Scholar]
- Shapiro, G.I.; McCallum, S.; Adams, L.M.; Sherman, L.; Weller, S.; Swann, S.; Keer, H.; Miles, D.; Muller, T.; Lorusso, P. A Phase 1 dose-escalation study of the safety and pharmacokinetics of once-daily oral foretinib, a multi-kinase inhibitor, in patients with solid tumors. Investig. New Drugs 2013, 31, 742–750. [Google Scholar] [CrossRef]
- Camidge, D.R.; Bang, Y.J.; Kwak, E.L.; Iafrate, A.J.; Varella-Garcia, M.; Fox, S.B.; Riely, G.J.; Solomon, B.; Ou, S.H.; Kim, D.W.; et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: Updated results from a phase 1 study. Lancet Oncol. 2012, 13, 1011–1019. [Google Scholar]
- Zhu, M.; Doshi, S.; Gisleskog, P.O.; Oliner, K.S.; Perez Ruixo, J.J.; Loh, E.; Zhang, Y. Population pharmacokinetics of rilotumumab, a fully human monoclonal antibody against hepatocyte growth factor, in cancer patients. J. Pharm. Sci 2014, 103, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, F.R.; Gandara, D.; Govindan, R.; Paton, V.E.; Yu, W. A randomised, Phase II, multicentre, double-blind, placebo-controlled study of onartuzumab (MetMAb) in combination with paclitaxel plus cisplatin (or carboplatin) as first-line treatment for patients (pts) with Stage IIIb or IV squamous non-small cell lung cancer (NSCLC). Ann. Oncol. 2012, 23, 445. [Google Scholar]
- Li, C.; Alvey, C.; Bello, A.; Wilner, K.D.; Tan, W. Pharmacokinetics (PK) of crizotinib (PF-02341066) in patients with advanced non-small cell lung cancer (NSCLC) and other solid tumors. J. Clin. Oncol. 2011, 29, e13065. [Google Scholar]
- Daniele, G.; Ranson, M.; Blanco-Codesido, M.; Dean, E.J.; Shah, K.J.; Krebs, M.; Brunetto, A.; Greystoke, A.; Johnston, C.; Kuznetsov, G.; et al. Phase I dose-finding study of golvatinib (E7050), a c-Met and Eph receptor targeted multi-kinase inhibitor, administered orally QD to patients with advanced solid tumors. ASCO Meet. Abstr. 2012, 30, 3030. [Google Scholar]
- Garber, K. MET inhibitors start on road to recovery. Nat. Rev. Drug Discov. 2014, 13, 563–565. [Google Scholar] [CrossRef] [PubMed]
- Spigel, D.R.; Edelman, M.J.; O’Byrne, K.; Paz-Ares, L.; Shames, D.S.; Yu, W.; Paton, V.E.; Mok, T. Onartuzumab plus erlotinib versus erlotinib in previously treated stage IIIb or IV NSCLC: Results from the pivotal phase III randomized, multicenter, placebo-controlled METLung (OAM4971g) global trial. J. Clin. Oncol. 2014, 32, 8000. [Google Scholar] [CrossRef]
- Two Faces of MET. 2014 ASCO Annual Meeting. Available online: http://meetinglibrary.asco.org/content/96406 (accessed on 5 March 2015).
- Wang, W.; Wang, E.Q.; Balthasar, J.P. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin. Pharmacol. Ther. 2008, 84, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Timm, A.; Kolesar, J.M. Crizotinib for the treatment of non-small-cell lung cancer. Am. J. Health Syst. Pharm. 2013, 70, 943–947. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Pal, S.K.; Cabanillas, M.E.; Ramies, D.A.; Tseng, L.; Holland, J.S.; Morrissey, S.; Dutcher, J.P. Antitumor activity observed in a phase I drug–drug interaction study of cabozantinib (XL184) and rosiglitazone in patients (pts) with renal cell carcinoma (RCC) and differentiated thyroid cancer (DTC). J. Clin. Oncol. 2011, 29, e13042. [Google Scholar] [CrossRef]
- Chia, S.K.L.; Ellard, S.; Mates, M.; Welch, S.; Mihalcioiu, C.L.D.; Miller, W.H.; Gelmon, K.A.; Lohrisch, C.A.; Kumar, V.; Taylor, S.K.; et al. A phase Ib study of an anti-HER2 inhibitor, lapatinib, in combination with a c-MET and VEGFR inhibitor, foretinib, in HER2-positive metastatic breast cancer (MBC): Results from NCIC CTG IND.198. ASCO Meet. Abstr. 2013, 31, 518. [Google Scholar]
- Zhou, H.; Mascelli, M.A. Mechanisms of monoclonal antibody-drug interactions. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; vande Woude, G. Targeting MET in cancer: Rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103. [Google Scholar] [CrossRef] [PubMed]
- You, W.K.; Sennino, B.; Williamson, C.W.; Falcon, B.; Hashizume, H.; Yao, L.C.; Aftab, D.T.; McDonald, D.M. VEGF and c-Met blockade amplify angiogenesis inhibition in pancreatic islet cancer. Cancer Res. 2011, 71, 4758–4768. [Google Scholar] [CrossRef] [PubMed]
- Sennino, B.; Ishiguro-Oonuma, T.; Wei, Y.; Naylor, R.M.; Williamson, C.W.; Bhagwandin, V.; Tabruyn, S.P.; You, W.K.; Chapman, H.A.; Christensen, J.G.; et al. Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discov. 2012, 2, 270–287. [Google Scholar]
- Xiang, Q.; Chen, W.; Ren, M.; Wang, J.; Zhang, H.; Deng, D.Y.; Zhang, L.; Shang, C.; Chen, Y. Cabozantinib suppresses tumor growth and metastasis in hepatocellular carcinoma by a dual blockade of VEGFR2 and MET. Clin. Cancer Res. 2014, 20, 2959–2970. [Google Scholar] [CrossRef] [PubMed]
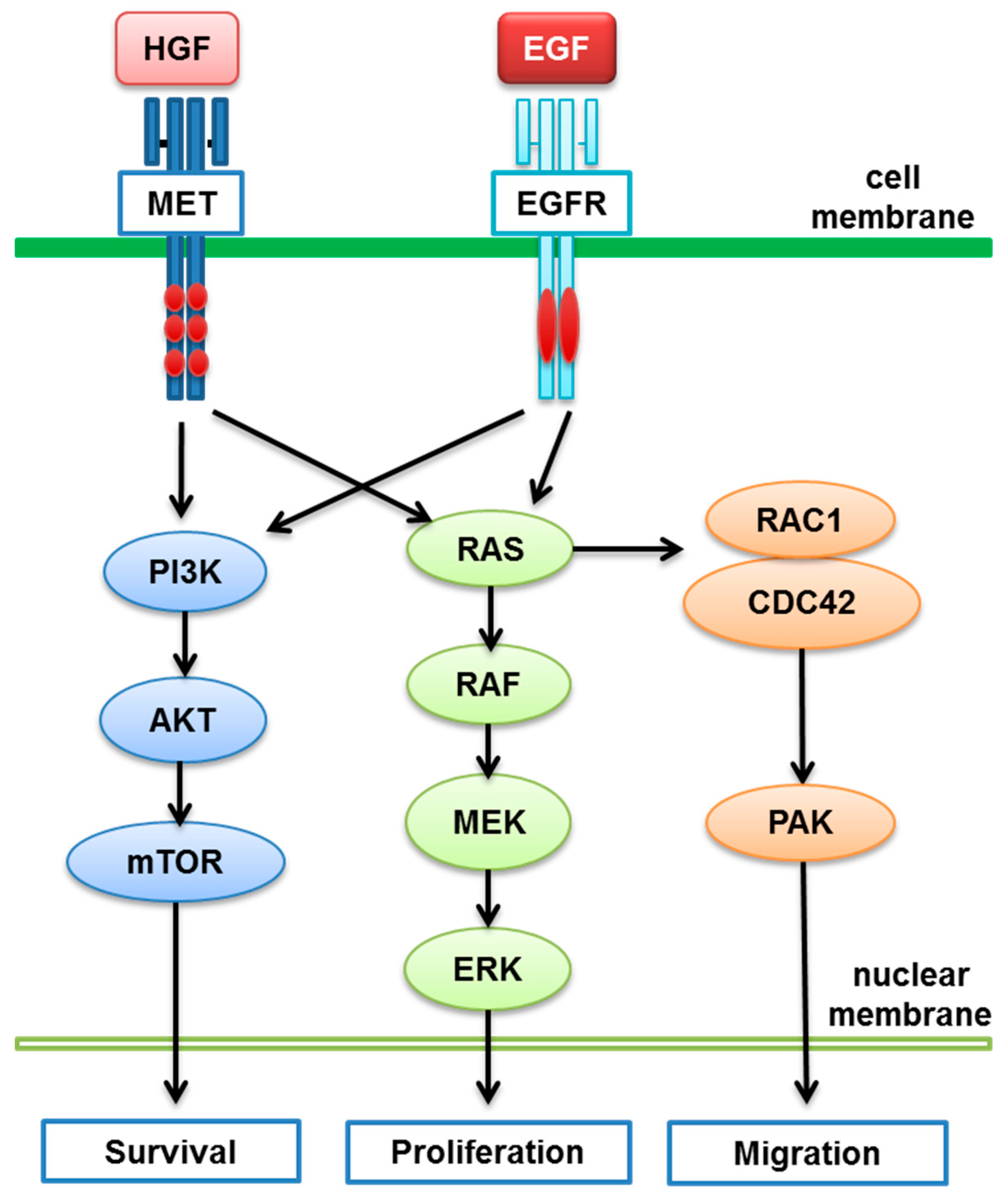
- Puri, N.; Salgia, R. Synergism of EGFR and c-Met pathways, cross-talk and inhibition, in non-small cell lung cancer. J. Carcinog. 2008, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Breindel, J.L.; Haskins, J.W.; Cowell, E.P.; Zhao, M.; Nguyen, D.X.; Stern, D.F. EGF receptor activates MET through MAPK to enhance non-small cell lung carcinoma invasion and brain metastasis. Cancer Res. 2013, 73, 5053–5065. [Google Scholar] [CrossRef] [PubMed]
- Chiari, R.; Duranti, S.; Ludovini, V.; Bellezza, G.; Pireddu, A.; Minotti, V.; Bennati, C.; Crino, L. Long-term response to gefitinib and crizotinib in lung adenocarcinoma harboring both epidermal growth factor receptor mutation and EML4-ALK fusion gene. J. Clin. Oncol. 2014, 32, e30–e32. [Google Scholar] [CrossRef] [PubMed]
- Mitamura, T.; Watari, H.; Wang, L.; Kanno, H.; Hassan, M.K.; Miyazaki, M.; Katoh, Y.; Kimura, T.; Tanino, M.; Nishihara, H.; et al. Downregulation of miRNA-31 induces taxane resistance in ovarian cancer cells through increase of receptor tyrosine kinase MET. Oncogenesis 2013, 2, e40. [Google Scholar]
- Bachleitner-Hofmann, T.; Sun, M.Y.; Chen, C.T.; Tang, L.; Song, L.; Zeng, Z.; Shah, M.; Christensen, J.G.; Rosen, N.; Solit, D.B.; et al. HER kinase activation confers resistance to MET tyrosine kinase inhibition in MET oncogene-addicted gastric cancer cells. Mol. Cancer Ther. 2008, 7, 3499–3508. [Google Scholar]
- Chiang, Y.Y.; Chow, K.C.; Lin, T.Y.; Chiang, I.P.; Fang, H.Y. Hepatocyte growth factor and HER2/neu downregulate expression of apoptosis-inducing factor in non-small cell lung cancer. Oncol. Rep. 2014, 31, 597–604. [Google Scholar] [PubMed]
- Penuel, E.; Li, C.; Parab, V.; Burton, L.; Cowan, K.J.; Merchant, M.; Yauch, R.L.; Patel, P.; Peterson, A.; Hampton, G.M.; et al. HGF as a circulating biomarker of onartuzumab treatment in patients with advanced solid tumors. Mol. Cancer Ther. 2013, 12, 1122–1130. [Google Scholar]
- Rosen, L.S.; Senzer, N.; Mekhail, T.; Ganapathi, R.; Chai, F.; Savage, R.E.; Waghorne, C.; Abbadessa, G.; Schwartz, B.; Dreicer, R. A phase I dose-escalation study of Tivantinib (ARQ 197) in adult patients with metastatic solid tumors. Clin. Cancer Res. 2011, 17, 7754–7764. [Google Scholar] [CrossRef] [PubMed]
- Mai, E.; Zheng, Z.; Chen, Y.; Peng, J.; Severin, C.; Filvaroff, E.; Romero, M.; Mallet, W.; Kaur, S.; Gelzleichter, T.; et al. Nonclinical evaluation of the serum pharmacodynamic biomarkers HGF and shed MET following dosing with the anti-MET monovalent monoclonal antibody onartuzumab. Mol. Cancer Ther. 2014, 13, 540–552. [Google Scholar]
- Rocci, A.; Gambella, M.; Aschero, S.; Baldi, I.; Trusolino, L.; Cavallo, F.; Gay, F.; Larocca, A.; Magarotto, V.; Omede, P.; et al. MET dysregulation is a hallmark of aggressive disease in multiple myeloma patients. Br. J. Haematol. 2014, 164, 841–850. [Google Scholar]
- Koeppen, H.; Rost, S.; Yauch, R.L. Developing biomarkers to predict benefit from HGF/MET pathway inhibitors. J. Pathol. 2014, 232, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Su, Y.; Dykema, K.; Johnson, J.; Koeman, J.; de Giorgi, V.; Huang, A.; Schlegel, R.; Essenburg, C.; Kang, L.; et al. Overexpression of HGF Promotes HBV-Induced Hepatocellular Carcinoma Progression and Is an Effective Indicator for Met-Targeting Therapy. Genes Cancer 2013, 4, 247–260. [Google Scholar]
- Xie, Q.; Su, Y.; Dykema, K.; Johnson, J.; Koeman, J.; de Giorgi, V.; Huang, A.; Schlegel, R.; Essenburg, C.; Kang, L.; et al. Prognostic impact of the c-MET polymorphism on the clinical outcome in locoregional gastric cancer patients. Pharmacogenet. Genomics 2014, 24, 588–586. [Google Scholar]
- Betts, G.; Valentine, H.; Pritchard, S.; Swindell, R.; Williams, V.; Morgan, S.; Griffiths, E.A.; Welch, I.; West, C.; Womack, C. FGFR2, HER2 and cMet in gastric adenocarcinoma: Detection, prognostic significance and assessment of downstream pathway activation. Virchows Arch. 2014, 464, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Sclafani, F.; Rimassa, L.; Colombo, P.; Destro, A.; Stinco, S.; Lutman, F.R.; Carnaghi, C.; Beretta, G.; Zanello, A.; Roncalli, M.; et al. An exploratory biomarker study in metastatic tumors from colorectal cancer patients treated with bevacizumab. Int. J. Biol. Markers 2015, 30, e73–e80. [Google Scholar]
- Matsui, S.; Osada, S.; Tomita, H.; Komori, S.; Mori, R.; Sanada, Y.; Takahashi, T.; Yamaguchi, K.; Yoshida, K. Clinical significance of aggressive hepatectomy for colorectal liver metastasis, evaluated from the HGF/c-Met pathway. Int. J. Oncol. 2010, 37, 289–297. [Google Scholar] [PubMed]
- Saigusa, S.; Tanaka, K.; Toiyama, Y.; Matsushita, K.; Kawamura, M.; Okugawa, Y.; Hiro, J.; Inoue, Y.; Uchida, K.; Mohri, Y.; et al. Gene expression profiles of tumor regression grade in locally advanced rectal cancer after neoadjuvant chemoradiotherapy. Oncol. Rep. 2012, 28, 855–861. [Google Scholar]
- Choueiri, T.K.; Vaishampayan, U.; Rosenberg, J.E.; Logan, T.F.; Harzstark, A.L.; Bukowski, R.M.; Rini, B.I.; Srinivas, S.; Stein, M.N.; Adams, L.M.; et al. Phase II and biomarker study of the dual MET/VEGFR2 inhibitor foretinib in patients with papillary renal cell carcinoma. J. Clin. Oncol. 2013, 31, 181–186. [Google Scholar]
- Xie, Q.; Bradley, R.; Kang, L.; Koeman, J.; Ascierto, M.L.; Worschech, A.; de Giorgi, V.; Wang, E.; Kefene, L.; Su, Y.; et al. Hepatocyte growth factor (HGF) autocrine activation predicts sensitivity to MET inhibition in glioblastoma. Proc. Natl. Acad. Sci. USA 2012, 109, 570–575. [Google Scholar]
- Koochekpour, S.; Jeffers, M.; Rulong, S.; Taylor, G.; Klineberg, E.; Hudson, E.A.; Resau, J.H.; vande Woude, G.F. Met and hepatocyte growth factor/scatter factor expression in human gliomas. Cancer Res. 1997, 57, 5391–5398. [Google Scholar] [PubMed]
- El-Attar, H.A.; Sheta, M.I. Hepatocyte growth factor profile with breast cancer. Indian J. Pathol. Microbiol. 2011, 54, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Sheen-Chen, S.M.; Liu, Y.W.; Eng, H.L.; Chou, F.F. Serum levels of hepatocyte growth factor in patients with breast cancer. Cancer Epidemiol. Biomark. Prev. 2005, 14, 715–717. [Google Scholar] [CrossRef]
- Raghav, K.P.; Wang, W.; Liu, S.; Chavez-MacGregor, M.; Meng, X.; Hortobagyi, G.N.; Mills, G.B.; Meric-Bernstam, F.; Blumenschein, G.R., Jr.; Gonzalez-Angulo, A.M. cMET and phospho-cMET protein levels in breast cancers and survival outcomes. Clin. Cancer Res. 2012, 18, 2269–2277. [Google Scholar] [CrossRef] [PubMed]
- Minuti, G.; Cappuzzo, F.; Duchnowska, R.; Jassem, J.; Fabi, A.; O’Brien, T.; Mendoza, A.D.; Landi, L.; Biernat, W.; Czartoryska-Arlukowicz, B.; et al. Increased MET and HGF gene copy numbers are associated with trastuzumab failure in HER2-positive metastatic breast cancer. Br. J. Cancer 2012, 107, 793–799. [Google Scholar]
- Hashem, M.; Essam, T. Hepatocyte growth factor as a tumor marker in the serum of patients with prostate cancer. J. Egypt Natl. Cancer Inst. 2005, 17, 114–120. [Google Scholar]
- Seidel, C.; Borset, M.; Turesson, I.; Abildgaard, N.; Sundan, A.; Waage, A. Elevated serum concentrations of hepatocyte growth factor in patients with multiple myeloma. The Nordic Myeloma Study Group. Blood 1998, 91, 806–812. [Google Scholar]
- Lim, Y.C.; Han, J.H.; Kang, H.J.; Kim, Y.S.; Lee, B.H.; Choi, E.C.; Kim, C.H. Overexpression of c-Met promotes invasion and metastasis of small oral tongue carcinoma. Oral Oncol. 2012, 48, 1114–1119. [Google Scholar] [CrossRef] [PubMed]
- Felix, A.S.; Edwards, R.P.; Stone, R.A.; Chivukula, M.; Parwani, A.V.; Bowser, R.; Linkov, F.; Weissfeld, J.L. Associations between hepatocyte growth factor, c-Met, and basic fibroblast growth factor and survival in endometrial cancer patients. Br. J. Cancer 2012, 106, 2004–2009. [Google Scholar] [CrossRef] [PubMed]
- Kakkar, T.; Ma, M.; Zhuang, Y.; Patton, A.; Hu, Z.; Mounho, B. Pharmacokinetics and safety of a fully human hepatocyte growth factor antibody, AMG 102, in cynomolgus monkeys. Pharm. Res. 2007, 24, 1910–1918. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.F.; Xie, Q.; Zhang, Y.W.; Su, Y.; Zhao, P.; Cao, B.; Furge, K.; Sun, J.; Rex, K.; Osgood, T.; et al. Therapeutic potential of hepatocyte growth factor/scatter factor neutralizing antibodies: Inhibition of tumor growth in both autocrine and paracrine hepatocyte growth factor/scatter factor: c-Met-driven models of leiomyosarcoma. Mol. Cancer Ther. 2009, 8, 2803–2810. [Google Scholar]
- Burgess, T.; Coxon, A.; Meyer, S.; Sun, J.; Rex, K.; Tsuruda, T.; Chen, Q.; Ho, S.Y.; Li, L.; Kaufman, S.; et al. Fully human monoclonal antibodies to hepatocyte growth factor with therapeutic potential against hepatocyte growth factor/c-Met-dependent human tumors. Cancer Res. 2006, 66, 1721–1729. [Google Scholar]
- Burgess, T.L.; Sun, J.; Meyer, S.; Tsuruda, T.S.; Sun, J.; Elliott, G.; Chen, Q.; Haniu, M.; Barron, W.F.; Juan, T.; et al. Biochemical characterization of AMG 102: A neutralizing, fully human monoclonal antibody to human and nonhuman primate hepatocyte growth factor. Mol. Cancer Ther. 2010, 9, 400–409. [Google Scholar]
- Wong, H.; Choo, E.F.; Alicke, B.; Ding, X.; La, H.; McNamara, E.; Theil, F.P.; Tibbitts, J.; Friedman, L.S.; Hop, C.E.; et al. Antitumor activity of targeted and cytotoxic agents in murine subcutaneous tumor models correlates with clinical response. Clin. Cancer Res. 2012, 18, 3846–3855. [Google Scholar]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209.
- Bang, Y.J.; van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar]
- Ohtsu, A.; Shah, M.A.; van Cutsem, E.; Rha, S.Y.; Sawaki, A.; Park, S.R.; Lim, H.Y.; Yamada, Y.; Wu, J.; Langer, B.; et al. Bevacizumab in combination with chemotherapy as first-line therapy in advanced gastric cancer: A randomized, double-blind, placebo-controlled phase III study. J. Clin. Oncol. 2011, 29, 3968–3976. [Google Scholar]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Jain, R.K.; Zhu, M. Recent Progress and Advances in HGF/MET-Targeted Therapeutic Agents for Cancer Treatment. Biomedicines 2015, 3, 149-181. https://doi.org/10.3390/biomedicines3010149
Zhang Y, Jain RK, Zhu M. Recent Progress and Advances in HGF/MET-Targeted Therapeutic Agents for Cancer Treatment. Biomedicines. 2015; 3(1):149-181. https://doi.org/10.3390/biomedicines3010149
Chicago/Turabian StyleZhang, Yilong, Rajul K. Jain, and Min Zhu. 2015. "Recent Progress and Advances in HGF/MET-Targeted Therapeutic Agents for Cancer Treatment" Biomedicines 3, no. 1: 149-181. https://doi.org/10.3390/biomedicines3010149
APA StyleZhang, Y., Jain, R. K., & Zhu, M. (2015). Recent Progress and Advances in HGF/MET-Targeted Therapeutic Agents for Cancer Treatment. Biomedicines, 3(1), 149-181. https://doi.org/10.3390/biomedicines3010149