CD64: An Attractive Immunotherapeutic Target for M1-type Macrophage Mediated Chronic Inflammatory Diseases


Abstract
:1. Introduction
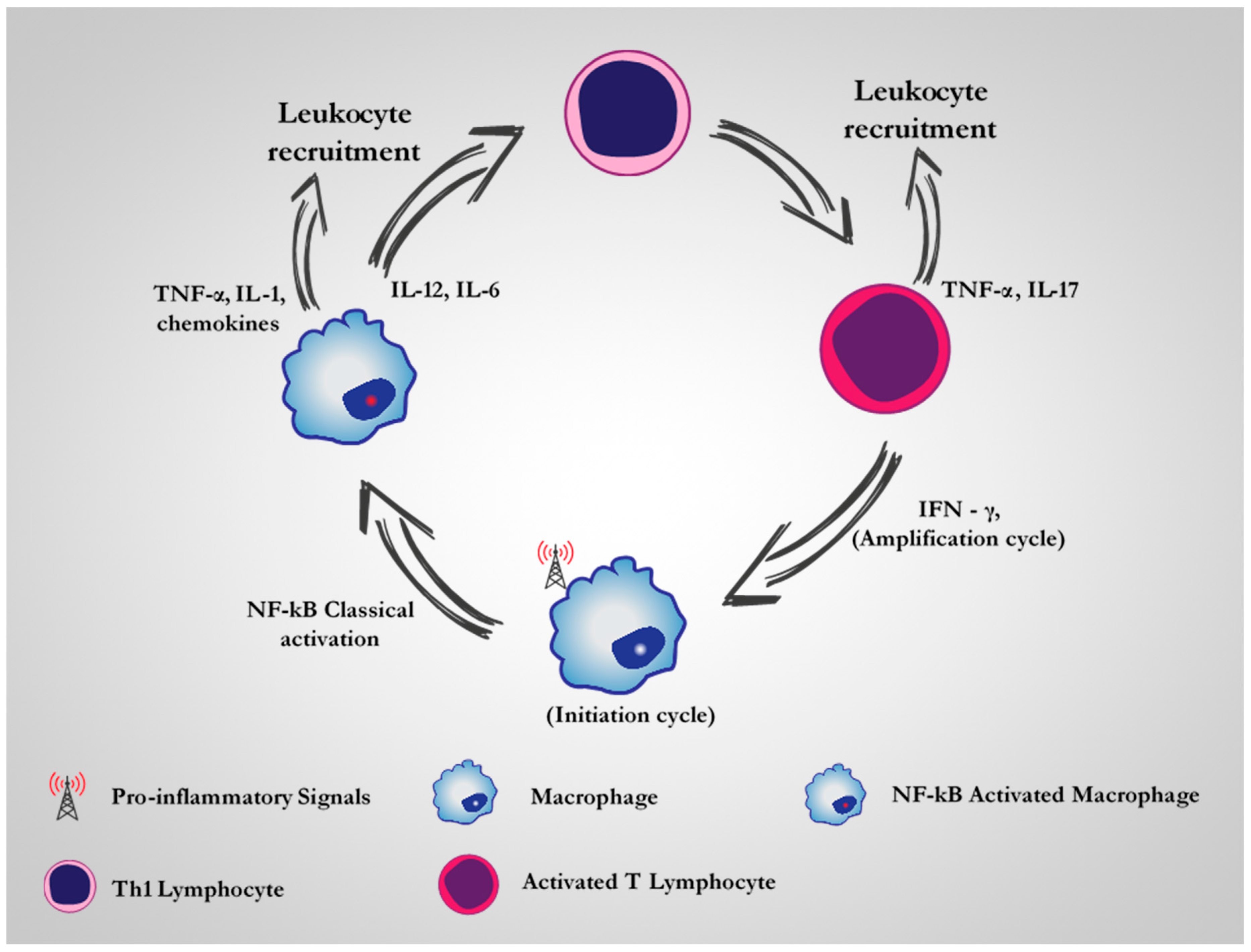
2. CD64 as A Therapeutic Target for M1 Dysregulated Macrophages in Chronic Inflammatory Diseases
3. CD64: Background
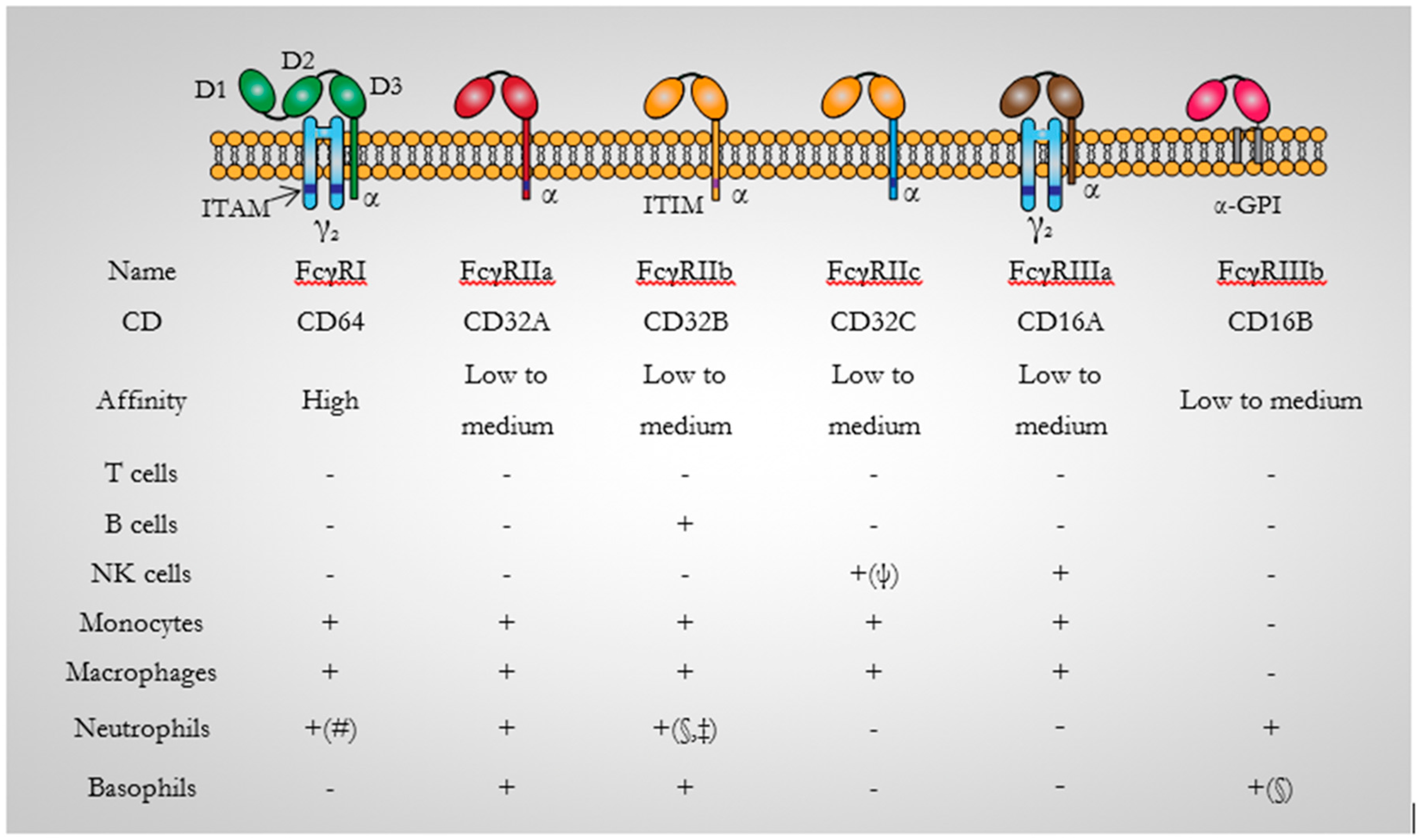
3.1. CD64 Structure
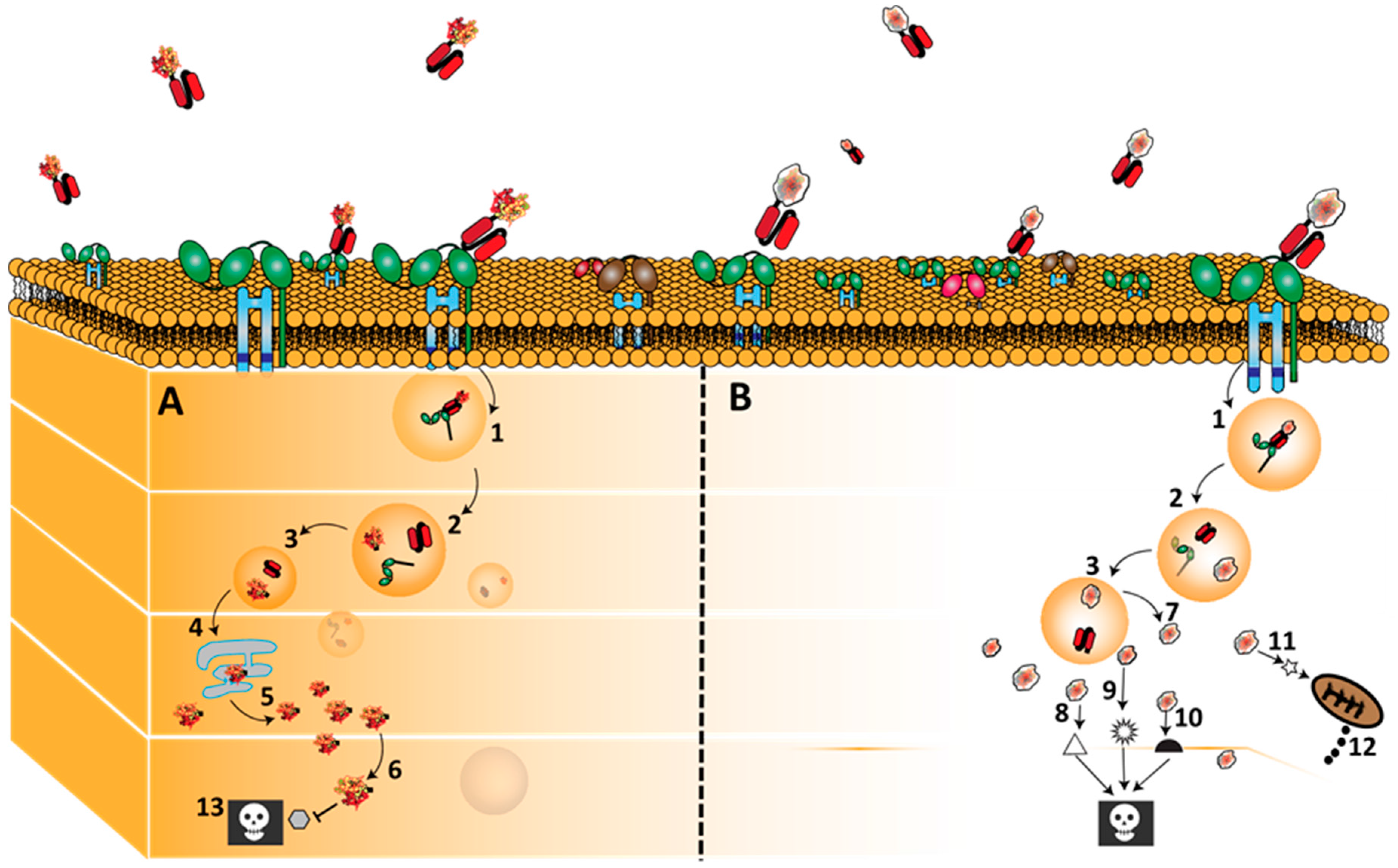
3.2. CD64 Signalling
3.3. CD64 Expression
3.4. CD64-Specific Antibodies
4. CD64 Based Immunotherapeutic Studies in Chronic Inflammatory Diseases
4.1. Anti-CD64(H22)-Ricin A
4.2. H22(scFv)-ETA’
4.3. Granzyme B-(H22)scFv
4.4. Granzyme M-(H22)scFv
4.5. H22(scFv)-Ang
4.6. H22(scFv)-MAP
5. Conclusions and Future Direction
Acknowledgments
Conflicts of Interest
References
- Hunter, P. The inflammation theory of disease: The growing realization that chronic inflammation is crucial in many diseases opens new avenues for treatment. EMBO Rep. 2012, 13, 968–970. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.K.; Mishra, V.; Mehra, N.K. Targeted drug delivery to macrophages. Expert Opin. Drug Deliv. 2013, 10, 353–367. [Google Scholar]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000prime Rep. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Galán, L.; Olleros, M.L.; Vesin, D.; Garcia, I. Much more than M1 and M2 macrophages, there are also CD169+ and TCR+ macrophages. Front. Immunol. 2015, 6. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; de Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Utispan, K.; Koontongkaew, S. Fibroblasts and macrophages: Key players in the head and neck cancer microenvironment. J. Oral Biosci. 2016, 59, 23–30. [Google Scholar] [CrossRef]
- Obeid, E.; Nanda, R.; Fu, Y.X.; Olopade, O.I. The role of tumor-associated macrophages in breast cancer progression (review). Int. J. Oncol. 2013, 43, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.; Jefferies, C.; Cryan, S.A. Targeted liposomal drug delivery to monocytes and macrophages. J. Drug Deliv. 2010, 2011. [Google Scholar] [CrossRef] [PubMed]
- Kasraie, S.; Werfel, T. Role of macrophages in the pathogenesis of atopic dermatitis. Med. Inflamm. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Helming, L. Inflammation: Cell recruitment versus local proliferation. Curr. Biol. 2011, 21, R548–R550. [Google Scholar] [CrossRef] [PubMed]
- Monsel, A.; Zhu, Y.-G.; Genna, S.; Hao, Q.; Liu, J.; Lee, J.W. Cell-based Therapy for Acute Organ InjuryPreclinical Evidence and Ongoing Clinical Trials Using Mesenchymal Stem Cells. J. Am. Soc. Anesthesiol. 2014, 121, 1099–1121. [Google Scholar] [CrossRef] [PubMed]
- Valledor, A.F.; Comalada, M.; Lloberas, J.; Celada, A. Macrophage Proinflammatory Activation and Deactivation: A Question of Balance. Adv. Immunol. 2010, 108. [Google Scholar] [CrossRef]
- Kinne, R.W.; Bräuer, R.; Stuhlmüller, B.; Palombo-Kinne, E.; Burmester, G.R. Macrophages in rheumatoid arthritis. Arthritis Res. Ther. 2000, 2, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmonds, R.E.; Foxwell, B.M. Signalling, inflammation and arthritis NF-κB and its relevance to arthritis and inflammation. Rheumatology 2008, 47, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.; Mayer, L. Current status of monoclonal antibody therapy for the treatment of inflammatory bowel disease. Expert Rev. Clin. Immunol. 2010, 6, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.G.; Hayden, M.S.; Ghosh, S. NF-κB, inflammation, and metabolic disease. Cell Metab. 2011, 13, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Kotsovilis, S.; Andreakos, E. Therapeutic human monoclonal antibodies in inflammatory diseases. Hum. Monoclon. Antib. 2014, 1060, 37–59. [Google Scholar]
- Hristodorov, D.; Mladenov, R.; Huhn, M.; Barth, S.; Thepen, T. Macrophage-targeted therapy: CD64-Based immunotoxins for treatment of chronic inflammatory diseases. Toxins 2012, 4, 676–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maini, R.N.; Feldmann, M. How does infliximab work in rheumatoid arthritis? Arthritis Res. Ther. 2002, 4, S22. [Google Scholar] [CrossRef] [PubMed]
- Derksen, R.; Bijlsma, J. The treatment of chronic inflammatory diseases with monoclonal antibodies against tumor necrosis factor: Side effects, contraindications and precautions. Ned. Tijdschr. Voor Geneeskd. 2002, 146, 1165–1168. [Google Scholar]
- Tas, S.W.; Vervoordeldonk, M.J.; Tak, P.P. Gene therapy targeting nuclear factor-κB: Towards clinical application in inflammatory diseases and cancer. Curr. Gene Ther. 2009, 9, 160–170. [Google Scholar] [CrossRef] [PubMed]
- McCormick, T.S.; Stevens, S.R.; Kang, K. Macrophages and cutaneous inflammation. Nat. Biotechnol. 2000, 18, 25–26. [Google Scholar] [CrossRef] [PubMed]
- Thepen, T.; van Vuuren, A.H.; Kiekens, R.C.; Damen, C.A.; Vooijs, W.C.; van de Winkel, J.G. Resolution of cutaneous inflammation after local elimination of macrophages. Nat. Biotechnol. 2000, 18, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Burke, B.; Lewis, C.E. The Macrophage, 2nd ed.; Oxford University Press: Oxford, UK, 2002. [Google Scholar]
- Hristodorov, D.; Mladenov, R.; von Felbert, V.; Huhn, M.; Fischer, R.; Barth, S.; Thepen, T. Targeting CD64 mediates elimination of M1 but not M2 macrophages in vitro and in cutaneous inflammation in mice and patient biopsies. mAbs 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Sun, P.D. Structural mechanism of high affinity FcγRI recognition of immunoglobulin G. Immunol. Rev. 2015, 268, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Kiyoshi, M.; Caaveiro, J.M.; Kawai, T.; Tashiro, S.; Ide, T.; Asaoka, Y.; Hatayama, K.; Tsumoto, K. Structural basis for binding of human IgG1 to its high-affinity human receptor FcγRI. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Bruhns, P.; Jönsson, F. Mouse and human FcR effector functions. Immunol. Rev. 2015, 268, 25–51. [Google Scholar] [PubMed]
- Hogarth, P.M.; Pietersz, G.A. Fc receptor-targeted therapies for the treatment of inflammation, cancer and beyond. Nat. Rev. Drug Discov. 2012, 11, 311–331. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, M.; Bjorkman, P.J. Fc receptors and their interactions with immunoglobulins. Annu. Rev. Cell Dev. Biol. 1996, 12, 181–220. [Google Scholar] [CrossRef] [PubMed]
- Mancardi, D.A.; Albanesi, M.; Jönsson, F.; Iannascoli, B.; Van Rooijen, N.; Kang, X.Q.; England, P.; Daëron, M.; Bruhns, P. The high-affinity human IgG receptor FcγRI (CD64) promotes IgG-mediated inflammation, anaphylaxis, and antitumor immunotherapy. Blood 2013, 121, 1563–1573. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Ellsworth, J.L.; Hamacher, N.; Oak, S.W.; Sun, P.D. Crystal structure of Fcγ receptor I and its implication in high affinity γ-immunoglobulin binding. J. Biol. Chem. 2011, 286, 40608–40613. [Google Scholar] [CrossRef] [PubMed]
- Bruhns, P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood 2012, 119, 5640–5649. [Google Scholar] [CrossRef] [PubMed]
- Dugast, A.S.; Tonelli, A.; Berger, C.T.; Ackerman, M.E.; Sciaranghella, G.; Liu, Q.; Sips, M.; Toth, I.; Piechocka-Trocha, A.; Ghebremichael, M.; et al. Decreased Fc receptor expression on innate immune cells is associated with impaired antibody-mediated cellular phagocytic activity in chronically HIV-1 infected individuals. Virology 2011, 415, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.T.; Davis, W.; Norman, J.C.; Hockaday, A.R.; Allen, J.M. Binding of monomeric immunoglobulin G triggers Fc γ RI-mediated endocytosis. J. Biol. Chem. 1994, 269, 24396–24402. [Google Scholar] [PubMed]
- Hulett, M.D.; Hogarth, P.M. The second and third extracellular domains of FcγRI (CD64) confer the unique high affinity binding of IgG2a. Mol. Immunol. 1998, 35, 989–996. [Google Scholar] [CrossRef]
- Guyre, P.; Graziano, R.; Vance, B.; Morganelli, P.; Fanger, M. Monoclonal antibodies that bind to distinct epitopes on Fc γ RI are able to trigger receptor function. J. Immunol. 1989, 143, 1650–1655. [Google Scholar] [PubMed]
- Van der Poel, C.E.; Spaapen, R.M.; van de Winkel, J.G.; Leusen, J.H. Functional characteristics of the high affinity IgG receptor, FcγRI. J. Immunol. 2011, 186, 2699–2704. [Google Scholar] [CrossRef] [PubMed]
- Wallace, P.K.; Keler, T.; Coleman, K.; Fisher, J.; Vitale, L.; Graziano, R.F.; Guyre, P.M.; Fanger, M.W. Humanized mAb H22 binds the human high affinity Fc receptor for IgG (FcγRI), blocks phagocytosis, and modulates receptor expression. J. Leukoc. Biol. 1997, 62, 469–479. [Google Scholar] [PubMed]
- Ericson, S.; Coleman, K.; Wardwell, K.; Baker, S.; Fanger, M.; Guyre, P.; Ely, P. Monoclonal antibody 197 (anti-FcγRI) infusion in a patient with immune thrombocytopenia purpura (ITP) results in down-modulation of FcγRI on circulating monocytes. Br. J. Haematol. 1996, 92, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Graziano, R.F.; Tempest, P.R.; White, P.; Keler, T.; Deo, Y.; Ghebremariam, H.; Coleman, K.; Pfefferkorn, L.C.; Fanger, M.W.; Guyre, P.M. Construction and characterization of a humanized anti-γ-Ig receptor type I (Fc γ RI) monoclonal antibody. J. Immunol. 1995, 155, 4996–5002. [Google Scholar] [PubMed]
- Heijnen, I.; van Vugt, M.J.; Fanger, N.A.; Graziano, R.F.; de Wit, T.; Hofhuis, F.; Guyre, P.M.; Cape, P.J.A.; Verbee, J.S.; Winkel van de, J.G.J. targeting to myeloid-specific human Fc γ RI/CD64 triggers enhanced antibody responses in transgenic mice. J. Clin. Investig. 1996, 97, 331. [Google Scholar] [CrossRef] [PubMed]
- de Kruif, J.; Tijmensen, M.; Goldsein, J.; Logtenberg, T. Recombinant lipid-tagged antibody fragments as functional cell-surface receptors. Nat. Med. 2000, 6, 223. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, S.; Rosinke, R.; Jost, E.; Hehmann-Titt, G.; Huhn, M.; Melmer, G.; Barth, S.; Thepen, T. Targeted ex vivo reduction of CD64-positive monocytes in chronic myelomonocytic leukemia and acute myelomonocytic leukemia using human granzyme B-based cytolytic fusion proteins. Int. J. Cancer 2014, 135, 1497–1508. [Google Scholar] [CrossRef] [PubMed]
- Thepen, T.; Huhn, M.; Melmer, G.; Tur, M.; Barth, S. Fcγ receptor 1 (CD64), a target beyond cancer. Curr. Pharm. Des. 2009, 15, 2712–2718. [Google Scholar] [CrossRef]
- Ortega-Gómez, A.; Perretti, M.; Soehnlein, O. Resolution of inflammation: An integrated view. EMBO Mol. Med. 2013, 5, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Mantegazza, A.R.; Magalhaes, J.G.; Amigorena, S.; Marks, M.S. Presentation of phagocytosed antigens by MHC class I and II. Traffic 2013, 14, 135–152. [Google Scholar] [CrossRef] [PubMed]
- Parikh, B.A.; Tortora, A.; Li, X.P.; Tumer, N.E. Ricin inhibits activation of the unfolded protein response by preventing splicing of the HAC1 mRNA. J. Biol. Chem. 2008, 283, 6145–6153. [Google Scholar] [CrossRef] [PubMed]
- Lord, J.M.; Roberts, L.M.; Robertus, J.D. Ricin: Structure, mode of action, and some current applications. FASEB J. 1994, 8, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Van Roon, J.A.; van Vuuren, A.J.; Wijngaarden, S.; Jacobs, K.M.; Bijlsma, J.W.; Lafeber, F.P.; Thepen, T.; van de Winkelet, J.G. Selective elimination of synovial inflammatory macrophages in rheumatoid arthritis by an Fcγ receptor I–Directed immunotoxin. Arthritis Rheum. 2003, 48, 1229–1238. [Google Scholar] [CrossRef] [PubMed]
- Van Vuuren, A.J.; van Roon, J.A.; Walraven, V.; Stuij, I.; Harmsen, M.C.; McLaughlin, P.M.; van de Winkelet, J.G.; Thepen, T. CD64-directed immunotoxin inhibits arthritis in a novel CD64 transgenic rat model. J. Immunol. 2006, 176, 5833–5838. [Google Scholar] [CrossRef] [PubMed]
- Wolf, P.; Elsässer-Beile, U. Pseudomonas exotoxin A: From virulence factor to anti-cancer agent. Int. J. Med. Microbiol. 2009, 299, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Becker, N.; Benhar, I. Antibody-based immunotoxins for the treatment of cancer. Antibodies 2012, 1, 39–69. [Google Scholar] [CrossRef]
- Wayne, A.S.; FitzGerald, D.J.; Kreitman, R.J.; Pastan, I. Immunotoxins for leukemia. Blood 2014, 123, 2470–2477. [Google Scholar] [CrossRef] [PubMed]
- Weldon, J.E.; Xiang, L.; Zhang, J.; Beers, R.; Walker, D.A.; Onda, M.; Hassan, R.; Pastan, I. A recombinant immunotoxin against the tumor-associated antigen mesothelin reengineered for high activity, low off-target toxicity, and reduced antigenicity. Mol. Cancer Ther. 2013, 12, 48–57. [Google Scholar] [CrossRef] [PubMed]
- West, S. Pseudomonas aeruginosa Exotoxin A: Structure/function, production, and intoxication of eukaryotic cells. Springer 2000, 145, 67–89. [Google Scholar]
- Wilson, B.; Collier, R. Diphtheria toxin and Pseudomonas aeruginosa exotoxin A: Active-site structure and enzymic mechanism. Curr. Top. Microb. Immunol. 1992, 175, 27. [Google Scholar]
- Ribbert, T.; Thepen, T.; Tur, M.; Fischer, R.; Huhn, M.; Barth, S. Recombinant, ETA′-based CD64 immunotoxins: Improved efficacy by increased valency, both in vitro and in vivo in a chronic cutaneous inflammation model in human CD64 transgenic mice. Br. J. Dermatol. 2010, 163, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Berges, N.; Hehmann-Titt, G.; Hristodorov, D.; Melmer, G.; Thepen, T.; Barth, S. Human cytolytic fusion proteins: Modified versions of human granzyme B and angiogenin have the potential to replace bacterial toxins in targeted therapies against CD64+ diseases. Antibodies 2014, 3, 92–115. [Google Scholar] [CrossRef]
- Bots, M.; Medema, J.P. Granzymes at a glance. J. Cell Sci. 2006, 119, 5011–5014. [Google Scholar] [CrossRef] [PubMed]
- Froelich, C.; Metkar, S.; Raja, S. Granzyme B-mediated apoptosis-the elephant and the blind men? Cell Death Differ. 2004, 11, 369. [Google Scholar] [CrossRef] [PubMed]
- Stahnke, B.; Thepen, T.; Stöcker, M.; Rosinke, R.; Jost, E.; Fischer, R.; Tur, M.K.; Barth, S. Granzyme B-H22 (scFv), a human immunotoxin targeting CD64 in acute myeloid leukemia of monocytic subtypes. Mol. Cancer Ther. 2008, 7, 2924–2932. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, S.; Hristodorov, D.; Mladenov, R.; Aslanian, E.; Huhn, M.; Fischer, R.; Barth, S.; Thepen, T. Species-dependent functionality of the human cytolytic fusion proteins granzyme B-H22 (scFv) and H22 (scFv)-angiogenin in macrophages. Antibodies 2013, 2, 9–18. [Google Scholar] [CrossRef]
- Hehmann-Titt, G.; Schiffer, S.; Berges, N.; Melmer, G.; Barth, S. Improving the therapeutic potential of human granzyme B for targeted cancer therapy. Antibodies 2013, 2, 19–49. [Google Scholar] [CrossRef]
- Losasso, V.; Schiffer, S.; Barth, S.; Carloni, P. Design of human granzyme B variants resistant to serpin B9. Proteins Struct. Funct. Bioinform. 2012, 80, 2514–2522. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, S.; Hansen, H.; Hehmann-Titt, G.; Huhn, M.; Fischer, R.; Barth, S.; Thepen, T. Efficacy of an adapted granzyme B-based anti-CD30 cytolytic fusion protein against PI-9-positive classical Hodgkin lymphoma cells in a murine model. Blood Cancer J. 2013, 3, e106. [Google Scholar] [CrossRef] [PubMed]
- Susanto, O.; Trapani, J.; Brasacchio, D. Controversies in granzyme biology. HLA 2012, 80, 477–487. [Google Scholar] [CrossRef] [PubMed]
- De Poot, S.; Bovenschen, N. Granzyme M: Behind enemy lines. Cell Death Differ. 2014, 21, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, S.; Letzian, S.; Jost, E.; Mladenov, R.; Hristodorov, D.; Huhn, M.; Fischer, R.; Barth, S.; Thepen, T. Granzyme M as a novel effector molecule for human cytolytic fusion proteins: CD64-Specific cytotoxicity of Gm-H22 (scFv) against leukemic cells. Cancer Lett. 2013, 341, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hu, G.F. Emerging role of angiogenin in stress response and cell survival under adverse conditions. J. Cell. Physiol. 2012, 227, 2822–2826. [Google Scholar] [CrossRef] [PubMed]
- Cremer, C.; Vierbuchen, T.; Hein, L.; Fischer, R.; Barth, S.; Nachreiner, T. Angiogenin mutants as novel effector molecules for the generation of fusion proteins with increased cytotoxic potential. J. Immunother. 2015, 38, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Tello-Montoliu, A.; Patel, J.; Lip, G. Angiogenin: A review of the pathophysiology and potential clinical applications. J. Thromb. Haemost. 2006, 4, 1864–1874. [Google Scholar] [CrossRef] [PubMed]
- St Clair, D.K.; Rybak, S.M.; Riordan, J.F.; Vallee, B.L. Angiogenin abolishes cell-free protein synthesis by specific ribonucleolytic inactivation of ribosomes. Proc. Natl. Acad. Sci. USA 1987, 84, 8330–8334. [Google Scholar] [CrossRef] [PubMed]
- Mathew, M.; Verma, R.S. Humanized immunotoxins: A new generation of immunotoxins for targeted cancer therapy. Cancer Sci. 2009, 100, 1359–1365. [Google Scholar] [CrossRef] [PubMed]
- Stöcker, M.; Tur, M.K.; Sasse, S.; Krüßmann, A.; Barth, S.; Engert, A. Secretion of functional anti-CD30-angiogenin immunotoxins into the supernatant of transfected 293T-cells. Protein Exp. Purif. 2003, 28, 211–219. [Google Scholar] [CrossRef]
- Cremer, C.; Braun, H.; Mladenov, R.; Schenke, L.; Cong, X.; Jost, E.; Brümmendorf, T.H.; Fischer, R.; Carloni, P.; Barth, S. Novel angiogenin mutants with increased cytotoxicity enhance the depletion of pro-inflammatory macrophages and leukemia cells ex vivo. Cancer Immunol. Immunother. 2015, 64, 1575–1586. [Google Scholar] [CrossRef] [PubMed]
- Cremer, C.; Hehmann-Titt, G.; Schiffer, S.; Melmer, G.; Carloni, P.; Barth, S.; et al. Engineered Versions of Granzyme B and Angiogenin Overcome Intrinsic Resistance to Apoptosis Mediated by Human Cytolytic Fusion Proteins. Resist. Immun. Cancer Ther. 2015, 6, 185–219. [Google Scholar]
- Hristodorov, D.; Mladenov, R.; Pardo, A.; Pham, A.; Huhn, M.; Fischer, R.; Thepen, T.; Barth, S. Microtubule-associated protein tau facilitates the targeted killing of proliferating cancer cells in vitro and in a xenograft mouse tumour model in vivo. Br. J. Cancer. 2013, 109, 1570–1578. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, R.; Hristodorov, D.; Cremer, C.; Gresch, G.; Grieger, E.; Schenke, L.; Klose, D.; Amoury, M.; Woitok, M.; Jost, E.; et al. CD64-directed microtubule associated protein tau kills leukemic blasts ex vivo. Oncotarge 2016, 7, 67166–67174. [Google Scholar] [CrossRef] [PubMed]
- Akinrinmade, O.A.; Jordaan, S.; Hristodorov, D.; Mladenov, R.; Mungra, N.; Chetty, S.; Barth, S. Human MAP tau based targeted cytolytic fusion proteins. Biomedicines 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Hristodorov, D.; Mladenov, R.; Fischer, R.; Barth, S.; Thepen, T. Fully human MAP-fusion protein selectively targets and eliminates proliferating CD64+ M1 macrophages. Immunol. Cell Biol. 2016, 94, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Van Ojik, H.; Repp, R.; Groenewegen, G.; Valerius, T.; van de Winkel, J.G. Clinical evaluation of the bispecific antibody MDX-H210 (anti-FcγRI× anti-HER-2/neu) in combination with granulocyte-colony-stimulating factor (Filgrastim) for treatment of advanced breast cancer. Cancer Immunol. Immunother. 1997, 45, 207–209. [Google Scholar] [CrossRef] [PubMed]
- Fury, M.G.; Lipton, A.; Smith, K.M.; Winston, C.B.; Pfister, D.G. A phase-I trial of the epidermal growth factor receptor directed bispecific antibody MDX-447 without and with recombinant human granulocyte-colony stimulating factor in patients with advanced solid tumors. Cancer Immunol. Immunother. 2008, 57, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Curnow, R.T. Clinical experience with CD64-directed immunotherapy. An overview. Cancer Immunol. Immunother. 1997, 45, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Borchmann, P.; Schnell, R.; Fuss, I.; Manzke, O.; Davis, T.; Lewis, L.D.; Behnke, D.; Wickenhauser, C.; Schiller, P.; Diehl, V.; et al. Phase 1 trial of the novel bispecific molecule H22xKi-4 in patients with refractory Hodgkin lymphoma. Blood 2002, 100, 3101–3107. [Google Scholar] [CrossRef] [PubMed]
- Repp, R.; van Ojik, H.; Valerius, T.; Groenewegen, G.; Wieland, G.; Oetzel, C.; Stockmeyer, B.; Becker, W.; Eisenhut, M.; Steininger, H.; et al. Phase I clinical trial of the bispecific antibody MDX-H210 (anti-FcγRI× anti-HER-2/neu) in combination with Filgrastim (G-CSF) for treatment of advanced breast cancer. Br. J. Cancer 2003, 89, 2234. [Google Scholar] [CrossRef] [PubMed]
- Tur, M.K.; Huhn, M.; Jost, E.; Thepen, T.; Brümmendorf, T.H.; Barth, S. In vivo efficacy of the recombinant anti-CD64 immunotoxin H22 (scFv)-ETA′ in a human acute myeloid leukemia xenograft tumor model. Int. J. Cancer 2011, 129, 1277–1282. [Google Scholar] [CrossRef] [PubMed]
- Van Weyenbergh, J.; Thepen, T.; Soares, G.; Khouri, R.; Barth, S.; Silva-Santos, G.; Costa, J.M.; Barral, A.; Barral-Netto, M. 267: Challenging the Th1 paradigm: A detrimental role for IFN-γ and IFN-regulated CD64 in human leishmaniasis. Cytokine 2013, 63, 306. [Google Scholar] [CrossRef]
- Fet, N.G.; Fiebeler, A.; Klinge, U.; Park, J.K.; Barth, S.; Thepen, T.; Tolba, R.H. Reduction of activated macrophages after ischaemia–Reperfusion injury diminishes oxidative stress and ameliorates renal damage. Nephrol. Dial. Transplant. 2012, 27, 3149–3155. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Upregulated on M1 Macrophages | ||||||||
| Murine | CD64 | CD14 | CD36 | CD25 | MHC I | CD39 | MHCII | CD204 |
| Human | CD64 | CD14 | CD16 | CD284 | CD80 | CD273 | - | - |
| Upregulated on M2 Macrophages | ||||||||
| Murine | CD206 | CD273 | CD284 | CD301 | CD11c | MOMA-1 | MOMA-2 | CD205 |
| Human | CD206 | CD200R | CD163 | CD301 | - | - | - | - |
| Disease Model | Construct | Application | Remark | Reference |
|---|---|---|---|---|
| AML | H22(scFv)-ETA’ | SCID mouse xenograft model for human AML | Potent anti-tumor activity against myeloid tumor cells, including a significantly prolonged the overall survival of AML xenograft animals | [90] |
| AML | Gb-H22(scFv) | In vitro and ex vivo | Specific binding to and elimination of CD64+ U937 cells as well as patient-derived CD64+ AML cells in vitro | [65] |
| AMML and CMML | Gbmut-H22(scFv) | In vitro and ex vivo | Induction of apoptosis in primary CD64+ AMML and CMML cells | [47] |
| AML, AMML, CMML, etc. | H22-Ang and mutants | In vitro and ex vivo | Induction of apoptosis in primary CD64+ leukemia cells isolated from patients | [79] |
| Leishmaniasis | H22(scFv)-ETA’ and H22-RiA | In vivo and ex vivo | Human: Selective killing of Leishmania infected monocytes. Mouse: arrest of cutaneous Leishmania model | [91] |
| Kidney transplantation | H22(scFv)-ETA’ | In vivo | Preservation of renal integrity and function | [92] |
| AML and CMML | H22(scFv)-MAP | Ex vivo | Specific binding to and elimination of CD64+ leukemic blasts with no cytotoxicity towards healthy CD64+ PBMC-derived cells and macrophages | [82] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akinrinmade, O.A.; Chetty, S.; Daramola, A.K.; Islam, M.-u.; Thepen, T.; Barth, S. CD64: An Attractive Immunotherapeutic Target for M1-type Macrophage Mediated Chronic Inflammatory Diseases. Biomedicines 2017, 5, 56. https://doi.org/10.3390/biomedicines5030056
Akinrinmade OA, Chetty S, Daramola AK, Islam M-u, Thepen T, Barth S. CD64: An Attractive Immunotherapeutic Target for M1-type Macrophage Mediated Chronic Inflammatory Diseases. Biomedicines. 2017; 5(3):56. https://doi.org/10.3390/biomedicines5030056
Chicago/Turabian StyleAkinrinmade, Olusiji A., Shivan Chetty, Adebukola K. Daramola, Mukit-ul Islam, Theo Thepen, and Stefan Barth. 2017. "CD64: An Attractive Immunotherapeutic Target for M1-type Macrophage Mediated Chronic Inflammatory Diseases" Biomedicines 5, no. 3: 56. https://doi.org/10.3390/biomedicines5030056
APA StyleAkinrinmade, O. A., Chetty, S., Daramola, A. K., Islam, M.-u., Thepen, T., & Barth, S. (2017). CD64: An Attractive Immunotherapeutic Target for M1-type Macrophage Mediated Chronic Inflammatory Diseases. Biomedicines, 5(3), 56. https://doi.org/10.3390/biomedicines5030056