Bringing the Next Generation of Immuno-Oncology Biomarkers to the Clinic

Abstract
:
{kind=link}
{kind=link}
1. Introduction
2. Required Features of an Ideal Immune-Oncology (IO) Patient-Enrichment-Selection (i.e., “Predictive”) Biomarker
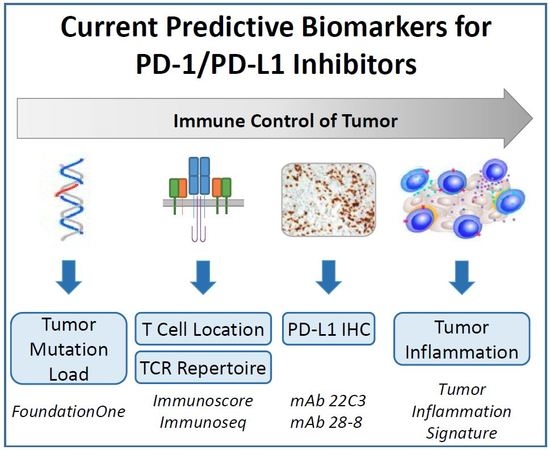
3. Approved and Candidate Biomarkers for IO Therapies
4. Measuring the Target by Immunohistochemistry (IHC)
5. Measuring Tumour Antigenicity: Tumour Mutation Load and Microsatellite Instability (Mismatch Repair Deficiency)
6. Quantification and Characterization of T cells
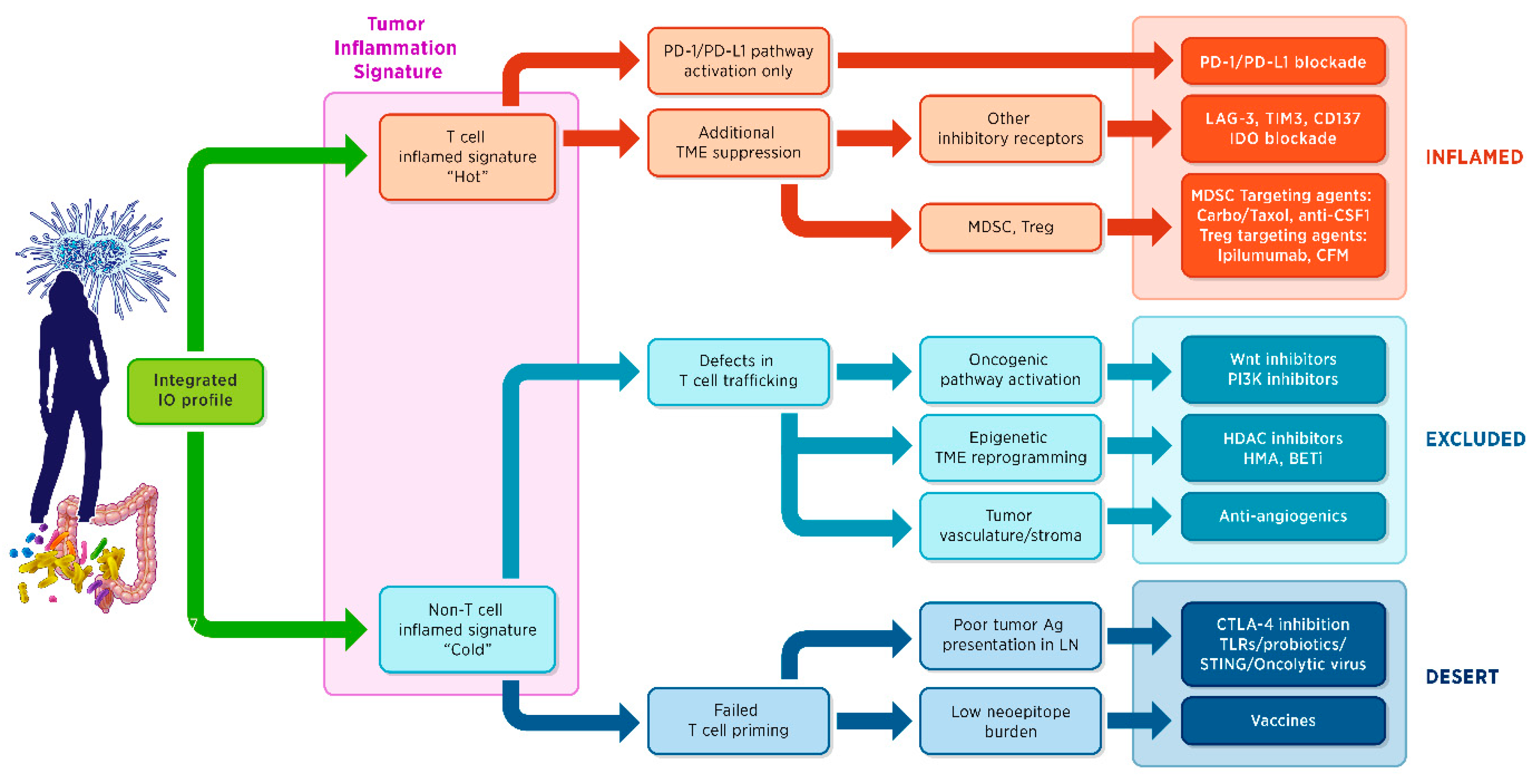
7. Gene Expression Signatures
- Approximately one-third of tumours have a T cell-inflamed tumour microenvironment signature (T cell markers, chemokines, macrophage-activated antigens, type I interferon (IFN) transcriptional profile) revealing a pre-existing adaptive immune response and thus suggesting that the expression of local inhibitory factors is at play; for this type of tumours, agents that help to brake the peripheral tolerance are more likely to be clinically successful;
- Non-T-cell-inflamed tumours have no T cell infiltration, suggesting a lack of innate immune activation or a block in T cell trafficking. These tumours require therapeutic approaches that activate the innate immune response by overcoming the central tolerance or by manipulating the oncogene signaling pathways interfering with T cell trafficking.
8. Challenges and Future Directions in the Clinical Development of IO Biomarkers
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer immunotherapy: The beginning of the end of cancer? BMC Med. 2016, 14. [Google Scholar] [CrossRef] [PubMed]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Merck KGaA, Darmstadt, Germany, and Pfizer Provide Update on Phase III JAVELIN Gastric 300 Study in Patients with Pre-Treated Advanced Gastric Cancer. Available online: https://www.pfizer.com/news/press-release/press-release-detail/merck_kgaa_darmstadt_germany_and_pfizer_provide_update_on_phase_iii_javelin_gastric_300_study_in_patients_with_pre_treated_advanced_gastric_cancer (accessed on 9 January 2018).
- Genentech Provides Update on Phase III Study of TECENTRIQ (Atezolizumab) in People with Previously Treated Advanced Bladder Cancer. Available online: https://www.gene.com/media/press-releases/14665/2017-05-09/genentech-provides-update-on-phase-iii-s (accessed on 9 January 2018).
- Bristol-Myers Squibb Announces Top-Line Results from CheckMate -026, a Phase 3 Study of Opdivo (nivolumab) in Treatment-Naïve Patients with Advanced Non-Small Cell Lung Cancer. Available online: https://news.bms.com/press-release/bristolmyers/bristol-myers-squibb-announces-top-line-results-checkmate-026-phase-3-stu (accessed on 9 January 2018).
- Bristol-Myers Squibb Announces Results from CheckMate-143, a Phase 3 Study of Opdivo (nivolumab) in Patients with Glioblastoma Multiforme. Available online: https://news.bms.com/press-release/bmy/bristol-myers-squibb-announces-results-checkmate-143-phase-3-study-opdivo-nivoluma (accessed on 9 January 2018).
- AstraZeneca Reports Initial Results from the Ongoing MYSTIC Trial in Stage IV Lung Cancer. Available online: https://www.astrazeneca.com/content/astraz/media-centre/press-releases/2017/astrazeneca-reports-initial-results-from-the-ongoing-mystic-trial-in-stage-iv-lung-cancer-27072017.html (accessed on 9 January 2018).
- Ott, P.A.; Hodi, F.S.; Kaufman, H.L.; Wigginton, J.M.; Wolchok, J.D. Combination immunotherapy: A road map. J. Immunother. Cancer 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Beasley, D. The Cost of Cancer: New Drugs Show Success at a Steep Price. Health News, 3 April 2017. Available online: https://www.reuters.com/article/us-usa-healthcare-cancer-costs/the-cost-of-cancer-new-drugs-show-success-at-a-steep-price-idUSKBN1750FU (accessed on 9 January 2018).
- FDA-NIH. Biomarker Working Group. BEST (Biomarkers, EndpointS, and Other Tools) Resource [Internet]. Food and Drug Administration: Silver Spring, MD, USA, 2016; Co-published by National Institutes of Health (US), Bethesda (MD). Available online: https://www.ncbi.nlm.nih.gov/books/NBK326791/ (accessed on 9 January 2018).
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Stieglmaier, J.; Benjamin, J.; Nagorsen, D. Utilizing the BiTE (bispecific T-cell engager) platform for immunotherapy of cancer. Expert Opin. Biol. Ther. 2015, 15, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors in cancer therapy: A focus on T-regulatory cells. Immunol. Cell Biol. 2018, 96, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, Z.; Liu, Y.; Han, W. New development in CAR-T cell therapy. J. Hematol. Oncol. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.; Pruteanu, J.; Gohil, M.; Wang, Y.; Boesteanu, A.; OConnor, R.; Ambrose, D.E.; Hwang, W.T. Identification of functional determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T-cell therapy of chronic lymphocytic leukemia. Blood 2017, 130, 3181. [Google Scholar]
- Ulloa-Montoya, F.; Louahed, J.; Dizier, B.; Gruselle, O.; Spiessens, B.; Lehmann, F.F.; Suciu, S.; Kruit, W.H.; Eggermont, A.M.; Vansteenkiste, J.; et al. Predictive gene signature in MAGE-A3 antigen-specific cancer immunotherapy. J. Clin. Oncol. 2013, 31, 2388–2395. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, F.; Liu, L. Prognostic significance of PD-L1 in solid tumour: An updated meta-analysis. Medicine 2017, 96, e6369. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N. Tumour Mutational Load: ESMO Biomarker Factsheet. Available online: http://oncologypro.esmo.org/Education-Library/Factsheets-on-Biomarkers/Tumour-Mutational-Load (accessed on 3 January 2018).
- Johnson, D.B.; Frampton, G.M.; Rioth, M.J.; Yusko, E.; Xu, Y.; Guo, X.; Ennis, R.C.; Fabrizio, D.; Chalmers, Z.R.; Greenbowe, J.; et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol. Res. 2016, 4, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Carbone, D.P.; Reck, M.; Paz-Arez, L.; Creelan, B.; Horn, L.; Steins, M.; Felip, E.; van den Heuvel, M.M.; Ciuleanu, T.E.; Badin, F.; et al. First-line nivolumab in stage IV or recurrent non-small-cell lung cancer. N. Engl. J. Med. 2017, 376, 2415–2426. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; Van Der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef]
- Champiat, S.; Ferté, C.; Lebel-Binay, S.; Eggermont, A.; Soria, J.C. Exomics and immunogenics: Bridging mutational load and immune checkpoints efficacy. Oncoimmunology 2014, 3, e27817. [Google Scholar] [CrossRef] [PubMed]
- Roszik, J.; Haydu, L.E.; Hess, K.R.; Oba, J.; Joon, A.Y.; Siroy, A.E.; Karpinets, T.V.; Stingo, F.C.; Baladandayuthapani, V.; Tetzlaff, M.T.; et al. Novel algorithmic approach predicts tumor mutation load and correlates with immunotherapy clinical outcomes using a defined gene mutation set. BMC Med. 2016, 14. [Google Scholar] [CrossRef] [PubMed]
- Campesato, L.F.; Barroso-Sousa, R.; Jimenez, L.; Correa, B.R.; Sabbaga, J.; Hoff, P.M.; Reis, L.F.; Galante, P.A.F.; Camargo, A.A. Comprehensive cancer-gene panels can be used to estimate mutational load and predict clinical benefit to PD-1 blockade in clinical practice. Oncotarget 2015, 6, 34221–34227. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.E.; Xiu, J.; Lenz, H.J.; Atkins, M.B.; Philip, P.A.; Hwang, J.J.; Gatalica, Z.; Xiao, N.; Gibney, G.T.; El-Deiry, W.S.; et al. Characterization of tumor mutation load (TML) in solid tumors. J. Clin. Oncol. 2017, 35 (Suppl. 15), 11517. [Google Scholar]
- Rosenberg, J.E.; Petrylak, D.P.; Van Der Heijden, M.S.; Necchi, A.; O’Donnell, P.H.; Loriot, Y.; Retz, M.; Perez-Gracia, J.L.; Bellmunt, J.; Grivas, P.; et al. PD-L1 expression, Cancer Genome Atlas (TCGA) subtype, and mutational load as independent predictors of response to atezolizumab (atezo) in metastatic urothelial carcinoma (mUC; IMvigor210). J. Clin. Oncol. 2016, 34 (Suppl. 15). [Google Scholar] [CrossRef]
- Johnson, D.B.; Frampton, G.M.; Rioth, M.J.; Yusko, E.; Ennis, R.; Fabrizio, D.; Greenbowe, J.R.; Ali, S.M.; Frederick, D.T.; Puzanov, I.; et al. Hybrid capture-based next-generation sequencing (HC NGS) in melanoma to identify markers of response to anti-PD-1/PD-L1. J. Clin. Oncol. 2016, 34 (Suppl. 15). [Google Scholar] [CrossRef]
- Spigel, D.R.; Schrock, A.B.; Fabrizio, D.; Frampton, G.M.; Sun, J.; He, J.; Gowen, K.; Johnson, M.L.; Bauer, T.M.; Kalemkerian, G.P.; et al. Total mutation burden (TMB) in lung cancer (LC) and relationship with response to PD-1/PD-L1 targeted therapies. J. Clin. Oncol. 2016, 34 (Suppl. 15). [Google Scholar] [CrossRef]
- George, T.J.; Frampton, G.M.; Sun, J.; Gowen, K.; Kennedy, M.; Greenbowe, J.R.; Schrock, A.B.; Ali, S.M.; Klempner, S.J.; Hezel, A.F.; et al. Tumor mutational burden as a potential biomarker for PD1/PD-L1 therapy in colorectal cancer. J. Clin. Oncol. 2016, 34 (Suppl. 15). [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch-repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.M.; Klein, A.P.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Gilligan, B.M.; Yuan, J.; Li, T. Current status and perspectives in translational biomarker research for PD-1/PD-L1 immune checkpoint blockade therapy. J. Hematol. Oncol. 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Rozeman, E.A.; Franchi, L.; Kuilman, T.; Krijgsman, O.; van Akkooi, A.C.J.; Kvistborg, P.; van Thienen, J.V.; Stegenga, B.; Lamon, B.; Haanen, J.B.A.G.; et al. Biomarker analysis from the OpACIN trial (neo-/adjuvant ipilimumab + nivolumab (IPI + NIVO) in palpable stage 3 melanoma). J. Immunother. Cancer 2017, 5 (Suppl. 2), 99. [Google Scholar]
- Warren, S.; Vallabhaneni, P.; El-Sawada, J.; Ren, X.; White, A.; Cesano, A.; Beechem, J.; Tarhini, A. Immunological profiling of baseline and resected biopsies from locally/regionally advanced/recurrent melanoma treated with neoadjuvant combination ipilimumab (3 mg/kg or 10 mg/kg) and high dose IFN-α2B. J. Immunother. Cancer 2017, 5 (Suppl. 2), 98. [Google Scholar]
- Haddad, R.I.; Seiwert, T.Y.; Chow, L.Q.; Gupta, S.; Weiss, J.; Gluck, I.; Eder, J.P.; Burtness, B.; Tahara, M.; Keam, B.; et al. Genomic determinants of response to pembrolizumab in head and neck squamous cell carcinoma (HNSCC). J. Clin. Oncol. 2017, 35 (Suppl. 15). [Google Scholar] [CrossRef]
- Cesano, A. nCounter® PanCancer Immune Profiling Panel (NanoString Technologies, Inc., Seattle, WA). J. Immunother. Cancer 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cesano, A.; Warren, S. Bringing the Next Generation of Immuno-Oncology Biomarkers to the Clinic. Biomedicines 2018, 6, 14. https://doi.org/10.3390/biomedicines6010014
Cesano A, Warren S. Bringing the Next Generation of Immuno-Oncology Biomarkers to the Clinic. Biomedicines. 2018; 6(1):14. https://doi.org/10.3390/biomedicines6010014
Chicago/Turabian StyleCesano, Alessandra, and Sarah Warren. 2018. "Bringing the Next Generation of Immuno-Oncology Biomarkers to the Clinic" Biomedicines 6, no. 1: 14. https://doi.org/10.3390/biomedicines6010014
APA StyleCesano, A., & Warren, S. (2018). Bringing the Next Generation of Immuno-Oncology Biomarkers to the Clinic. Biomedicines, 6(1), 14. https://doi.org/10.3390/biomedicines6010014