The Use of Tricyclo-DNA Oligomers for the Treatment of Genetic Disorders



Abstract
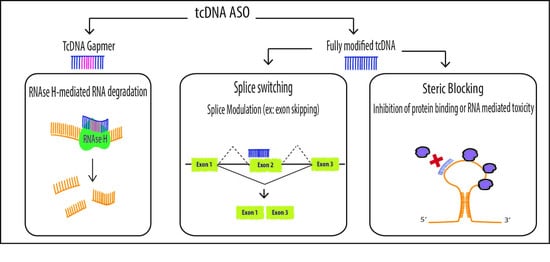
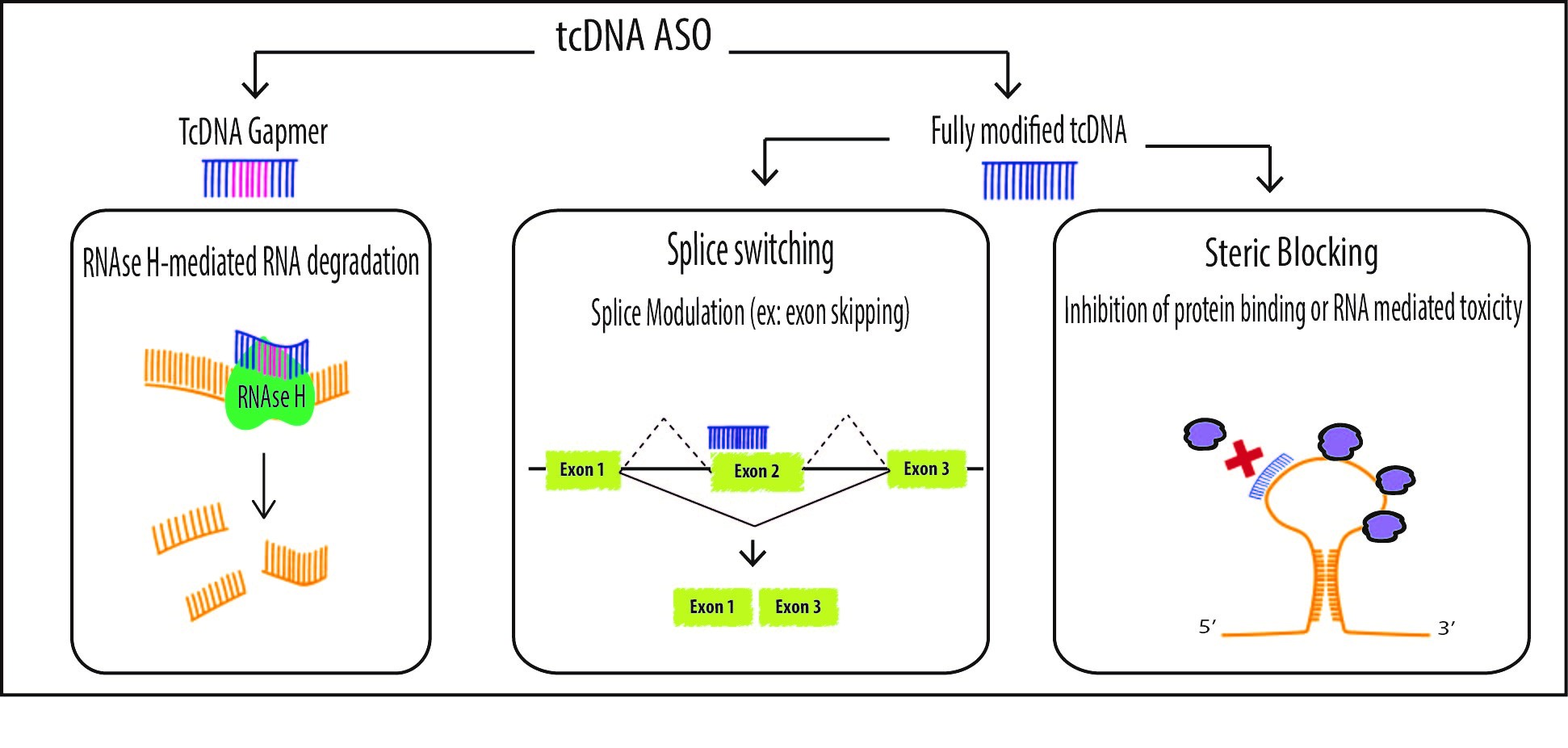
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Mechanisms of Action
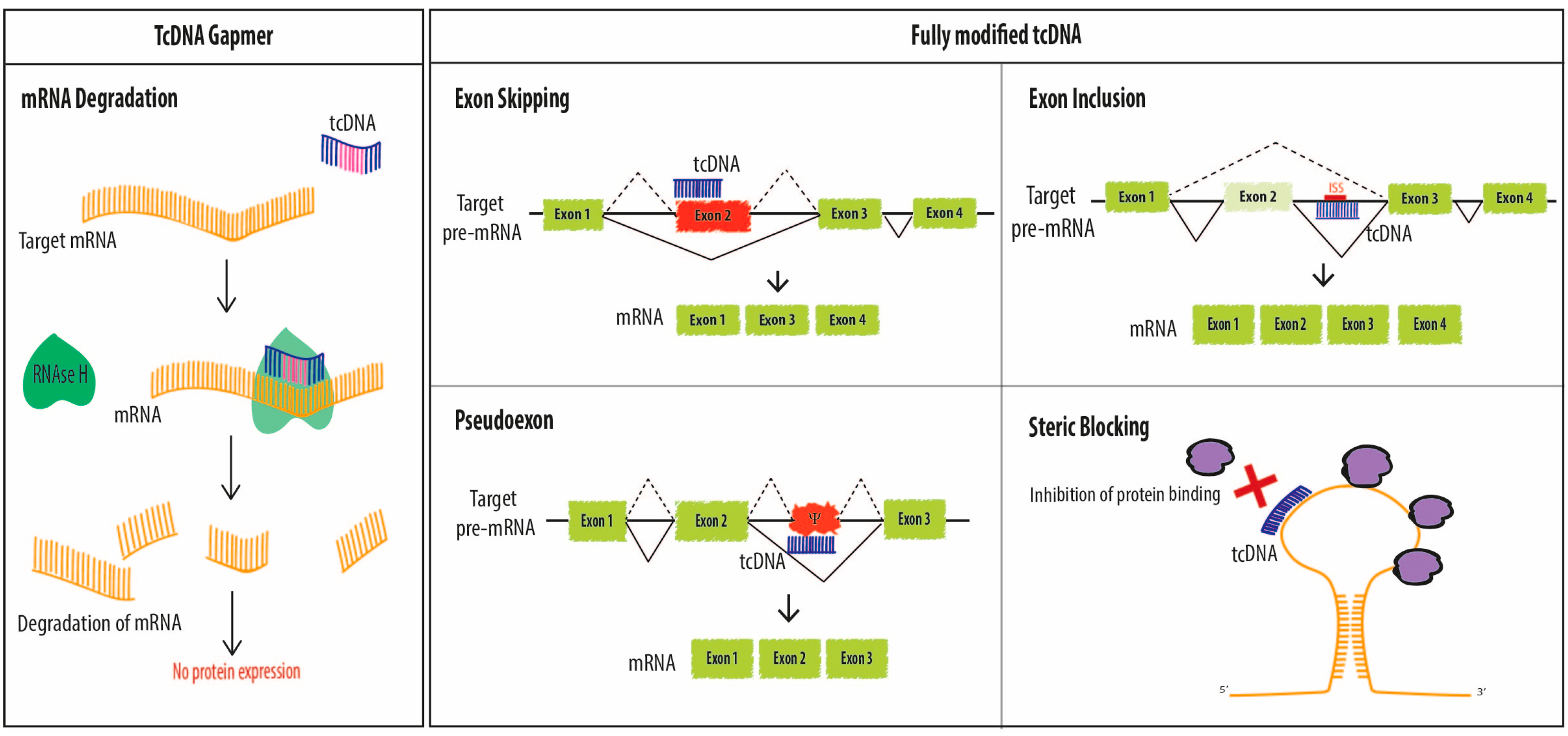
2.1. Splice Switching
2.1.1. Exon-Skipping
2.1.2. Exon Inclusion
2.2. Steric Blocking Applications
2.2.1. Inhibition of Trans-Activation
2.2.2. Reduction of mRNA Induced Toxicity
2.3. Degradation of mRNA
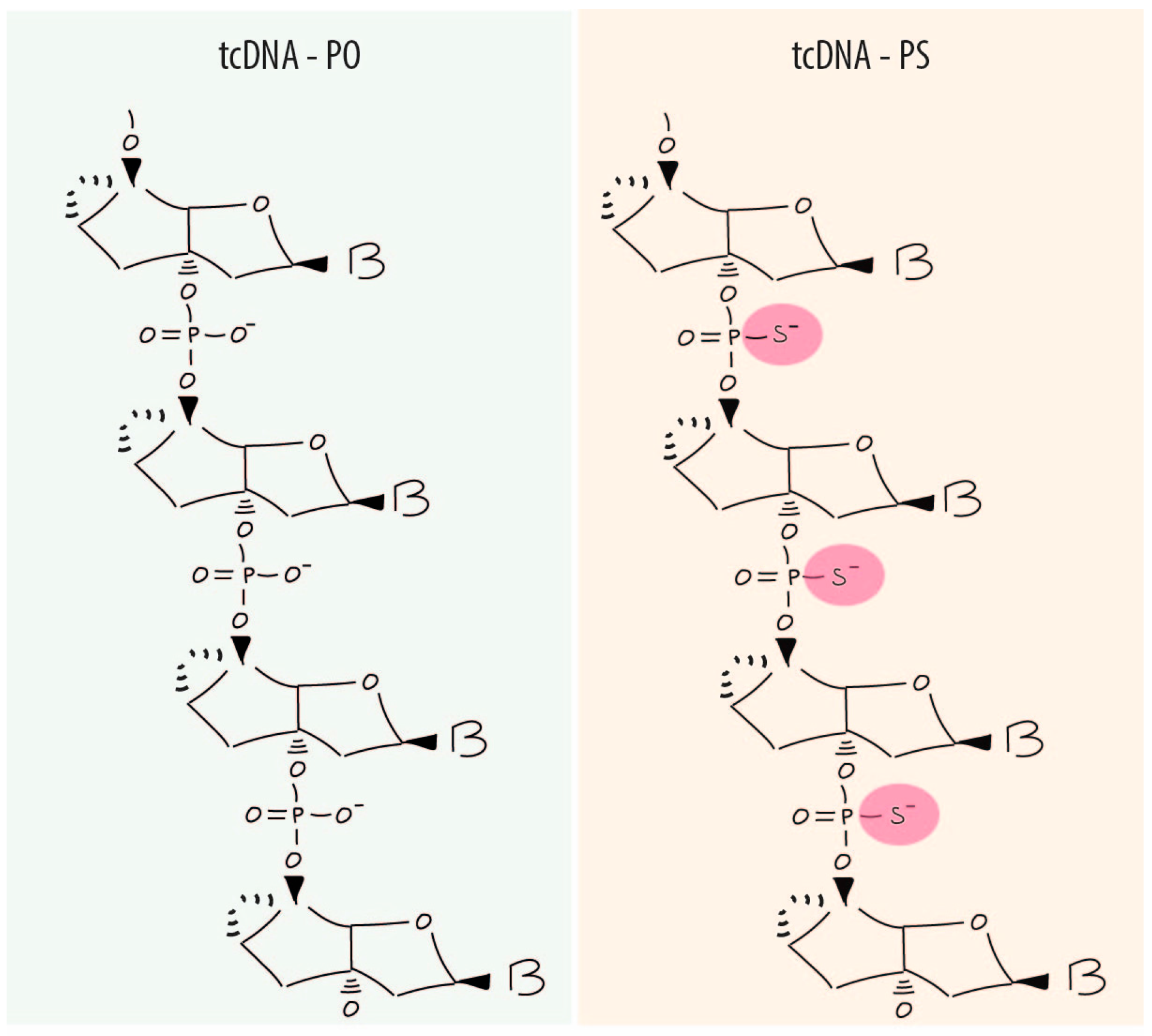
3. Properties and Advantages
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kaur, H.; Wengel, J.; Maiti, S. LNA-modified oligonucleotides effectively drive intramolecular-stable hairpin to intermolecular-duplex state. Biochem. Biophys. Res. Commun. 2007, 352, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Griepenburg, J.C.; Rapp, T.L.; Carroll, P.J.; Eberwine, J.; Dmochowski, I.J. Ruthenium-Caged Antisense Morpholinos for Regulating Gene Expression in Zebrafish Embryos. Chem. Sci. 2015, 6, 2342–2346. [Google Scholar] [CrossRef] [PubMed]
- Gilar, M.; Belenky, A.; Smisek, D.L.; Bourque, A.; Cohen, A.S. Kinetics of phosphorothioate oligonucleotide metabolism in biological fluids. Nucleic Acids Res. 1997, 25, 3615–3620. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F.; Swayze, E.E. RNA targeting therapeutics: Molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 259–293. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T. Molecular Mechanisms of Antisense Oligonucleotides. Nucleic Acid Ther. 2017, 27, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Schoch, K.M.; Miller, T.M. Antisense Oligonucleotides: Translation from Mouse Models to Human Neurodegenerative Diseases. Neuron 2017, 94, 1056–1070. [Google Scholar] [CrossRef] [PubMed]
- Wickstrom, E. Oligodeoxynucleotide stability in subcellular extracts and culture media. J. Biochem. Biophys. Methods 1986, 13, 97–102. [Google Scholar] [CrossRef]
- Benimetskaya, L.; Tonkinson, J.L.; Koziolkiewicz, M.; Karwowski, B.; Guga, P.; Zeltser, R.; Stec, W.; Stein, C.A. Binding of phosphorothioate oligodeoxynucleotides to basic fibroblast growth factor, recombinant soluble CD4, laminin and fibronectin is P-chirality independent. Nucleic Acids Res. 1995, 23, 4239–4245. [Google Scholar] [CrossRef] [PubMed]
- Iannitti, T.; Morales-Medina, J.C.; Palmieri, B. Phosphorothioate oligonucleotides: Effectiveness and toxicity. Curr. Drug Targets 2014, 15, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Goyenvalle, A.; Leumann, C.; Garcia, L. Therapeutic Potential of Tricyclo-DNA antisense oligonucleotides. J. Neuromuscul. Dis. 2016, 3, 157–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, S.; Stecker, K.; Brooks, D.; Monteith, D.; Conklin, B.; Bennett, C.F. Chemically modified oligonucleotides exhibit decreased immune stimulation in mice. J. Pharmacol. Exp. Ther. 2000, 292, 468–479. [Google Scholar] [PubMed]
- Summerton, J.; Weller, D. Morpholino antisense oligomers: Design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997, 7, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Hagedorn, P.H.; Persson, R.; Funder, E.D.; Albæk, N.; Diemer, S.L.; Hansen, D.J.; Møller, M.R.; Papargyri, N.; Christiansen, H.; Hansen, B.R.; et al. Locked nucleic acid: Modality, diversity, and drug discovery. Drug Discov. Today 2017. [Google Scholar] [CrossRef] [PubMed]
- Seth, P.P.; Siwkowski, A.; Allerson, C.R.; Vasquez, G.; Lee, S.; Prakash, T.P.; Kinberger, G.; Migawa, M.T.; Gaus, H.; Bhat, B.; et al. Design, synthesis and evaluation of constrained methoxyethyl (cMOE) and constrained ethyl (cEt) nucleoside analogues. Nucleic Acids Symp. Ser. 2004 2008, 553–554. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Mishra, A.; Puri, N. Peptide nucleic acids: Advanced tools for biomedical applications. J. Biotechnol. 2017, 259, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A. New Momentum for the Field of Oligonucleotide Therapeutics. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 193–194. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, C.; Desviat, L.R.; Smedsrød, B.; Piétri-Rouxel, F.; Denti, M.A.; Disterer, P.; Lorain, S.; Nogales-Gadea, G.; Sardone, V.; Anwar, R.; et al. Delivery is key: Lessons learnt from developing splice-switching antisense therapies. EMBO Mol. Med. 2017, 9, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. [Google Scholar] [CrossRef] [PubMed]
- Steffens, R.; Leumann, C.J. Tricyclo-DNA: A Phosphodiester-Backbone Based DNA Analog Exhibiting Strong Complementary Base-Pairing Properties. J. Am. Chem. Soc. 1997, 119, 11548–11549. [Google Scholar] [CrossRef]
- Steffens, R.; Leumann, C.J. Synthesis and Thermodynamic and Biophysical Properties of Tricyclo-DNA. J. Am. Chem. Soc. 1999, 121, 3249–3255. [Google Scholar] [CrossRef]
- Renneberg, D.; Bouliong, E.; Reber, U.; Schümperli, D.; Leumann, C.J. Antisense properties of tricyclo-DNA. Nucleic Acids Res. 2002, 30, 2751–2757. [Google Scholar] [CrossRef] [PubMed]
- Murray, S.; Ittig, D.; Koller, E.; Berdeja, A.; Chappell, A.; Prakash, T.P.; Norrbom, M.; Swayze, E.E.; Leumann, C.J.; Seth, P.P. TricycloDNA-modified oligo-2′-deoxyribonucleotides reduce scavenger receptor B1 mRNA in hepatic and extra-hepatic tissues—A comparative study of oligonucleotide length, design and chemistry. Nucleic Acids Res. 2012, 40, 6135–6143. [Google Scholar] [CrossRef] [PubMed]
- Ittig, D.; Liu, S.; Renneberg, D.; Schumperli, D.; Leumann, C.J. Nuclear antisense effects in cyclophilin A pre-mRNA splicing by oligonucleotides: A comparison of tricyclo-DNA with LNA. Nucleic Acids Res. 2004, 32, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Fokkema, I.; Verschuuren, J.; Ginjaar, I.; van Deutekom, J.; van Ommen, G.J.; den Dunnen, J.T. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum. Mutat. 2009, 30, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Pramono, Z.A.; Takeshima, Y.; Alimsardjono, H.; Ishii, A.; Takeda, S.; Matsuo, M. Induction of exon skipping of the dystrophin transcript in lymphoblastoid cells by transfecting an antisense oligodeoxynucleotide complementary to an exon recognition sequence. Biochem. Biophys. Res. Commun. 1996, 226, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.L.; Mann, C.J.; Lou, F.; Bou-Gharios, G.; Morris, G.E.; Xue, S.A.; Fletcher, S.; Partridge, T.A.; Wilton, S.D. Functional amounts of dystrophin produced by skipping the mutated exon in the mdx dystrophic mouse. Nat. Med. 2003, 9, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.; Honeyman, K.; Fall, A.M.; Harding, P.L.; Johnsen, R.D.; Wilton, S.D. Dystrophin expression in the mdx mouse after localised and systemic administration of a morpholino antisense oligonucleotide. J. Gene Med. 2006, 8, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.; Honeyman, K.; Fall, A.M.; Harding, P.L.; Johnsen, R.D.; Steinhaus, J.P.; Moulton, H.M.; Iversen, P.L.; Wilton, S.D. Morpholino oligomer-mediated exon skipping averts the onset of dystrophic pathology in the mdx mouse. Mol. Ther. 2007, 15, 1587–1592. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.L.; Rabinowitz, A.; Chen, Y.C.; Yokota, T.; Yin, H.; Alter, J.; Jadoon, A.; Bou-Gharios, G.; Partridge, T. Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles. Proc. Natl. Acad. Sci. USA 2005, 102, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, G.D.; Arzumanov, A.; Abes, R.; Yin, H.; Wood, M.J.; Lebleu, B.; Gait, M.J. Improved cell-penetrating peptide-PNA conjugates for splicing redirection in HeLa cells and exon skipping in mdx mouse muscle. Nucleic Acids Res. 2008, 36, 6418–6428. [Google Scholar] [CrossRef] [PubMed]
- van Deutekom, J.C.; Janson, A.A.; Ginjaar, I.B.; Frankhuizen, W.S.; Aartsma-Rus, A.; Bremmer-Bout, M.; den Dunnen, J.T.; Koop, K.; van der Kooi, A.J.; Goemans, N.M.; et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N. Engl. J. Med. 2007, 357, 2677–2686. [Google Scholar] [CrossRef] [PubMed]
- Goemans, N.M.; Tulinius, M.; van den Akker, J.T.; Burm, B.E.; Ekhart, P.F.; Heuvelmans, N.; Holling, T.; Janson, A.A.; Platenburg, G.J.; Sipkens, J.A.; et al. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N. Engl. J. Med. 2011, 364, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Voit, T.; Topaloglu, H.; Straub, V.; Muntoni, F.; Deconinck, N.; Campion, G.; De Kimpe, S.J.; Eagle, M.; Guglieri, M.; Hood, S.; et al. Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): An exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol. 2014, 13, 987–996. [Google Scholar] [CrossRef]
- Flanigan, K.M.; Voit, T.; Rosales, X.Q.; Servais, L.; Kraus, J.E.; Wardell, C.; Morgan, A.; Dorricott, S.; Nakielny, J.; Quarcoo, N.; et al. Pharmacokinetics and safety of single doses of drisapersen in non-ambulant subjects with Duchenne muscular dystrophy: Results of a double-blind randomized clinical trial. Neuromuscul. Disord. 2014, 24, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Goemans, N.M.; Tulinius, M.; van den Hauwe, M.; Kroksmark, A.-K.; Buyse, G.; Wilson, R.J.; van Deutekom, J.C.; de Kimpe, S.J.; Lourbakos, A.; Campion, G. Long-Term Efficacy, Safety, and Pharmacokinetics of Drisapersen in Duchenne Muscular Dystrophy: Results from an Open-Label Extension Study. PLoS ONE 2016, 11, e0161955. [Google Scholar] [CrossRef] [PubMed]
- Shimizu-Motohashi, Y.; Miyatake, S.; Komaki, H.; Takeda, S.; Aoki, Y. Recent advances in innovative therapeutic approaches for Duchenne muscular dystrophy: From discovery to clinical trials. Am. J. Transl. Res. 2016, 8, 2471–2489. [Google Scholar] [PubMed]
- Cirak, S.; Feng, L.; Anthony, K.; Arechavala-Gomeza, V.; Torelli, S.; Sewry, C.; Morgan, J.E.; Muntoni, F. Restoration of the dystrophin-associated glycoprotein complex after exon skipping therapy in Duchenne muscular dystrophy. Mol. Ther. 2012, 20, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Cirak, S.; Arechavala-Gomeza, V.; Guglieri, M.; Feng, L.; Torelli, S.; Anthony, K.; Abbs, S.; Garralda, M.E.; Bourke, J.; Wells, D.J.; et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: An open-label, phase 2, dose-escalation study. Lancet 2011, 378, 595–605. [Google Scholar] [CrossRef]
- Mendell, J.R.; Rodino-Klapac, L.R.; Sahenk, Z.; Roush, K.; Bird, L.; Lowes, L.P.; Alfano, L.; Gomez, A.M.; Lewis, S.; Kota, J.; et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 2013, 74, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Goemans, N.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Shao, J.; Kaye, E.M.; Mercuri, E. Eteplirsen Study Group and Telethon Foundation DMD Italian Network Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann. Neurol. 2016, 79, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Goyenvalle, A.; Griffith, G.; Babbs, A.; Andaloussi, S.E.; Ezzat, K.; Avril, A.; Dugovic, B.; Chaussenot, R.; Ferry, A.; Voit, T.; et al. Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers. Nat. Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Relizani, K.; Griffith, G.; Echevarría, L.; Zarrouki, F.; Facchinetti, P.; Vaillend, C.; Leumann, C.; Garcia, L.; Goyenvalle, A. Efficacy and Safety Profile of Tricyclo-DNA Antisense Oligonucleotides in Duchenne Muscular Dystrophy Mouse Model. Mol. Ther. Nucleic Acids 2017, 8, 144–157. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, S.; Burglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef]
- Feldkotter, M.; Schwarzer, V.; Wirth, R.; Wienker, T.F.; Wirth, B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: Fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet. 2002, 70, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Lorson, C.L.; Hahnen, E.; Androphy, E.J.; Wirth, B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 1999, 96, 6307–6311. [Google Scholar] [CrossRef] [PubMed]
- Monani, U.R.; Lorson, C.L.; Parsons, D.W.; Prior, T.W.; Androphy, E.J.; Burghes, A.H.; McPherson, J.D. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999, 8, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Butchbach, M.E.R. Copy Number Variations in the Survival Motor Neuron Genes: Implications for Spinal Muscular Atrophy and Other Neurodegenerative Diseases. Front. Mol. Biosci. 2016, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Sahashi, K.; Hung, G.; Rigo, F.; Passini, M.A.; Bennett, C.F.; Krainer, A.R. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010, 24, 1634–1644. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Sahashi, K.; Rigo, F.; Hung, G.; Horev, G.; Bennett, C.F.; Krainer, A.R. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 2011, 478, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Passini, M.A.; Bu, J.; Richards, A.M.; Kinnecom, C.; Sardi, S.P.; Stanek, L.M.; Hua, Y.; Rigo, F.; Matson, J.; Hung, G.; et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci. Transl. Med. 2011, 3, 72ra18. [Google Scholar] [CrossRef] [PubMed]
- Chiriboga, C.A.; Swoboda, K.J.; Darras, B.T.; Iannaccone, S.T.; Montes, J.; De Vivo, D.C.; Norris, D.A.; Bennett, C.F.; Bishop, K.M. Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology 2016, 86, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Garber, K. Big win possible for Ionis/Biogen antisense drug in muscular atrophy. Nat. Biotechnol. 2016, 34, 1002–1003. [Google Scholar] [CrossRef] [PubMed]
- Robin, V.; Griffith, G.; Carter, J.-P.L.; Leumann, C.J.; Garcia, L.; Goyenvalle, A. Efficient SMN Rescue following Subcutaneous Tricyclo-DNA Antisense Oligonucleotide Treatment. Mol. Ther. Nucleic Acids 2017, 7, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.K.; Singh, N.N.; Androphy, E.J.; Singh, R.N. Splicing of a critical exon of human Survival Motor Neuron is regulated by a unique silencer element located in the last intron. Mol. Cell Biol. 2006, 26, 1333–1346. [Google Scholar] [CrossRef] [PubMed]
- Brady, J.; Kashanchi, F. Tat gets the “green” light on transcription initiation. Retrovirology 2005, 2, 69. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Iwata, A.; Nyhuis, J.; Nitta, Y.; Miller, A.D.; Halbert, C.L.; Allen, M.D. Adeno-associated virus vector transduction of vascular smooth muscle cells in vivo. Physiol. Genom. 2000, 2, 117–127. [Google Scholar]
- Turner, J.J.; Fabani, M.; Arzumanov, A.A.; Ivanova, G.; Gait, M.J. Targeting the HIV-1 RNA leader sequence with synthetic oligonucleotides and siRNA: Chemistry and cell delivery. Biochim. Biophys. Acta 2006, 1758, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, G.; Arzumanov, A.A.; Turner, J.J.; Reigadas, S.; Toulmé, J.-J.; Brown, D.E.; Lever, A.M.L.; Gait, M.J. Anti-HIV activity of steric block oligonucleotides. Ann. N. Y. Acad. Sci. 2006, 1082, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Wahlestedt, C.; Salmi, P.; Good, L.; Kela, J.; Johnsson, T.; Hökfelt, T.; Broberger, C.; Porreca, F.; Lai, J.; Ren, K.; et al. Potent and nontoxic antisense oligonucleotides containing locked nucleic acids. Proc. Natl. Acad. Sci. USA 2000, 97, 5633–5638. [Google Scholar] [CrossRef] [PubMed]
- Arzumanov, A.; Walsh, A.P.; Rajwanshi, V.K.; Kumar, R.; Wengel, J.; Gait, M.J. Inhibition of HIV-1 Tat-dependent trans activation by steric block chimeric 2′-O-methyl/LNA oligoribonucleotides. Biochemistry (Mosc.) 2001, 40, 14645–14654. [Google Scholar] [CrossRef]
- Mestre, B.; Arzumanov, A.; Singh, M.; Boulmé, F.; Litvak, S.; Gait, M.J. Oligonucleotide inhibition of the interaction of HIV-1 Tat protein with the trans-activation responsive region (TAR) of HIV RNA. Biochim. Biophys. Acta 1999, 1445, 86–98. [Google Scholar] [CrossRef]
- Arzumanov, A.; Stetsenko, D.A.; Malakhov, A.D.; Reichelt, S.; Sørensen, M.D.; Babu, B.R.; Wengel, J.; Gait, M.J. A structure-activity study of the inhibition of HIV-1 Tat-dependent trans-activation by mixmer 2′-O-methyl oligoribonucleotides containing locked nucleic acid (LNA), alpha-L-LNA, or 2′-thio-LNA residues. Oligonucleotides 2003, 13, 435–453. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, G.; Reigadas, S.; Ittig, D.; Arzumanov, A.; Andreola, M.L.; Leumann, C.; Toulme, J.J.; Gait, M.J. Tricyclo-DNA containing oligonucleotides as steric block inhibitors of human immunodeficiency virus type 1 tat-dependent trans-activation and HIV-1 infectivity. Oligonucleotides 2007, 17, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, M.; Tsilfidis, C.; Sabourin, L.; Shutler, G.; Amemiya, C.; Jansen, G.; Neville, C.; Narang, M.; Barcelo, J.; O’Hoy, K.; et al. Myotonic dystrophy mutation: An unstable CTG repeat in the 3′ untranslated region of the gene. Science 1992, 255, 1253–1255. [Google Scholar] [CrossRef] [PubMed]
- Harper, P.S.; Brook, J.D. Myotonic Dystrophy, 3rd ed.; Major Problems in Neurology, 1st publ.; Saunders: London, UK, 2001; ISBN 978-0-7020-2152-7. [Google Scholar]
- Brook, J.D.; McCurrach, M.E.; Harley, H.G.; Buckler, A.J.; Church, D.; Aburatani, H.; Hunter, K.; Stanton, V.P.; Thirion, J.P.; Hudson, T.; et al. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 1992, 69, 385. [Google Scholar] [CrossRef]
- Liquori, C.L.; Ricker, K.; Moseley, M.L.; Jacobsen, J.F.; Kress, W.; Naylor, S.L.; Day, J.W.; Ranum, L.P. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 2001, 293, 864–867. [Google Scholar] [CrossRef] [PubMed]
- Mankodi, A.; Logigian, E.; Callahan, L.; McClain, C.; White, R.; Henderson, D.; Krym, M.; Thornton, C.A. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 2000, 289, 1769–1773. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.W.; Urbinati, C.R.; Teng-Umnuay, P.; Stenberg, M.G.; Byrne, B.J.; Thornton, C.A.; Swanson, M.S. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. Embo J. 2000, 19, 4439–4448. [Google Scholar] [CrossRef] [PubMed]
- Mankodi, A.; Urbinati, C.R.; Yuan, Q.P.; Moxley, R.T.; Sansone, V.; Krym, M.; Henderson, D.; Schalling, M.; Swanson, M.S.; Thornton, C.A. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum. Mol. Genet. 2001, 10, 2165–2170. [Google Scholar] [CrossRef] [PubMed]
- Kuyumcu-Martinez, N.M.; Wang, G.S.; Cooper, T.A. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol. Cell 2007, 28, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.S.; Kearney, D.L.; De Biasi, M.; Taffet, G.; Cooper, T.A. Elevation of RNA-binding protein CUGBP1 is an early event in an inducible heart-specific mouse model of myotonic dystrophy. J. Clin. Investig. 2007, 117, 2802–2811. [Google Scholar] [CrossRef] [PubMed]
- Kapsa, R.; Kornberg, A.J.; Byrne, E. Novel therapies for Duchenne muscular dystrophy. Lancet Neurol. 2003, 2, 299–310. [Google Scholar] [CrossRef]
- Savkur, R.S.; Philips, A.V.; Cooper, T.A. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat. Genet. 2001, 29, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Fugier, C.; Klein, A.F.; Hammer, C.; Vassilopoulos, S.; Ivarsson, Y.; Toussaint, A.; Tosch, V.; Vignaud, A.; Ferry, A.; Messaddeq, N.; et al. Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat. Med. 2011, 17, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, T.M.; Sobczak, K.; Lueck, J.D.; Osborne, R.J.; Lin, X.; Dirksen, R.T.; Thornton, C.A. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science 2009, 325, 336–339. [Google Scholar] [CrossRef] [PubMed]
- Mulders, S.A.; van den Broek, W.J.; Wheeler, T.M.; Croes, H.J.; van Kuik-Romeijn, P.; de Kimpe, S.J.; Furling, D.; Platenburg, G.J.; Gourdon, G.; Thornton, C.A.; et al. Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc. Natl. Acad. Sci. USA 2009, 106, 13915–13920. [Google Scholar] [CrossRef] [PubMed]
- Leger, A.J.; Mosquea, L.M.; Clayton, N.P.; Wu, I.H.; Weeden, T.; Nelson, C.A.; Phillips, L.; Roberts, E.; Piepenhagen, P.A.; Cheng, S.H.; et al. Systemic delivery of a Peptide-linked morpholino oligonucleotide neutralizes mutant RNA toxicity in a mouse model of myotonic dystrophy. Nucleic Acid Ther. 2013, 23, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Huntington, G. On chorea. J. Neuropsychiatry Clin. Neurosci. 2003, 15, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Leggo, J.; Coles, R.; Almqvist, E.; Biancalana, V.; Cassiman, J.J.; Chotai, K.; Connarty, M.; Crauford, D.; Curtis, A.; et al. Phenotypic characterization of individuals with 30–40 CAG repeats in the Huntington disease (HD) gene reveals HD cases with 36 repeats and apparently normal elderly individuals with 36–39 repeats. Am. J. Hum. Genet. 1996, 59, 16–22. [Google Scholar] [PubMed]
- Finkbeiner, S. Huntington’s Disease. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Kordasiewicz, H.B.; Stanek, L.M.; Wancewicz, E.V.; Mazur, C.; McAlonis, M.M.; Pytel, K.A.; Artates, J.W.; Weiss, A.; Cheng, S.H.; Shihabuddin, L.S.; et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron 2012, 74, 1031–1044. [Google Scholar] [CrossRef] [PubMed]
- Imbert, M.; Blandel, F. Lowering mutant Huntingtin using tricyclo-DNA antisense oligonucleotides as a therapeutic approach for Huntington’s disease. (Manuscript in preparation).
- Carroll, J.B.; Bates, G.P.; Steffan, J.; Saft, C.; Tabrizi, S.J. Treating the whole body in Huntington’s disease. Lancet Neurol. 2015, 14, 1135–1142. [Google Scholar] [CrossRef]
- Ezzat, K.; Aoki, Y.; Koo, T.; McClorey, G.; Benner, L.; Coenen-Stass, A.; O’Donovan, L.; Lehto, T.; Garcia-Guerra, A.; Nordin, J.; et al. Self-Assembly into Nanoparticles Is Essential for Receptor Mediated Uptake of Therapeutic Antisense Oligonucleotides. Nano Lett. 2015, 15, 4364–4373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nel, A.E.; Madler, L.; Velegol, D.; Xia, T.; Hoek, E.M.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding biophysicochemical interactions at the nano-bio interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Boutin, S.R.; Shen, Z.; Roesch, P.L.; Stiefel, S.M.; Sanderson, A.E.; Multari, H.M.; Pridhoko, E.A.; Smith, J.C.; Taylor, N.S.; Lohmiller, J.J.; et al. Helicobacter pullorum outbreak in C57BL/6NTac and C3H/HeNTac barrier-maintained mice. J. Clin. Microbiol. 2010, 48, 1908–1910. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Castro, J.E.; Motta, M.; Cottam, H.B.; Kyburz, D.; Kipps, T.J.; Corr, M.; Carson, D.A. Selection of oligonucleotide aptamers with enhanced uptake and activation of human leukemia B cells. Hum. Gene Ther. 2003, 14, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Frazier, K.S. Antisense oligonucleotide therapies: The promise and the challenges from a toxicologic pathologist’s perspective. Toxicol. Pathol. 2015, 43, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Henry, S.P.; Beattie, G.; Yeh, G.; Chappel, A.; Giclas, P.; Mortari, A.; Jagels, M.A.; Kornbrust, D.J.; Levin, A.A. Complement activation is responsible for acute toxicities in rhesus monkeys treated with a phosphorothioate oligodeoxynucleotide. Int. Immunopharmacol. 2002, 2, 1657–1666. [Google Scholar] [CrossRef]
- Agrawal, S.; Kandimalla, E.R. Role of Toll-like receptors in antisense and siRNA [corrected]. Nat. Biotechnol. 2004, 22, 1533–1537. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, V.S.; Ferguson, M.A.; Bonventre, J.V. Biomarkers of acute kidney injury. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 463–493. [Google Scholar] [CrossRef] [PubMed]
- Frazier, K.S.; Sobry, C.; Derr, V.; Adams, M.J.; Besten, C.D.; De Kimpe, S.; Francis, I.; Gales, T.L.; Haworth, R.; Maguire, S.R.; et al. Species-specific inflammatory responses as a primary component for the development of glomerular lesions in mice and monkeys following chronic administration of a second-generation antisense oligonucleotide. Toxicol. Pathol. 2014, 42, 923–935. [Google Scholar] [CrossRef] [PubMed]
- Echevarria, L.; Aupy, P. Evaluation of the Phosphorothioate content within tricyclo-DNA splice-switching oligonucleotides in DMD mouse model. In preparation.


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aupy, P.; Echevarría, L.; Relizani, K.; Goyenvalle, A. The Use of Tricyclo-DNA Oligomers for the Treatment of Genetic Disorders. Biomedicines 2018, 6, 2. https://doi.org/10.3390/biomedicines6010002
Aupy P, Echevarría L, Relizani K, Goyenvalle A. The Use of Tricyclo-DNA Oligomers for the Treatment of Genetic Disorders. Biomedicines. 2018; 6(1):2. https://doi.org/10.3390/biomedicines6010002
Chicago/Turabian StyleAupy, Philippine, Lucía Echevarría, Karima Relizani, and Aurélie Goyenvalle. 2018. "The Use of Tricyclo-DNA Oligomers for the Treatment of Genetic Disorders" Biomedicines 6, no. 1: 2. https://doi.org/10.3390/biomedicines6010002
APA StyleAupy, P., Echevarría, L., Relizani, K., & Goyenvalle, A. (2018). The Use of Tricyclo-DNA Oligomers for the Treatment of Genetic Disorders. Biomedicines, 6(1), 2. https://doi.org/10.3390/biomedicines6010002