CD117/c-kit in Cancer Stem Cell-Mediated Progression and Therapeutic Resistance

 , ,
, ,
Abstract
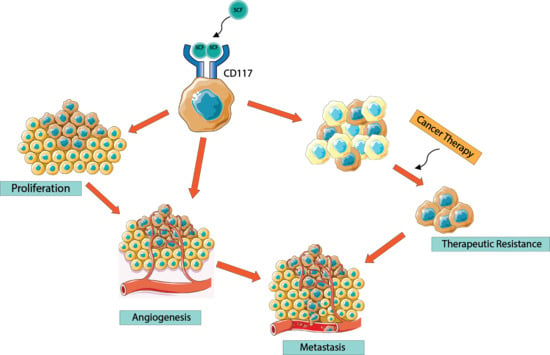
:
1. Introduction
2. The CD117 Receptor
2.1. CD117 Splice Variants
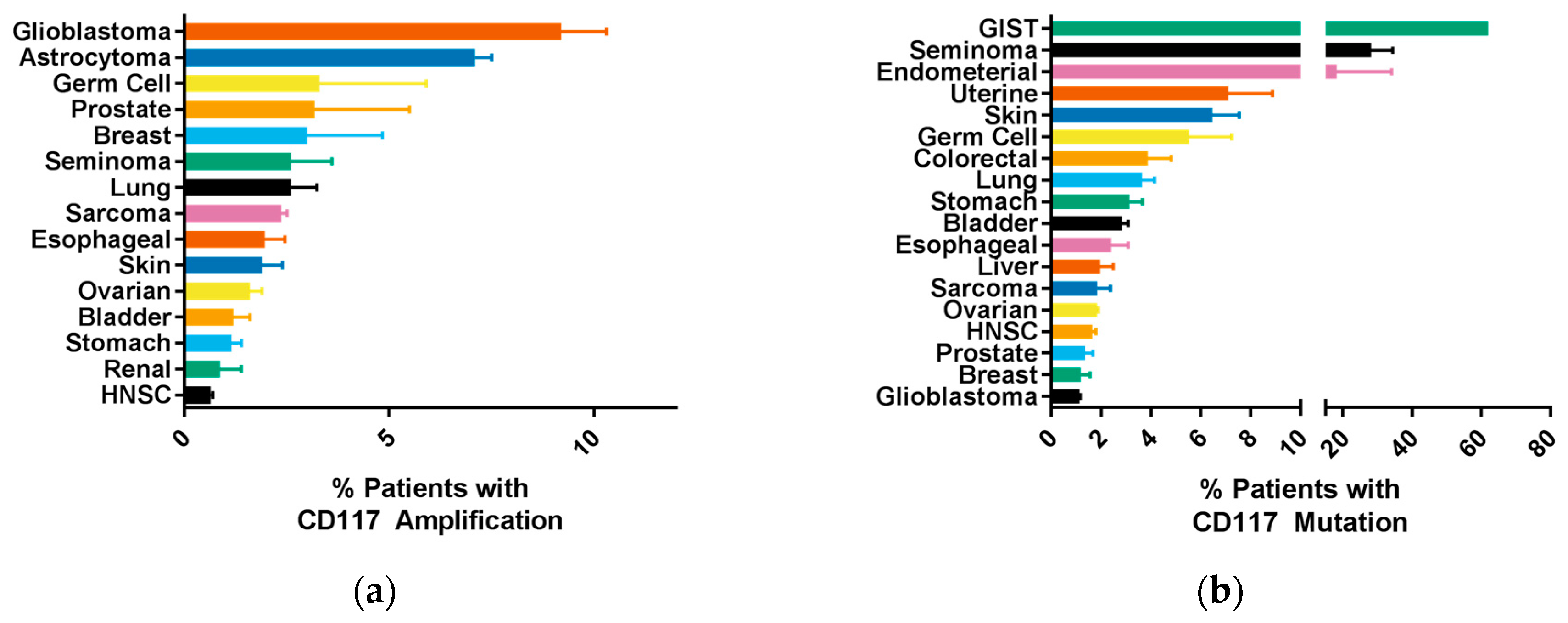
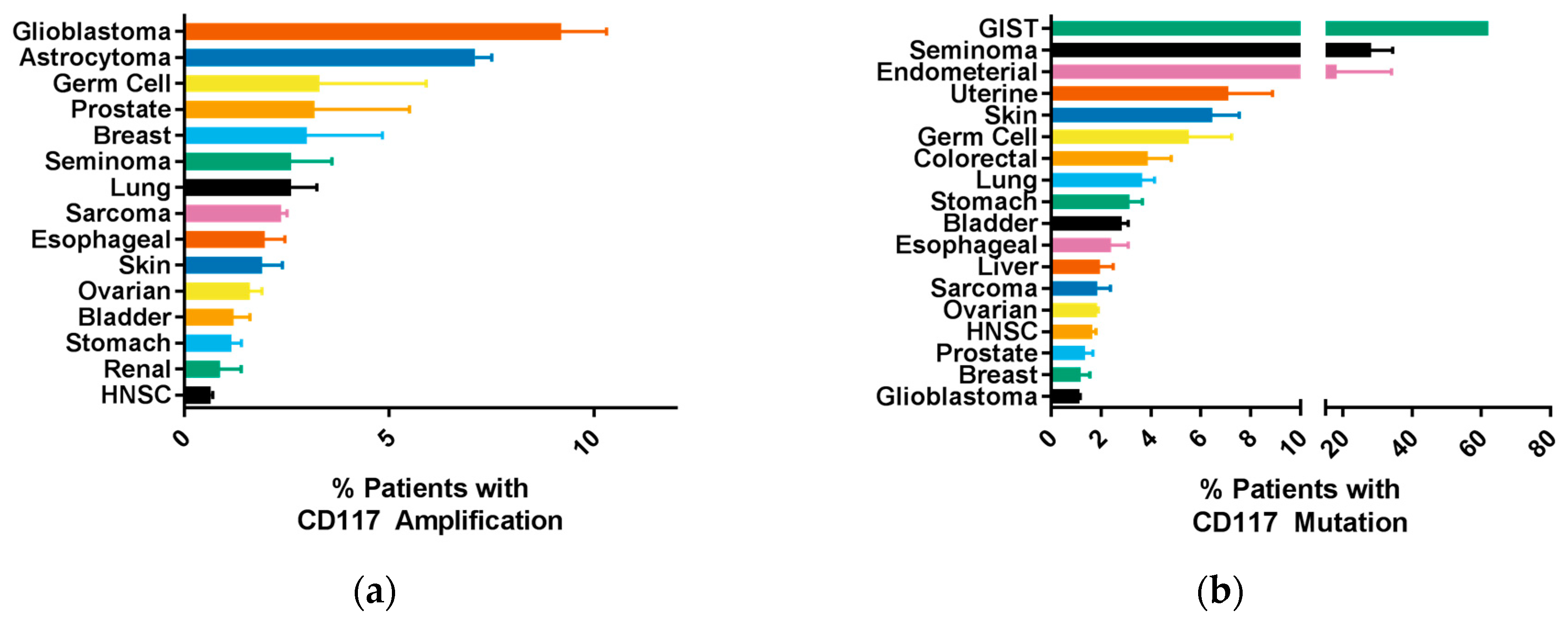
2.2. Common CD117 Oncogenic Mutations
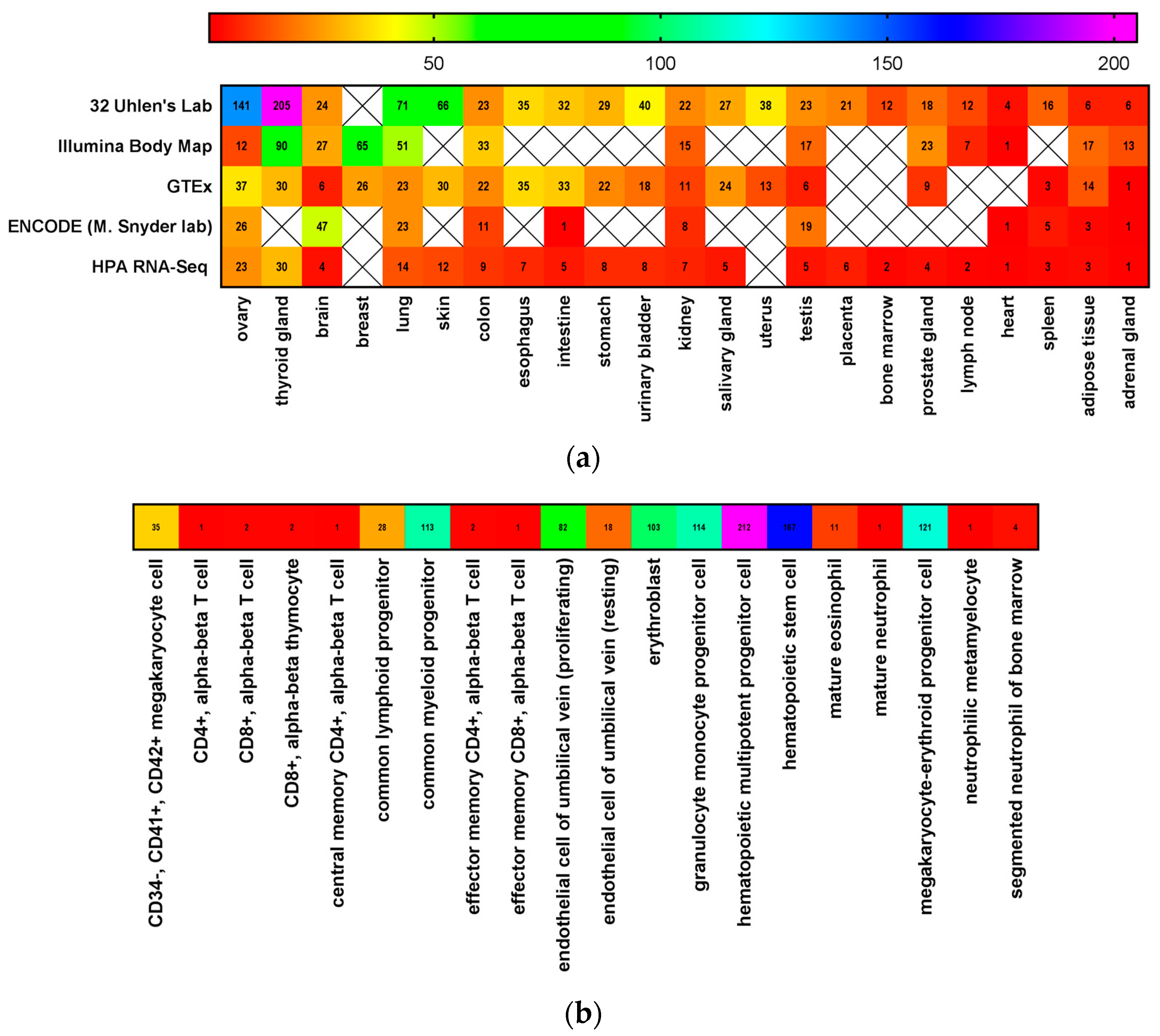
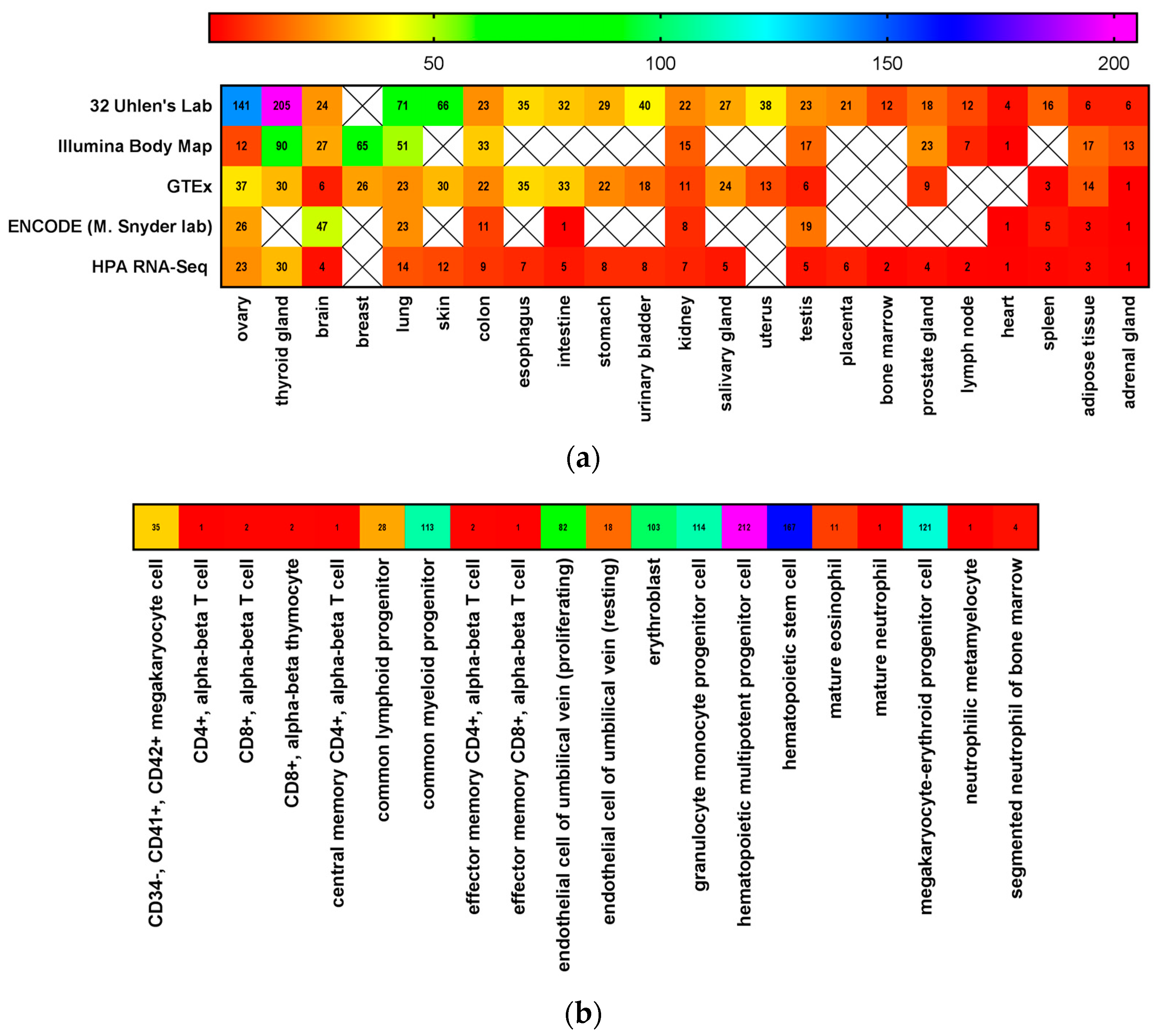
3. CD117 Expression in Normal Stem Cells
4. SCF Expression in Stem Cell Niches
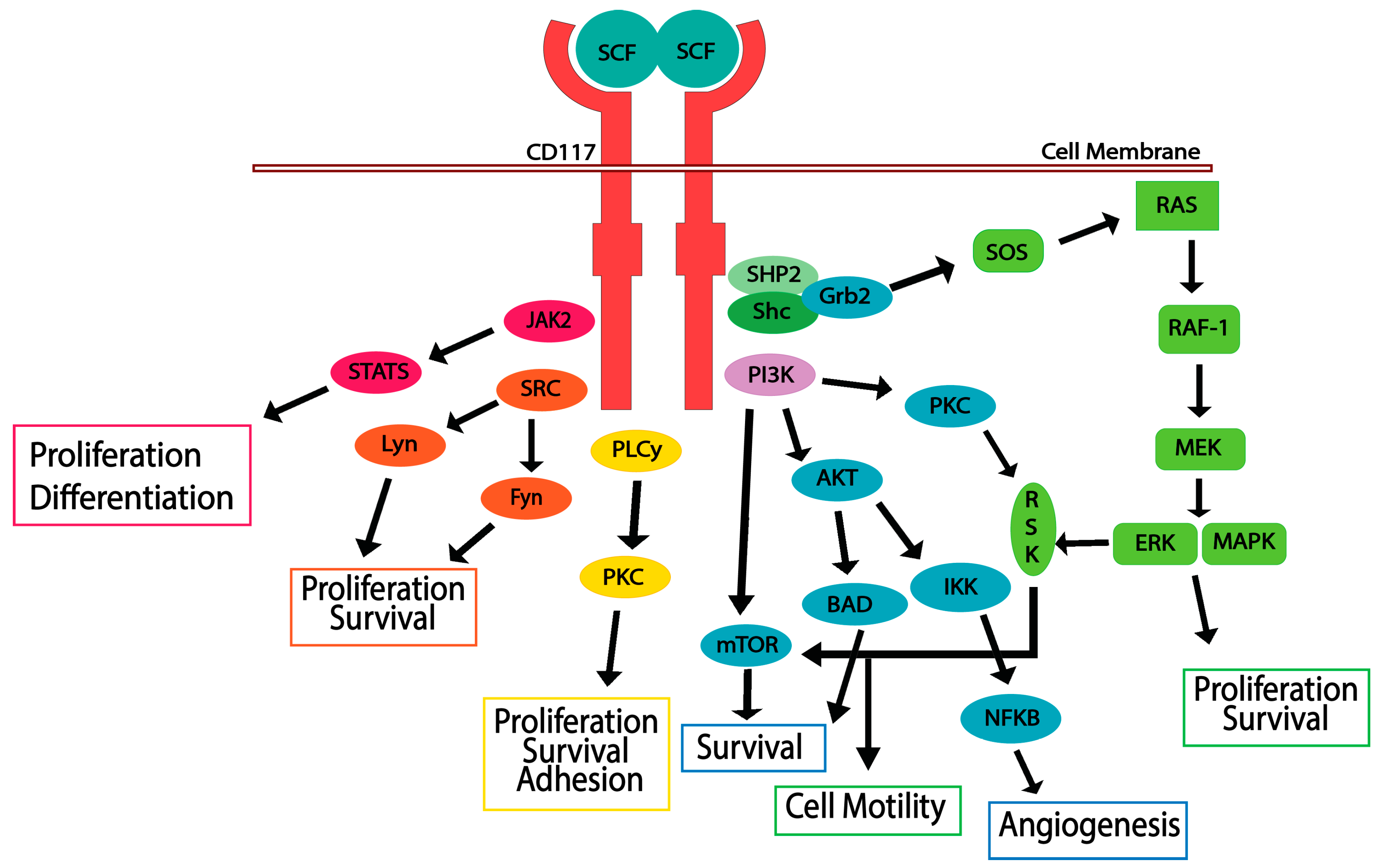
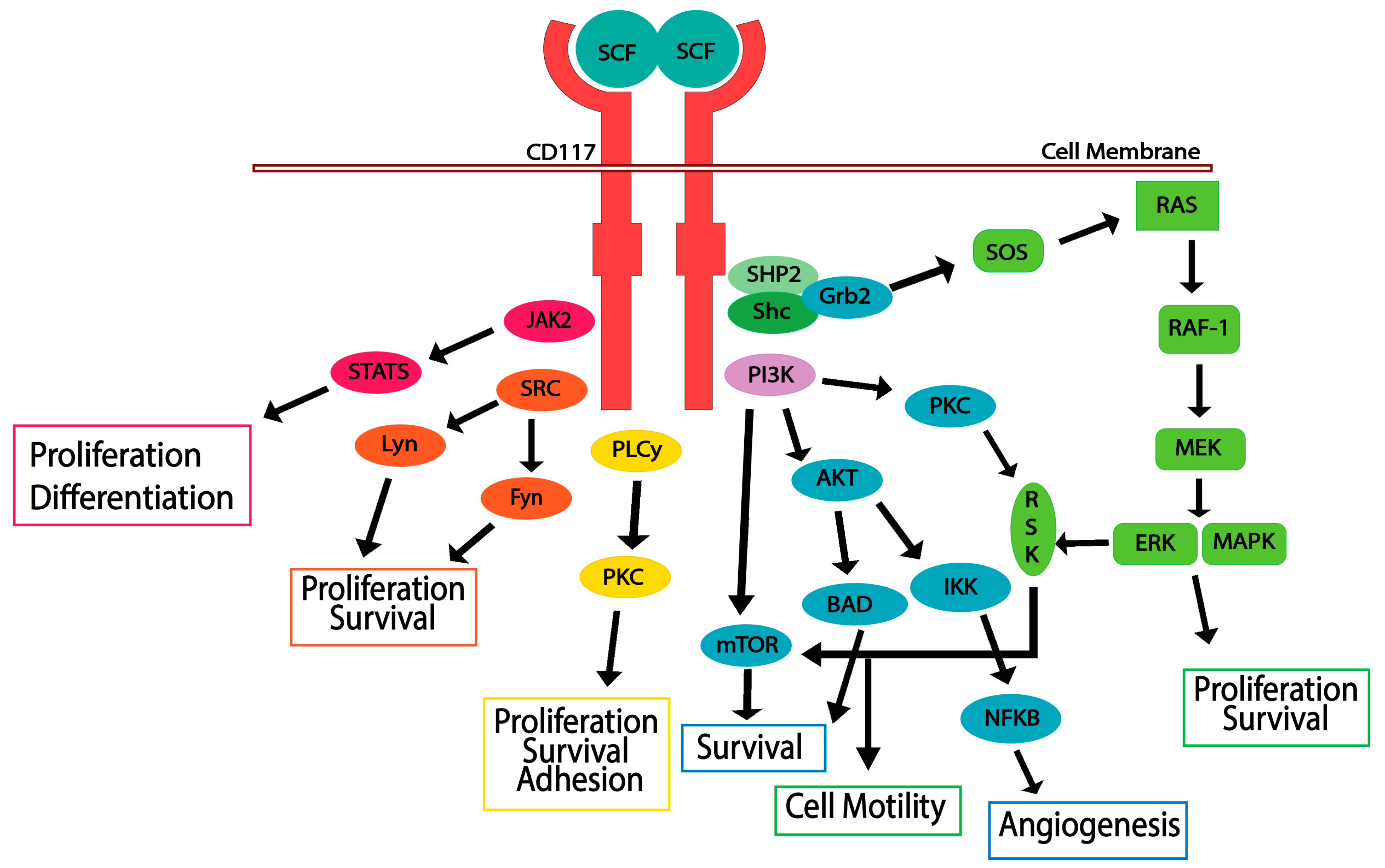
5. CD117 Activated Signaling Pathways
5.1. JAK/STAT Pathway
5.2. RAS/MAP Kinase Pathway
5.3. PI3-Kinase/Akt Pathway
5.4. SRC Family Kinase Pathways
5.5. PLCγ Pathway
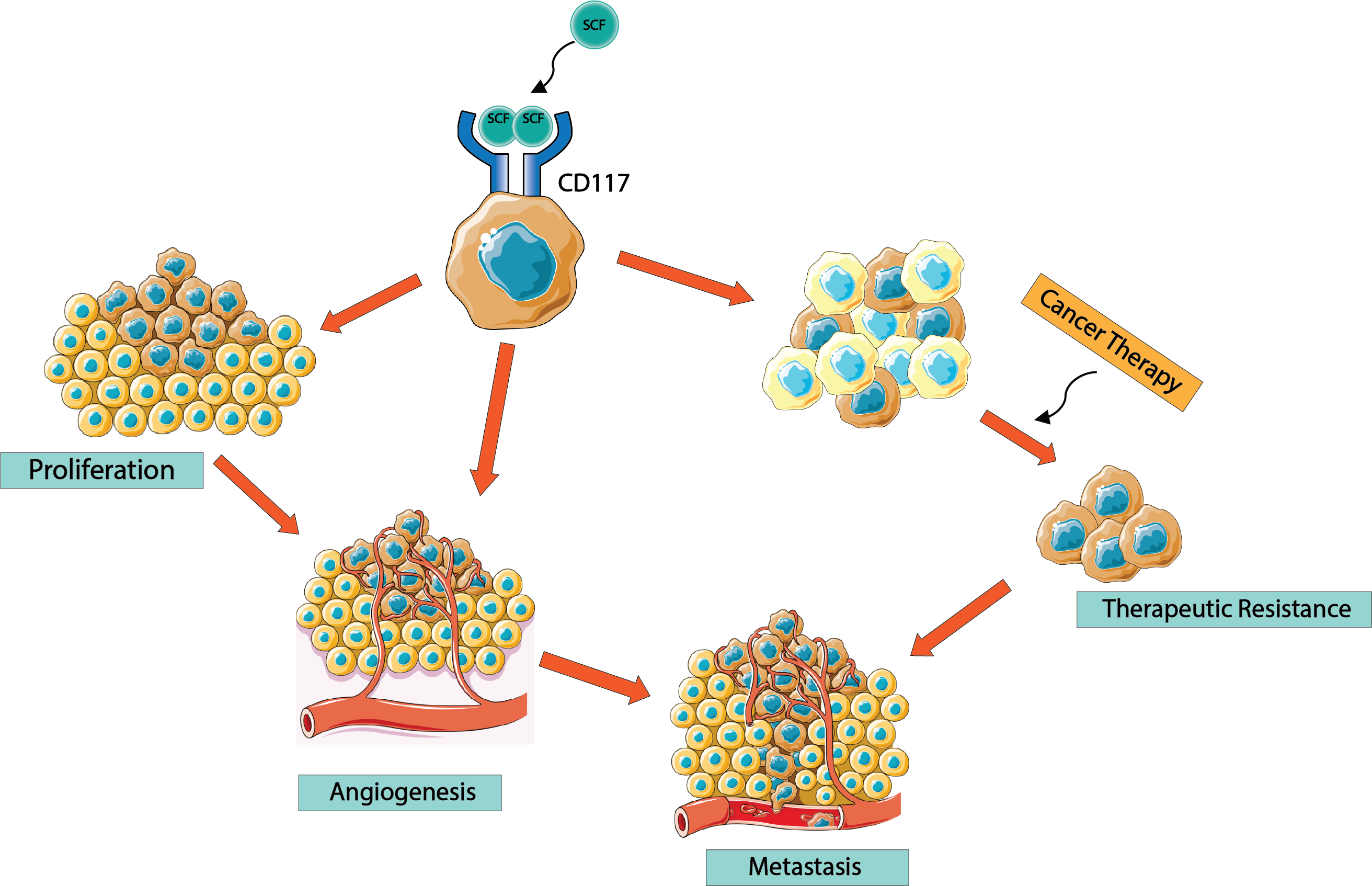
6. CD117 Regulation of Cancer Progression
7. CD117 Regulation of Cancer Cell “Stemness”
8. CD117 Resistance to Tyrosine Kinase Inhibitors
9. The Future of the SCF/CD117 Signaling Axis in Cancer Treatment
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lobo, N.A.; Shimono, Y.; Qian, D.; Clarke, M.F. The Biology of Cancer Stem Cells. Annu. Rev. Cell Dev. Biol. 2007, 23, 675–699. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.M.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells—Perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef] [PubMed]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Moltzahn, F.R.; Volkmer, J.P.; Rottke, D.; Ackermann, R. “Cancer stem cells”-lessons from Hercules to fight the Hydra. Urol. Oncol. 2008, 26, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.V.; Vanner, R.; Dirks, P.; Eaves, C.J. Cancer stem cells: An evolving concept. Nat. Rev. Cancer 2012, 12, 133. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Bonnet, D.; De Maria, R.; Lapidot, T.; Copland, M.; Melo, J.V.; Chomienne, C.; Ishikawa, F.; Schuringa, J.J.; Stassi, G.; et al. Cancer stem cell definitions and terminology: The devil is in the details. Nat. Rev. Cancer 2012, 12, 767–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, S.S.; He, J.Q.; Esparza, R.; Hutter, G.; Cheshier, S.H.; Weissman, I. Introduction: Cancer Stem Cells. In Cancer Stem Cells; Liu, H., Lathia, J.D., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 3–24. [Google Scholar]
- Kyjacova, L.; Hubackova, S.; Krejcikova, K.; Strauss, R.; Hanzlikova, H.; Dzijak, R.; Imrichova, T.; Simova, J.; Reinis, M.; Bartek, J.; et al. Radiotherapy-induced plasticity of prostate cancer mobilizes stem-like non-adherent, Erk signaling-dependent cells. Cell Death Differ. 2015, 22, 898–911. [Google Scholar] [CrossRef] [PubMed]
- Schilling, D.; Todenhöfer, T.; Hennenlotter, J.; Schwentner, C.; Fehm, T.; Stenzl, A. Isolated, disseminated and circulating tumour cells in prostate cancer. Nat. Rev. Urol. 2012, 9, 448–463. [Google Scholar] [CrossRef] [PubMed]
- Butler, T.P.; Gullino, P.M. Quantitation of cell shedding into efferent blood of mammary adenocarcinoma. Cancer Res. 1975, 35, 512–516. [Google Scholar] [PubMed]
- Chopra, A.S.; Liu, X.; Liu, H. Cancer Stem Cells: Metastasis and Evasion from the Host Immune System. In Cancer Stem Cells; Liu, H., Lathia, J.D., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 341–366. [Google Scholar]
- Van der Toom, E.E.; Verdone, J.E.; Pienta, K.J. Disseminated tumor cells and dormancy in prostate cancer metastasis. Curr. Opin. Biotechnol. 2016, 40, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.S.; Kerr, B.A. Prostate Cancer Stem Cell Markers Drive Progression, Therapeutic Resistance, and Bone Metastasis. Stem Cells Int. 2017, 2017, 8629234. [Google Scholar] [CrossRef] [PubMed]
- Kerr, B.A.; Miocinovic, R.; Smith, A.K.; West, X.Z.; Watts, K.E.; Alzayed, A.W.; Klink, J.C.; Mir, M.C.; Sturey, T.; Hansel, D.E.; et al. CD117+ cells in the circulation are predictive of advanced prostate cancer. Oncotarget 2015, 6, 1889–1897. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.S.; Foster, B.M.; Shi, L.; Mobley, M.E.; Elliot, P.; Kerr, B.A. CTC Marker CD117/c-kit Represents a Prostate Cancer Stem-Like Subpopulation Driving Progression, Migration, and TKI Resistance. bioRxiv 2018. [Google Scholar] [CrossRef]
- Wiesner, C.; Nabha, S.M.; Dos Santos, E.B.; Yamamoto, H.; Meng, H.; Melchior, S.W.; Bittinger, F.; Thüroff, J.W.; Vessella, R.L.; Cher, M.L.; et al. C-kit and its ligand stem cell factor: Potential contribution to prostate cancer bone metastasis. Neoplasia 2008, 10, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Landuzzi, L.; De Giovanni, C.; Nicoletti, G.; Rossi, I.; Ricci, C.; Astolfi, A.; Scopece, L.; Scotlandi, K.; Serra, M.; Bagnara, G.P.; et al. The metastatic ability of Ewing’s sarcoma cells is modulated by stem cell factor and by its receptor c-kit. Am. J. Pathol. 2000, 157, 2123–2131. [Google Scholar] [CrossRef]
- D’Auriol, L.; Mattei, M.G.; Andre, C.; Galibert, F. Localization of the human c-kit protooncogene on the q11-q12 region of chromosome 4. Hum. Genet. 1988, 78, 374–376. [Google Scholar] [CrossRef] [PubMed]
- Rönnstrand, L.; Lennartsson, J. KIT (v-Kit Hardy-Zuckerman 4 Feline Sarcoma Viral Oncogene Homolog). Available online: http://atlasgeneticsoncology.org/Genes/KITID127.html (accessed on 25 February 2018).
- Andre, C.; Hampe, A.; Lachaume, P.; Martin, E.; Wang, X.-P.; Manus, V.; Hu, W.-X.; Galibert, F. Sequence Analysis of Two Genomic Regions Containing the KIT and the FMS Receptor Tyrosine Kinase Genes. Genomics 1997, 39, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Kuang, W.J.; Yang-Feng, T.; Coussens, L.; Munemitsu, S.; Dull, T.J.; Chen, E.; Schlessinger, J.; Francke, U.; Ullrich, A. Human proto-oncogene c-kit: A new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J. 1987, 6, 3341–3351. [Google Scholar] [PubMed]
- Liang, J.; Wu, Y.L.; Chen, B.J.; Zhang, W.; Tanaka, Y.; Sugiyama, H. The C-Kit receptor-mediated signal transduction and tumor-related diseases. Int. J. Biol. Sci. 2013, 9, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.; Kazi, J.U.; Zhao, H.; Sun, J. Germline mutations of KIT in gastrointestinal stromal tumor (GIST) and mastocytosis. Cell Biosci. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Furitsu, T.; Tsujimura, T.; Tono, T.; Ikeda, H.; Kitayama, H.; Koshimizu, U.; Sugahara, H.; Butterfield, J.H.; Ashman, L.K.; Kanayama, Y.; et al. Identification of mutations in the coding sequence of the proto-oncogene c-kit in a human mast cell leukemia cell line causing ligand-independent activation of c-kit product. J. Clin. Investig. 1993, 92, 1736–1744. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Hirota, S. Kit as a human oncogenic tyrosine kinase. Cell. Mol. Life Sci. 2004, 61, 2924–2931. [Google Scholar] [CrossRef] [PubMed]
- Ashman, L.K. The biology of stem cell factor and its receptor C-kit. Int. J. Biochem. Cell Biol. 1999, 31, 1037–1051. [Google Scholar] [CrossRef]
- Agarwal, S.; Kazi, J.U.; Rönnstrand, L. Phosphorylation of the activation loop tyrosine 823 in c-Kit is crucial for cell survival and proliferation. J. Biol. Chem. 2013, 288, 22460–22468. [Google Scholar] [CrossRef] [PubMed]
- Lammie, A.; Drobnjak, M.; Gerald, W.; Saad, A.; Cote, R.; Cordon-Cardo, C. Expression of c-kit and kit ligand proteins in normal human tissues. J. Histochem. Cytochem. 1994, 42, 1417–1425. [Google Scholar] [CrossRef] [PubMed]
- Stankov, K.; Popovic, S.; Mikov, M. C-KIT signaling in cancer treatment. Curr. Pharm. Des. 2014, 20, 2849–2880. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.L.; Yarden, Y. Receptor Tyrosine Kinases: Family and Subfamilies; Springer International Publishing: Basel, Switzerland, 2015. [Google Scholar]
- Wheeler, D.L.; Yarden, Y. Receptor Tyrosine Kinases: Structure, Functions and Role in Human Disease; Springer: New York, NY, USA, 2015. [Google Scholar]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, D36–D42. [Google Scholar] [CrossRef] [PubMed]
- KIT GenBank Page. Available online: https://www.ncbi.nlm.nih.gov/gene/3815 (accessed on 26 February 2018).
- Voytyuk, O.; Lennartsson, J.; Mogi, A.; Caruana, G.; Courtneidge, S.; Ashman, L.K.; Rö, L. Src Family Kinases Are Involved in the Differential Signaling from Two Splice Forms of c-Kit. J. Biol. Chem. 2003, 278, 9159–9166. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.C.; Bai, Y.; Bandara, G.; Simakova, O.; Brittain, E.; Scott, L.; Dyer, K.D.; Klion, A.D.; Maric, I.; Gilfillan, A.M.; et al. KIT GNNK splice variants: Expression in systemic mastocytosis and influence on the activating potential of the D816V mutation in mast cells. Exp. Hematol. 2013, 41, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Caruana, G.; Cambareri, A.C.; Ashman, L.K. Isoforms of c-KIT differ in activation of signalling pathways and transformation of NIH3T3 Fibroblasts. Oncogene 1999, 18, 5573–5581. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, Y.; Sakurai, S.; Oguni, S.; Hironaka, M.; Saito, K. Alterations of the c-kit gene in testicular germ cell tumors. Cancer Sci. 2003, 94, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Crosier, P.S.; Ricciardi, S.T.; Hall, L.R.; Vitas, M.R.; Clark, S.C.; Crosier, K.E. Expression of Isoforms of the Human Receptor Tyrosine Kinase c-kit in Leukemic Cell Lines and Acute Myeloid Leukemia. Blood 1993, 82, 1151–1158. [Google Scholar] [PubMed]
- Longley, B.J.; Reguera, M.J.; Ma, Y. Classes of c-KIT activating mutations: Proposed mechanisms of action and implications for disease classification and therapy. Leuk. Res. 2001, 25, 571–576. [Google Scholar] [CrossRef]
- Sanlorenzo, M.; Vujic, I.; Posch, C.; Ma, J.; Lin, K.; Lai, K.; Oses-Prieto, J.A.; Chand, S.; Rodriguez-Peralto, J.L.; Burlingame, A.; et al. Oncogenic KIT mutations in different exons lead to specific changes in melanocyte phospho-proteome HHS Public Access. J. Proteom. 2016, 144, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, H.J.; Figueira, M.I.; Socorro, S. The stem cell factor (SCF)/c-KIT signalling in testis and prostate cancer. J. Cell Commun. Signal. 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.; Lasota, J. KIT (CD117): A review on expression in normal and neoplastic tissues, and mutations and their clinicopathologic correlation. Appl. Immunohistochem. Mol. Morphol. 2005, 13, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Stem Cell Basics I. Available online: https://stemcells.nih.gov/ (accessed on 26 February 2018).
- Burns, C.E.; Zon, L.I. Portrait of a stem cell. Dev. Cell 2002, 3, 612–613. [Google Scholar] [CrossRef]
- Durand, C.; Charbord, P. Stem Cell Biology and Regenerative Medicine; River Publishers: Gistrup, Denmark, 2015. [Google Scholar]
- Lin, H.; Schagat, T. Neuroblasts: A model for the asymmetric division of stem cells. Trends Genet. 1997, 13, 33–39. [Google Scholar] [CrossRef]
- Petritsch, C.; Shen, X. Asymmetric Division of Cancer Stem Cells. In Cancer Stem Cells; Elsevier: Amsterdam, The Netherlands, 2016; pp. 285–315. [Google Scholar]
- Loeffler, D.; Schroeder, T. Asymmetric cell division of hematopoietic stem cells. Exp. Hematol. 2015, 43, S77. [Google Scholar] [CrossRef]
- Klonisch, T.; Wiechec, E.; Hombach-Klonisch, S.; Ande, S.R.; Wesselborg, S.; Schulze-Osthoff, K.; Los, M. Cancer stem cell markers in common cancers—Therapeutic implications. Trends Mol. Med. 2008, 14, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Leong, K.G.; Wang, B.E.; Johnson, L.; Gao, W.Q. Generation of a prostate from a single adult stem cell. Nature 2008, 456, 804–808. [Google Scholar] [CrossRef] [PubMed]
- Petryszak, R.; Keays, M.; Tang, Y.A.; Fonseca, N.A.; Barrera, E.; Burdett, T.; Füllgrabe, A.; Fuentes, A.M.-P.; Jupp, S.; Koskinen, S.; et al. Expression Atlas update—An integrated database of gene and protein expression in humans, animals and plants. Nucleic Acids Res. 2016, 44, D746–D752. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the Human Tissue-specific Expression by Genome-wide Integration of Transcriptomics and Antibody-based Proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Ashman, B.L.K.; Cambareri, A.C.; To, L.B.; Levinsky, R.J.; Juttner, C.A. Expression of the YB5.BS Antigen (c-kit proto-oncogene product) in Normal Human Bone Marrow. Blood 2017, 78, 30–37. [Google Scholar]
- Acar, M.; Kocherlakota, K.S.; Murphy, M.M.; Peyer, J.G.; Oguro, H.; Inra, C.N.; Jaiyeola, C.; Zhao, Z.; Luby-Phelps, K.; Morrison, S.J. Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature 2015, 526, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Ema, H.; Takano, H.; Sudo, K.; Nakauchi, H. In vitro self-renewal division of hematopoietic stem cells. J. Exp. Med. 2000, 192, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Matsui, J.; Wakabayashi, T.; Asada, M.; Yoshimatsu, K.; Okada, M. Stem Cell Factor/c-kit Signaling Promotes the Survival, Migration, and Capillary Tube Formation of Human Umbilical Vein Endothelial Cells. J. Biol. Chem. 2004, 279, 18600–18607. [Google Scholar] [CrossRef] [PubMed]
- Linnekin, D. Early signaling pathways activated by c-Kit in hematopoietic cells. Int. J. Biochem. Cell Biol. 1999, 31, 1053–1074. [Google Scholar] [CrossRef]
- Kimura, Y.; Ding, B.; Imai, N.; Nolan, D.J.; Butler, J.M.; Rafii, S. c-Kit-mediated functional positioning of stem cells to their niches is essential for maintenance and regeneration of adult hematopoiesis. PLoS ONE 2011, 6, e26918. [Google Scholar] [CrossRef] [PubMed]
- KITLG GenBank Page. Available online: https://www.ncbi.nlm.nih.gov/gene/4254 (accessed on 26 February 2018).
- Reber, L.; Da Silva, C.A.; Frossard, N. Stem cell factor and its receptor c-Kit as targets for inflammatory diseases. Eur. J. Pharmacol. 2006, 533, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, K.; Williams, D.A.; Gotoh, A.; Nishimaki, J.; Broxmeyer, H.E.; Toyama, K. Membrane-bound Steel factor induces more persistent tyrosine kinase activation and longer life span of c-kit gene-encoded protein than its soluble form. Blood 1995, 85, 641–649. [Google Scholar] [PubMed]
- Longley, B.J.; Tyrrell, L.; Ma, Y.; Williams, D.A.; Halaban, R.; Langley, K.; Lu, H.S.; Schechter, N.M. Chymase cleavage of stem cell factor yields a bioactive, soluble product. Proc. Natl. Acad. Sci. USA 1997, 94, 9017–9021. [Google Scholar] [CrossRef] [PubMed]
- Clarke, K.; Basserb, R.L. The Role of Platelet Growth Factors in Cancer Therapy. Stem Cells 1996, 14, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Mizuochi, C.; Horio, Y.; Nakao, K.; Akashi, K.; Sugiyama, D. Regulation of hematopoietic cell clusters in the placental niche through SCF/Kit signaling in embryonic mouse. Development 2010, 137, 3941–3952. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Saunders, T.L.; Enikolopov, G.; Morrison, S.J. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 2012, 481, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Broudy, V.C. Stem cell factor and hematopoiesis. Blood 1997, 90, 1345–1364. [Google Scholar] [PubMed]
- Calvi, L.M.; Link, D.C. The hematopoietic stem cell niche in homeostasis and disease. Blood 2015, 126, 2443–2451. [Google Scholar] [CrossRef] [PubMed]
- Asada, N.; Kunisaki, Y.; Pierce, H.; Wang, Z.; Fernandez, N.F.; Birbrair, A.; Ma’ayan, A.; Frenette, P.S. Differential cytokine contributions of perivascular haematopoietic stem cell niches. Nat. Cell Biol. 2017, 19, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Kacena, M.A.; Gundberg, C.M.; Horowitz, M.C. A reciprocal regulatory interaction between megakaryocytes, bone cells, and hematopoietic stem cells. Bone 2006, 39, 978–984. [Google Scholar] [CrossRef] [PubMed]
- Inra, C.N.; Zhou, B.O.; Acar, M.; Murphy, M.M.; Richardson, J.; Zhao, Z.; Morrison, S.J. A perisinusoidal niche for extramedullary haematopoiesis in the spleen. Nature 2015, 527, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Kollet, O.; Shivtiel, S.; Chen, Y.-Q.; Suriawinata, J.; Thung, S.N.; Dabeva, M.D.; Kahn, J.; Spiegel, A.; Dar, A.; Samira, S.; et al. HGF, SDF-1, and MMP-9 are involved in stress-induced human CD34+ stem cell recruitment to the liver. J. Clin. Investig. 2003, 112, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Philo, J.S.; Wen, J.; Wypych, J.; Schwartz, M.G.; Mendiaz, E.A.; Langley, K.E. Human stem cell factor dimer forms a complex with two molecules of the extracellular domain of its receptor, Kit. J. Biol. Chem. 1996, 271, 6895–6902. [Google Scholar] [CrossRef] [PubMed]
- Blume-Jensen, P.; Claesson-Welsh, L.; Siegbahn, A.; Zsebo, K.M.; Westermark, B.; Heldin, C.H. Activation of the human c-kit product by ligand-induced dimerization mediates circular actin reorganization and chemotaxis. EMBO J. 1991, 10, 4121–4128. [Google Scholar] [PubMed]
- Blechman, J.M.; Lev, S.; Givol, D.; Yarden, Y. Structure-function analyses of the kit receptor for the steel factor. Stem Cells 1993, 11 (Suppl. S2), 12–21. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.; Wu, G.; Mendiaz, E.A.; Syed, R.; Wypych, J.; Toso, R.; Mann, M.B.; Boone, T.C.; Narhi, L.O.; Lu, H.S.; et al. The Majority of Stem Cell Factor Exists as Monomer under Physiological Conditions. J. Biol. Chem. 1997, 272, 6406–6415. [Google Scholar] [CrossRef] [PubMed]
- Weiler, S.R.; Mou, S.; Deberry, C.S.; Keller, J.R.; Ruscetti, F.W.; Ferris, D.K.; Longo, D.L.; Linnekin, D. JAK2 Is Associated With the c-kit Proto-oncogene Product and Is Phosphorylated in Response to Stem Cell Factor. Blood 1996, 87, 3688–3693. [Google Scholar] [PubMed]
- MOUb, S.; Deberry, C.S.; Weiler, S.R.; KELLERb, J.R.; Rusce, F.W.; Longo, D.L. Stem Cell Factor, the JAK-STAT Pathway and Signal Transduction. Leuk. Lymphoma 1997, 27, 439–444. [Google Scholar]
- Thömmes, K.; Lennartsson, J.; Carlberg, M.; Rönnstrand, L. Identification of Tyr-703 and Tyr-936 as the primary association sites for Grb2 and Grb7 in the c-Kit/stem cell factor receptor. Biochem. J. 1999, 341 Pt 1, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Duronio, V.; Welham, M.J.; Abraham, S.; Dryden, P.; Schrader, J.W. p21ras activation via hemopoietin receptors and c-kit requires tyrosine kinase activity but not tyrosine phosphorylation of p21ras {GTPase-activating} protein. Proc. Natl. Acad. Sci. USA 1992, 89, 1587–1591. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Kurosaki, T. Regulation of lymphocyte fate by Ras/ERK signals. Cell Cycle 2008, 7, 3634–3640. [Google Scholar] [CrossRef] [PubMed]
- Kuang, D.; Zhao, X.; Xiao, G.; Ni, J.; Feng, Y.; Wu, R.; Wang, G. Stem cell factor/c-kit signaling mediated cardiac stem cell migration via activation of p38 MAPK. Basic Res. Cardiol. 2008, 103, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Pedersen, M.; Rönnstrand, L. Gab2 is involved in differential phosphoinositide 3-kinase signaling by two splice forms of c-Kit. J. Biol. Chem. 2008, 283, 27444–27451. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Mizuki, M.; Ikeda, H.; Tsujimura, T.; Matsumura, I.; Nakano, K.; Daino, H.; Honda, Z.-I.; Sonoyama, J.; Shibayama, H.; et al. Critical roles of c-Kit tyrosine residues 567 and 719 in stem cell factor–induced chemotaxis: Contribution of src family kinase and PI3-kinase on calcium mobilization and cell migration. Blood 2002, 99, 3342–3349. [Google Scholar] [CrossRef] [PubMed]
- Linnekin, D.; DeBerry, C.S.; Mou, S. Lyn associates with the juxtamembrane region of c-Kit and is activated by stem cell factor in hematopoietic cell lines and normal progenitor cells. J. Biol. Chem. 1997, 272, 27450–27455. [Google Scholar] [CrossRef]
- Saleem, M.; Babaei, A.; Press, D. Receptor tyrosine kinase (c-Kit) inhibitors: A potential therapeutic target in cancer cells. Drug Des. Dev. Ther. 2016, 10, 2443–2459. [Google Scholar] [CrossRef]
- Shivakrupa, R.; Linnekin, D. Lyn contributes to regulation of multiple Kit-dependent signaling pathways in murine bone marrow mast cells. Cell Signal. 2005, 17, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Sette, C.; Paronetto, M.P.; Barchi, M.; Bevilacqua, A.; Geremia, R.; Rossi, P. Tr-kit-induced resumption of the cell cycle in mouse eggs requires activation of a Src-like kinase. EMBO J. 2002, 21, 5386–5395. [Google Scholar] [CrossRef] [PubMed]
- Phane Maddens, S.; Charruyer, A.; Plo, I.; Dubreuil, P.; Berger, S.; Salles, B.; Laurent, G.; Jaffré, J.-P. Kit signaling inhibits the sphingomyelin-ceramide pathway through PLC gamma 1: Implication in stem cell factor radioprotective effect. Blood 2002, 100, 1294–1301. [Google Scholar]
- Lennartsson, J.; Wernstedt, C.; Engström, U.; Hellman, U.; Rönnstrand, L. Identification of Tyr900 in the kinase domain of c-Kit as a Src-dependent phosphorylation site mediating interaction with c-Crk. Exp. Cell Res. 2003, 288, 110–118. [Google Scholar] [CrossRef]
- Gommerman, J.L.; Sittaro, D.; Klebasz, N.Z.; Williams, D.A.; Berger, S.A. Differential stimulation of c-Kit mutants by membrane-bound and soluble Steel Factor correlates with leukemic potential. Blood 2000, 96, 3734–3742. [Google Scholar] [PubMed]
- Lennartsson, J.; Jelacic, T.; Linnekin, D.; Shivakrupa, R. Normal and Oncogenic Forms of the Receptor Tyrosine Kinase Kit. Stem Cells 2005, 23, 16–43. [Google Scholar] [CrossRef] [PubMed]
- Tay, C.M.; Ong, C.W.; Lee, V.K.M.; Pang, B. {KIT} gene mutation analysis in solid tumours: Biology, clincial applications and trends in diagnostic reporting. Pathology 2013, 45, 127–137. [Google Scholar] [PubMed]
- Medinger, M.; Kleinschmidt, M.; Mross, K.; Wehmeyer, B.; Unger, C.; Schaefer, H.-E.E.; Weber, R.; Azemar, M. c-kit (CD117) expression in human tumors and its prognostic value: An immunohistochemical analysis. Pathol. Oncol. Res. 2010, 16, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.-Y.; Grabellus, F.; Weber, F.; Zhou, Y.; Tan, Y.-S.; Li, J.; Shen, K.-T.; Qin, J.; Sun, Y.-H.; Qin, X.-Y.; et al. Impact of {KIT} and {PDGFRA} gene mutations on prognosis of patients with gastrointestinal stromal tumors after complete primary tumor resection. J. Gastrointest. Surg. 2009, 13, 1583–1592. [Google Scholar] [CrossRef] [PubMed]
- Andersson, J.; Bümming, P.; Meis-Kindblom, J.M.; Sihto, H.; Nupponen, N.; Joensuu, H.; Odén, A.; Gustavsson, B.; Kindblom, L.-G.; Nilsson, B. Gastrointestinal stromal tumors with {KIT} exon 11 deletions are associated with poor prognosis. Gastroenterology 2006, 130, 1573–1581. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, A.; Rutkowski, P.; Piskorz, A.; Ciwoniuk, M.; Osuch, C.; Bylina, E.; Sygut, J.; Chosia, M.; Rys, J.; Urbanczyk, K.; et al. Prognostic value of {KIT/PDGFRA} mutations in gastrointestinal stromal tumours ({GIST)}: Polish Clinical {GIST} Registry experience. Ann. Oncol. 2012, 23, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Søreide, K.; Sandvik, O.M.; Søreide, J.A.; Gudlaugsson, E.; Mangseth, K.; Haugland, H.K. Tyrosine-kinase mutations in {c-KIT} and {PDGFR-alpha} genes of imatinib na{“\i}ve adult patients with gastrointestinal stromal tumours ({GISTs}) of the stomach and small intestine: Relation to tumour-biological risk-profile and long-term outcome. Clin. Transl. Oncol. 2012, 14, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Kosemehmetoglu, K.; Kaygusuz, G.; Fritchie, K.; Aydin, O.; Yapicier, O.; Coskun, O.; Karatayli, E.; Boyacigil, S.; Guler, G.; Dervisoglu, S.; et al. Clinical and pathological characteristics of gastrointestinal stromal tumor (GIST) metastatic to bone. Virchows Arch. 2017, 471, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Penzel, R.; Aulmann, S.; Moock, M.; Schwarzbach, M.; Rieker, R.J.; Mechtersheimer, G. The location of {KIT} and {PDGFRA} gene mutations in gastrointestinal stromal tumours is site and phenotype associated. J. Clin. Pathol. 2005, 58, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Burger, K.; den Bakker, M.A.; Kros, J.M.; de Bruin, A.M.; Oosterhuis, W.; van den Ingh, H.F.G.M.; van der Harst, E.; de Schipper, H.P.; Wiemer, E.A.C.; Nooter, K. Activating mutations in {c-KIT} and {PDGFR$α$} are exclusively found in gastrointestinal stromal tumors and not in other tumors overexpressing these imatinib mesylate target genes. Cancer Biol. Ther. 2005, 4, 1270–1274. [Google Scholar] [CrossRef] [PubMed]
- Stemberger-Papić, S.; Vrdoljak-Mozetic, D.; Ostojić, D.V.; Rubesa-Mihaljević, R.; Krigtofić, I.; Brncić-Fisher, A.; Kragević, M.; Eminović, S. Expression of {CD133} and {CD117} in 64 Serous Ovarian Cancer Cases. Coll. Antropol. 2015, 39, 745–753. [Google Scholar] [PubMed]
- Burgos-Ojeda, D.; Rueda, B.R.; Buckanovich, R.J. Ovarian cancer stem cell markers: Prognostic and therapeutic implications. Cancer Lett. 2012, 322, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Zeng, J.; Liang, B.; Zhao, Z.; Sun, L.; Cao, D.; Yang, J.; Shen, K. Ovarian cancer cells with the {CD117} phenotype are highly tumorigenic and are related to chemotherapy outcome. Exp. Mol. Pathol. 2011, 91, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Foroozan, M.; Roudi, R.; Abolhasani, M.; Gheytanchi, E.; Mehrazma, M. Clinical significance of endothelial cell marker CD34 and mast cell marker CD117 in prostate adenocarcinoma. Pathol. Res. Pract. 2017, 213, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Mainetti, L.E.; Zhe, X.; Diedrich, J.; Saliganan, A.D.; Cho, W.J.; Cher, M.L.; Heath, E.; Fridman, R.; Kim, H.-R.C.; Bonfil, R.D. Bone-induced c-kit expression in prostate cancer: A driver of intraosseous tumor growth. Int. J. Cancer 2015, 136, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Atala, A. Re: {Bone-Induced} c-Kit Expression in Prostate Cancer: A Driver of Intraosseous Tumor Growth. J. Urol. 2015, 194, 260. [Google Scholar] [CrossRef] [PubMed]
- Hines, S.J.; Organ, C.; Kornstein, M.J.; Krystal, G.W. Coexpression of the c-kit and stem cell factor genes in breast carcinomas. Cell Growth Differ. 1995, 6, 769–779. [Google Scholar] [PubMed]
- Krystal, G.W.; Hines, S.J.; Organ, C.P. Autocrine growth of small cell lung cancer mediated by coexpression of c-kit and stem cell factor. Cancer Res. 1996, 56, 370–376. [Google Scholar] [PubMed]
- Schmidt-Hieber, M.; Perez-Andres, M.; Paiva, B.; Flores-Montero, J.; Perez, J.J.; Gutierrez, N.C.; Vidriales, M.-B.; Matarraz, S.; San Miguel, J.F.; Orfao, A. {CD117} expression in gammopathies is associated with an altered maturation of the myeloid and lymphoid hematopoietic cell compartments and favorable disease features. Haematologica 2010, 96, 328–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bataille, R.; Pellat-Deceunynck, C.; Robillard, N.; Avet-Loiseau, H.; Harousseau, J.-L.; Moreau, P. {CD117} (c-kit) is aberrantly expressed in a subset of {MGUS} and multiple myeloma with unexpectedly good prognosis. Leuk. Res. 2008, 32, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Wang, H.; Tao, Q.; Zhang, C.; Yang, D.; Qin, H.; Xiong, S.; Tao, L.; Wu, F.; Zhang, J.; et al. Absence of both {CD56} and {CD117} expression on malignant plasma cells is related with a poor prognosis in patients with newly diagnosed multiple myeloma. Leuk. Res. 2016, 40, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Chen, Y.; Wu, Q.; Wang, Z.; Lu, J. Prognostic value of {CD117} in cancer: A meta-analysis. Int. J. Clin. Exp. Pathol. 2014, 7, 1012–1021. [Google Scholar] [PubMed]
- Kuonen, F.; Laurent, J.; Secondini, C.; Lorusso, G.; Stehle, J.-C.; Rausch, T.; Faes-Van’t Hull, E.; Bieler, G.; Alghisi, G.-C.; Schwendener, R.; et al. Inhibition of the Kit ligand/c-Kit axis attenuates metastasis in a mouse model mimicking local breast cancer relapse after radiotherapy. Clin. Cancer Res. 2012, 18, 4365–4374. [Google Scholar] [CrossRef] [PubMed]
- Sakabe, T.; Azumi, J.; Haruki, T.; Umekita, Y.; Nakamura, H.; Shiota, G. {CD117} expression is a predictive marker for poor prognosis in patients with non-small cell lung cancer. Oncol. Lett. 2017, 13, 3703–3708. [Google Scholar] [CrossRef] [PubMed]
- Foster, R.; Buckanovich, R.J.; Rueda, B.R. Ovarian cancer stem cells: Working towards the root of stemness. Cancer Lett. 2013, 338, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.; Krishnamoorthy, N.; Oriss, T.B.; Ray, A. Signaling of c-kit in dendritic cells influences adaptive immunity. Ann. N. Y. Acad. Sci. 2010, 1183, 104–122. [Google Scholar] [CrossRef] [PubMed]
- Vanegas, N.-D.P.; Vernot, J.-P. Loss of quiescence and self-renewal capacity of hematopoietic stem cell in an in vitro leukemic niche. Exp. Hematol. Oncol. 2017, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-Y.; Jiang, L.-J.; Chen, L.; Ding, M.-L.; Guo, H.-Z.; Zhang, W.; Zhang, H.-X.; Ma, X.-D.; Liu, X.-Z.; Xi, X.-D.; et al. {RIG-I} modulates Src-mediated {AKT} activation to restrain leukemic stemness. Mol. Cell 2014, 53, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Anonymous. {RIG-I} inhibits {SRC-mediated} {AKT/mTOR} signaling and stemness in {AML}. Cancer Discov. 2014, 4, OF19. [Google Scholar]
- Jiang, T.; Qiu, Y. Interaction between Src and a C-terminal Proline-rich Motif of Akt Is Required for Akt Activation. J. Biol. Chem. 2003, 278, 15789–15793. [Google Scholar] [CrossRef] [PubMed]
- Tabu, K.; Kimura, T.; Sasai, K.; Wang, L.; Bizen, N.; Nishihara, H.; Taga, T.; Tanaka, S. Analysis of an alternative human {CD133} promoter reveals the implication of {Ras/ERK} pathway in tumor stem-like hallmarks. Mol. Cancer 2010, 9, 39. [Google Scholar] [CrossRef] [PubMed]
- Ivanovic, Z.; Vlaski-Lafarge, M. Evolutionary Origins of Stemness. In Anaerobiosis and Stemness; Elsevier Science & Technology Books: Amsterdam, The Netherlands, 2016; pp. 177–209. [Google Scholar]
- Cai, J.; Weiss, M.L.; Rao, M.S. In search of “stemness”. Exp. Hematol. 2004, 32, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, N.; Iqbal, N. Imatinib: A breakthrough of targeted therapy in cancer. Chemother. Res. Pract. 2014, 2014, 357027. [Google Scholar] [CrossRef] [PubMed]
- Al-Hadiya, B.M.H.; Bakheit, A.H.H.; Abd-Elgalil, A.A. Imatinib mesylate. Profiles Drug Subst. Excip. Relat. Methodol. 2014, 39, 265–297. [Google Scholar] [PubMed]
- Joensuu, H.; Roberts, P.J.; Sarlomo-Rikala, M.; Andersson, L.C.; Tervahartiala, P.; Tuveson, D.; Silberman, S.L.; Capdeville, R.; Dimitrijevic, S.; Druker, B.; et al. Effect of the Tyrosine Kinase Inhibitor {STI571} in a Patient with a Metastatic Gastrointestinal Stromal Tumor. N. Engl. J. Med. 2001, 344, 1052–1056. [Google Scholar] [CrossRef] [PubMed]
- Van Oosterom, A.T.; Judson, I.; Verweij, J.; Stroobants, S.; di Paola, E.; Dimitrijevic, S.; Martens, M.; Webb, A.; Sciot, R.; Van Glabbeke, M.; et al. Safety and efficacy of imatinib ({STI571}) in metastatic gastrointestinal stromal tumours: A phase {I} study. Lancet 2001, 358, 1421–1423. [Google Scholar] [CrossRef]
- Tuveson, D.A.; Willis, N.A.; Jacks, T.; Griffin, J.D.; Singer, S.; Fletcher, C.D.; Fletcher, J.A.; Demetri, G.D. {STI571} inactivation of the gastrointestinal stromal tumor {c-KIT} oncoprotein: Biological and clinical implications. Oncogene 2001, 20, 5054–5058. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, V.P.; Cavnar, M.J.; Zeng, S.; Bamboat, Z.M.; Ocuin, L.M.; Obaid, H.; Sorenson, E.C.; Popow, R.; Ariyan, C.; Rossi, F.; et al. Imatinib potentiates antitumor {T} cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat. Med. 2011, 17, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
- Seifert, A.M.; Zeng, S.; Zhang, J.Q.; Kim, T.S.; Cohen, N.A.; Beckman, M.J.; Medina, B.D.; Maltbaek, J.H.; Loo, J.K.; Crawley, M.H.; et al. {PD-1/PD-L1} Blockade Enhances T-cell Activity and Antitumor Efficacy of Imatinib in Gastrointestinal Stromal Tumors. Clin. Cancer Res. 2017, 23, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Seifert, A.M.; Kim, T.S.; Greer, J.B.; Cohen, N.A.; Beckman, M.J.; Santamaria-Barria, J.A.; Zeng, S.; Crawley, M.H.; Green, B.L.; DeMatteo, R.P. {PD-1/PD-L1} Blockade Enhances the Efficacy of Imatinib in Gastrointestinal Stromal Tumor ({GIST}). J. Am. Coll. Surg. 2014, 219, S129. [Google Scholar] [CrossRef]
- Edris, B.; Willingham, S.B.; Weiskopf, K.; Volkmer, A.K.; Volkmer, J.-P.; Mühlenberg, T.; Montgomery, K.D.; Contreras-Trujillo, H.; Czechowicz, A.; Fletcher, J.A.; et al. {Anti-KIT} monoclonal antibody inhibits imatinib-resistant gastrointestinal stromal tumor growth. Proc. Natl. Acad. Sci. USA 2013, 110, 3501–3506. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; van Oosterom, A.T.; Garrett, C.R.; Blackstein, M.E.; Shah, M.H.; Verweij, J.; McArthur, G.; Judson, I.R.; Heinrich, M.C.; Morgan, J.A.; et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial. Lancet 2006, 368, 1329–1338. [Google Scholar] [CrossRef]
- Demetri, G.D.; Reichardt, P.; Kang, Y.-K.; Blay, J.-Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; von Mehren, M.; Joensuu, H.; et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib ({GRID)}: An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 295–302. [Google Scholar] [CrossRef]
- Parikh, P.M.; Gupta, S. Management of gastrointestinal stromal tumor: The imatinib era and beyond. Indian J. Cancer 2013, 50, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; Heinrich, M.C.; Fletcher, J.A.; Fletcher, C.D.M.; den Abbeele, A.D.; Corless, C.L.; Antonescu, C.R.; George, S.; Morgan, J.A.; Chen, M.H.; et al. Molecular target modulation, imaging, and clinical evaluation of gastrointestinal stromal tumor patients treated with sunitinib malate after imatinib failure. Clin. Cancer Res. 2009, 15, 5902–5909. [Google Scholar] [CrossRef] [PubMed]
- Gajiwala, K.S.; Wu, J.C.; Christensen, J.; Deshmukh, G.D.; Diehl, W.; DiNitto, J.P.; English, J.M.; Greig, M.J.; He, Y.-A.; Jacques, S.L.; et al. KIT kinase mutants show unique mechanisms of drug resistance to imatinib and sunitinib in gastrointestinal stromal tumor patients. Proc. Natl. Acad. Sci. USA 2009, 106, 1542–1547. [Google Scholar] [CrossRef] [PubMed]
- Saglio, G.; Kim, D.-W.; Issaragrisil, S.; le Coutre, P.; Etienne, G.; Lobo, C.; Pasquini, R.; Clark, R.E.; Hochhaus, A.; Hughes, T.P.; et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N. Engl. J. Med. 2010, 362, 2251–2259. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; Hochhaus, A.; Saglio, G.; De Souza, C.; Flinn, I.W.; Stenke, L.; Goh, Y.-T.; Rosti, G.; Nakamae, H.; Gallagher, N.J.; et al. Nilotinib versus imatinib for the treatment of patients with newly diagnosed chronic phase, Philadelphia chromosome-positive, chronic myeloid leukaemia: 24-month minimum follow-up of the phase 3 randomised {ENESTnd} trial. Lancet Oncol. 2011, 12, 841–851. [Google Scholar] [CrossRef]
- Reichardt, P.; Blay, J.-Y.; Gelderblom, H.; Schlemmer, M.; Demetri, G.D.; Bui-Nguyen, B.; McArthur, G.A.; Yazji, S.; Hsu, Y.; Galetic, I.; et al. Phase {III} study of nilotinib versus best supportive care with or without a {TKI} in patients with gastrointestinal stromal tumors resistant to or intolerant of imatinib and sunitinib. Ann. Oncol. 2012, 23, 1680–1687. [Google Scholar] [CrossRef] [PubMed]
- Drugbank Database. Available online: https://www.drugbank.ca/ (accessed on 24 November 2017).
- Juurikivi, A.; Sandler, C.; Lindstedt, K.A.; Kovanen, P.T.; Juutilainen, T.; Leskinen, M.J.; Mäki, T.; Eklund, K.K. Inhibition of c-kit tyrosine kinase by imatinib mesylate induces apoptosis in mast cells in rheumatoid synovia: A potential approach to the treatment of arthritis. Ann. Rheum. Dis. 2005, 64, 1126–1131. [Google Scholar] [CrossRef] [PubMed]
- Galanis, A.; Levis, M. Inhibition of c-Kit by tyrosine kinase inhibitors. Haematologica 2015, 100, e77-9. [Google Scholar] [CrossRef] [PubMed]
- Di Gion, P.; Kanefendt, F.; Lindauer, A.; Scheffler, M.; Doroshyenko, O.; Fuhr, U.; Wolf , J.; Jaehde, U. Clinical Pharmacokinetics of Tyrosine Kinase Inhibitors. Clin. Pharmacokinet. 2011, 50, 551–603. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Niu, H.; Minkin, P.; Orwick, S.; Shimada, A.; Inaba, H.; Dahl, G.V.H.; Rubnitz, J.; Baker, S.D. Comparison of antitumor effects of multitargeted tyrosine kinase inhibitors in acute myelogenous leukemia. Mol. Cancer Ther. 2008, 7, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Andersson, P.; Von Euler, M.; Beckert, M. Comparable pharmacokinetics of 85 mg RightSize nilotinib (XS003) and 150 mg Tasigna in healthy volunteers using a hybrid nanoparticle-based formulation platform for protein kinase inhibitors. J. Clin. Oncol. 2014, 32 (Suppl. S15), e13551. [Google Scholar] [CrossRef]
- Wong, S.-F. New dosing schedules of dasatinib for CML and adverse event management. J. Hematol. Oncol. 2009, 2, 10. [Google Scholar] [CrossRef] [PubMed]
- Santos, F.P.S.; Ravandi, F. Advances in treatment of chronic myelogenous leukemia—New treatment options with tyrosine kinase inhibitors. Leuk. Lymphoma 2009, 50 (Suppl. S2), 16–26. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tortorici, M.A.; Garrett, M.; Hee, B.; Klamerus, K.J.; Pithavala, Y.K. Clinical Pharmacology of Axitinib. Clin. Pharmacokinet. 2013, 52, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Bellesoeur, A.; Carton, E.; Alexandre, J.; Goldwasser, F.; Huillard, O. Axitinib in the treatment of renal cell carcinoma: Design, development, and place in therapy. Drug Des. Dev. Ther. 2017, 11, 2801–2811. [Google Scholar] [CrossRef] [PubMed]
- Masitinib (Also Known as Kinavet® and Masivet®)—MSAA: The Multiple Sclerosis Association of America. Available online: https://mymsaa.org/publications/msresearch-update-2017/masitinib (accessed on 18 January 2018).
- Bellamy, F.; Bader, T.; Moussy, A.; Hermine, O. Pharmacokinetics of masitinib in cats. Vet. Res. Commun. 2009, 33, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Dubreuil, P.; Letard, S.; Ciufolini, M.; Gros, L.; Humbert, M.; Castéran, N.; Borge, L.; Hajem, B.; Lermet, A.; Sippl, W.; et al. Masitinib (AB1010), a Potent and Selective Tyrosine Kinase Inhibitor Targeting KIT. PLoS ONE 2009, 4, e7258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Y.; Sychterz, C.; Suttle, A.B.; Dar, M.M.; Bershas, D.; Negash, K.; Qian, Y.; Chen, E.P.; Gorycki, P.D.; Ho, M.Y.K. Bioavailability, metabolism and disposition of oral pazopanib in patients with advanced cancer. Xenobiotica 2013, 43, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Pazopanib HCl (GW786034 HCl). VEGFR Inhibitor. Read Reviews & Product Use Citations. Available online: http://www.selleckchem.com/products/Pazopanib-Hydrochloride.html (accessed on 18 January 2018).
- Yancey, M.F.; Merritt, D.A.; Lesman, S.P.; Boucher, J.F.; Michels, G.M. Pharmacokinetic properties of toceranib phosphate (Palladia, SU11654), a novel tyrosine kinase inhibitor, in laboratory dogs and dogs with mast cell tumors. J. Vet. Pharmacol. Ther. 2010, 33, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Halsey, C.H.; Gustafson, D.L.; Rose, B.J.; Wolf-Ringwall, A.; Burnett, R.C.; Duval, D.L.; Avery, A.C.; Thamm, D.H. Development of an in vitro model of acquired resistance to toceranib phosphate (Palladia®) in canine mast cell tumor. BMC Vet. Res. 2014, 10, 105. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Quan, H.; Xu, Y.; Kong, X.; Jin, L.; Lou, L. Flumatinib, a selective inhibitor of BCR-ABL/PDGFR/KIT, effectively overcomes drug resistance of certain KIT mutants. Cancer Sci. 2014, 105, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Quan, H.; Xie, C.; Xu, Y.; Fu, L.; Lou, L. HH-GV-678, a novel selective inhibitor of Bcr-Abl, outperforms imatinib and effectively overrides imatinib resistance. Leukemia 2010, 24, 1807–1809. [Google Scholar] [CrossRef] [PubMed]
- Blay, J.-Y.; Shen, L.; Kang, Y.-K.; Rutkowski, P.; Qin, S.; Nosov, D.; Wan, D.; Trent, J.; Srimuninnimit, V.; Pápai, Z.; et al. Nilotinib versus imatinib as first-line therapy for patients with unresectable or metastatic gastrointestinal stromal tumours ({ENESTg1)}: A randomised phase 3 trial. Lancet Oncol. 2015, 16, 550–560. [Google Scholar] [CrossRef]
- Kanda, T.; Ishikawa, T.; Takahashi, T.; Nishida, T. Nilotinib for treatment of gastrointestinal stromal tumors: Out of the equation? Expert Opin. Pharmacother. 2013, 14, 1859–1867. [Google Scholar] [CrossRef] [PubMed]
- Koon, H.B.; Krown, S.E.; Lee, J.Y.; Honda, K.; Rapisuwon, S.; Wang, Z.; Aboulafia, D.; Reid, E.G.; Rudek, M.A.; Dezube, B.J.; et al. Phase {II} trial of imatinib in {AIDS-associated} Kaposi’s sarcoma: {AIDS} Malignancy Consortium Protocol 042. J. Clin. Oncol. 2014, 32, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Koon, H.B.; Bubley, G.J.; Pantanowitz, L.; Masiello, D.; Smith, B.; Crosby, K.; Proper, J.; Weeden, W.; Miller, T.E.; Chatis, P.; et al. Imatinib-induced regression of {AIDS-related} Kaposi’s sarcoma. J. Clin. Oncol. 2005, 23, 982–989. [Google Scholar] [CrossRef] [PubMed]
- Scheinfeld, N.; Schienfeld, N. A comprehensive review of imatinib mesylate (Gleevec) for dermatological diseases. J. Drugs Dermatol. 2006, 5, 117–122. [Google Scholar] [PubMed]
- Hodi, F.S.; Corless, C.L.; Giobbie-Hurder, A.; Fletcher, J.A.; Zhu, M.; Marino-Enriquez, A.; Friedlander, P.; Gonzalez, R.; Weber, J.S.; Gajewski, T.F.; et al. Imatinib for melanomas harboring mutationally activated or amplified {KIT} arising on mucosal, acral, and chronically sun-damaged skin. J. Clin. Oncol. 2013, 31, 3182–3190. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D. Another option in our {KIT} of effective therapies for advanced melanoma. J. Clin. Oncol. 2013, 31, 3173–3175. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.L.; Eisenhauer, E.L.; Herzog, T.J. Emerging therapies: Angiogenesis inhibitors for ovarian cancer. Expert Opin. Emerg. Drugs 2015, 20, 331–346. [Google Scholar] [CrossRef] [PubMed]
- Leone Roberti Maggiore, U.; Valenzano Menada, M.; Venturini, P.L.; Ferrero, S. The potential of sunitinib as a therapy in ovarian cancer. Expert Opin. Investig. Drugs 2013, 22, 1671–1686. [Google Scholar] [CrossRef] [PubMed]
- Mahipal, A.; Tijani, L.; Chan, K.; Laudadio, M.; Mastrangelo, M.J.; Sato, T. A pilot study of sunitinib malate in patients with metastatic uveal melanoma. Melanoma Res. 2012, 22, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Penel, N.; Le Cesne, A.; Bui, B.N.; Perol, D.; Brain, E.G.; Ray-Coquard, I.; Guillemet, C.; Chevreau, C.; Cupissol, D.; Chabaud, S.; et al. Imatinib for progressive and recurrent aggressive fibromatosis (desmoid tumors): An {FNCLCC/French} Sarcoma Group phase {II} trial with a long-term follow-up. Ann. Oncol. 2011, 22, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Chugh, R.; Wathen, J.K.; Patel, S.R.; Maki, R.G.; Meyers, P.A.; Schuetze, S.M.; Priebat, D.A.; Thomas, D.G.; Jacobson, J.A.; Samuels, B.L.; et al. Efficacy of imatinib in aggressive fibromatosis: Results of a phase {II} multicenter Sarcoma Alliance for Research through Collaboration ({SARC}) trial. Clin. Cancer Res. 2010, 16, 4884–4891. [Google Scholar] [CrossRef] [PubMed]
- Koch, C.A.; Gimm, O.; Vortmeyer, A.O.; Al-Ali, H.K.; Lamesch, P.; Ott, R.; Kluge, R.; Bierbach, U.; Tannapfel, A. Does the expression of c-kit ({CD117}) in neuroendocrine tumors represent a target for therapy? Ann. N. Y. Acad. Sci. 2006, 1073, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Kostoula, V.; Khan, K.; Savage, K.; Stubbs, M.; Quaglia, A.; Dhillon, A.; Hochhauser, D.; Caplin, M. Expression of c-kit ({CD117}) in neuroendocrine tumours—A target for therapy? Oncol. Rep. 2005, 13, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Skardal, A.; Devarasetty, M.; Forsythe, S.; Atala, A.; Soker, S. A reductionist metastasis-on-a-chip platform for in vitro tumor progression modeling and drug screening. Biotechnol. Bioeng. 2016, 113, 2020–2032. [Google Scholar] [CrossRef] [PubMed]
- Skardal, A.; Devarasetty, M.; Soker, S.; Hall, A.R. In situ patterned micro 3D liver constructs for parallel toxicology testing in a fluidic device. Biofabrication 2015, 7, 31001. [Google Scholar] [CrossRef] [PubMed]
- Kozminsky, M.; Nagrath, S. Circulating Tumor Cells, Cancer Stem Cells, and Emerging Microfluidic Detection Technologies With Clinical Applications. In Cancer Stem Cells; Elsevier: Amsterdam, The Netherlands, 2016; pp. 473–497. [Google Scholar]
- Srinivasaraghavan, V.; Strobl, J.; Agah, M. Microelectrode bioimpedance analysis distinguishes basal and claudin-low subtypes of triple negative breast cancer cells. Biomed. Microdevices 2015, 17, 80. [Google Scholar] [CrossRef] [PubMed]
- Na, Y.J.; Baek, H.S.; Ahn, S.M.; Shin, H.J.; Chang, I.-S.; Hwang, J.S. [4-t-Butylphenyl]-N-(4-imidazol-1-yl phenyl)sulfonamide (ISCK03) inhibits SCF/c-kit signaling in 501mel human melanoma cells and abolishes melanin production in mice and brownish guinea pigs. Biochem. Pharmacol. 2007, 74, 780–786. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | Trade Name | Select Targets (Other than CD117) | Bioavailability | Specificity for CD117 | References |
|---|---|---|---|---|---|
| Imatinib | Gleevec/Glivec, STI571 | BCR-Abl, RET, PDGF-R | 98% | 0.1 μM | [145,146,147] |
| Sunitinib | Sutent, SU11248 | JAK/STAT, PDGF-R, Ras/MAPK, VEGFR | 50% (fasting) | 26 nM | [145,147,148,149] |
| Nilotinib | Tasigna | BCR-Abl, Lck | 30% | N.A. | [145,150] |
| Dasatinib | Sprycel | BCR-Abl, Src | 14–34% | 13 nM | [145,147,151,152] |
| Axitinib | Inlyta | BCR-Abl, PDGFR, VEGFR | 58% | 1.7 nM | [145,153,154] |
| Masitinib | Masivet, Kinavet | FGFR, PDGFR | 60% (animals) | 200 ± 40 nM | [155,156,157] |
| Pazopanib | Votrient | FGFR, PDGFR, VEGFR | 14–39% | 146 nM | [145,147,158,159] |
| Toceranib | Palladia | PDGFR, VEGFR | 77% | <10 nM | [160,161] |
| Cabozantinib | XL184 | VEGFR, c-Met | 74–93% | 4.6 nM | [162] |
| Flumatinib | HH-GV-678 | c-Abl, PDGFR | N.A. | 2.66 μM | [163,164] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Foster, B.M.; Zaidi, D.; Young, T.R.; Mobley, M.E.; Kerr, B.A. CD117/c-kit in Cancer Stem Cell-Mediated Progression and Therapeutic Resistance. Biomedicines 2018, 6, 31. https://doi.org/10.3390/biomedicines6010031
Foster BM, Zaidi D, Young TR, Mobley ME, Kerr BA. CD117/c-kit in Cancer Stem Cell-Mediated Progression and Therapeutic Resistance. Biomedicines. 2018; 6(1):31. https://doi.org/10.3390/biomedicines6010031
Chicago/Turabian StyleFoster, Brittni M., Danish Zaidi, Tyler R. Young, Mary E. Mobley, and Bethany A. Kerr. 2018. "CD117/c-kit in Cancer Stem Cell-Mediated Progression and Therapeutic Resistance" Biomedicines 6, no. 1: 31. https://doi.org/10.3390/biomedicines6010031
APA StyleFoster, B. M., Zaidi, D., Young, T. R., Mobley, M. E., & Kerr, B. A. (2018). CD117/c-kit in Cancer Stem Cell-Mediated Progression and Therapeutic Resistance. Biomedicines, 6(1), 31. https://doi.org/10.3390/biomedicines6010031