Cell-Penetrating Peptides to Enhance Delivery of Oligonucleotide-Based Therapeutics

Abstract
:1. Introduction
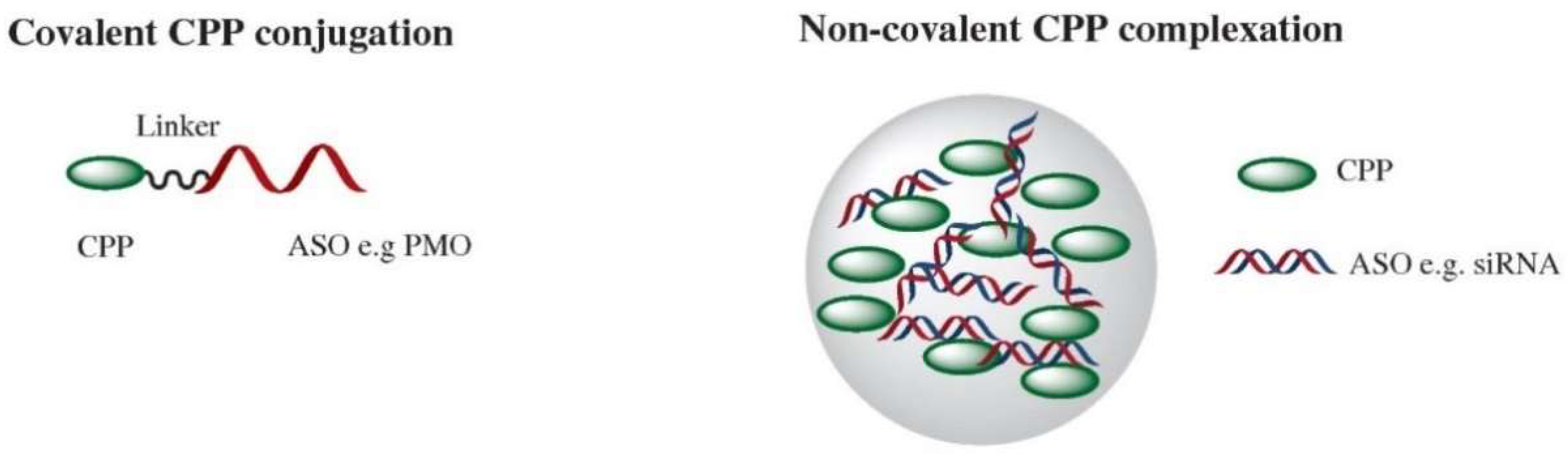
2. Direct CPP Conjugation for Nucleic Acid Delivery
3. Noncovalent CPP Delivery of Nucleic acids
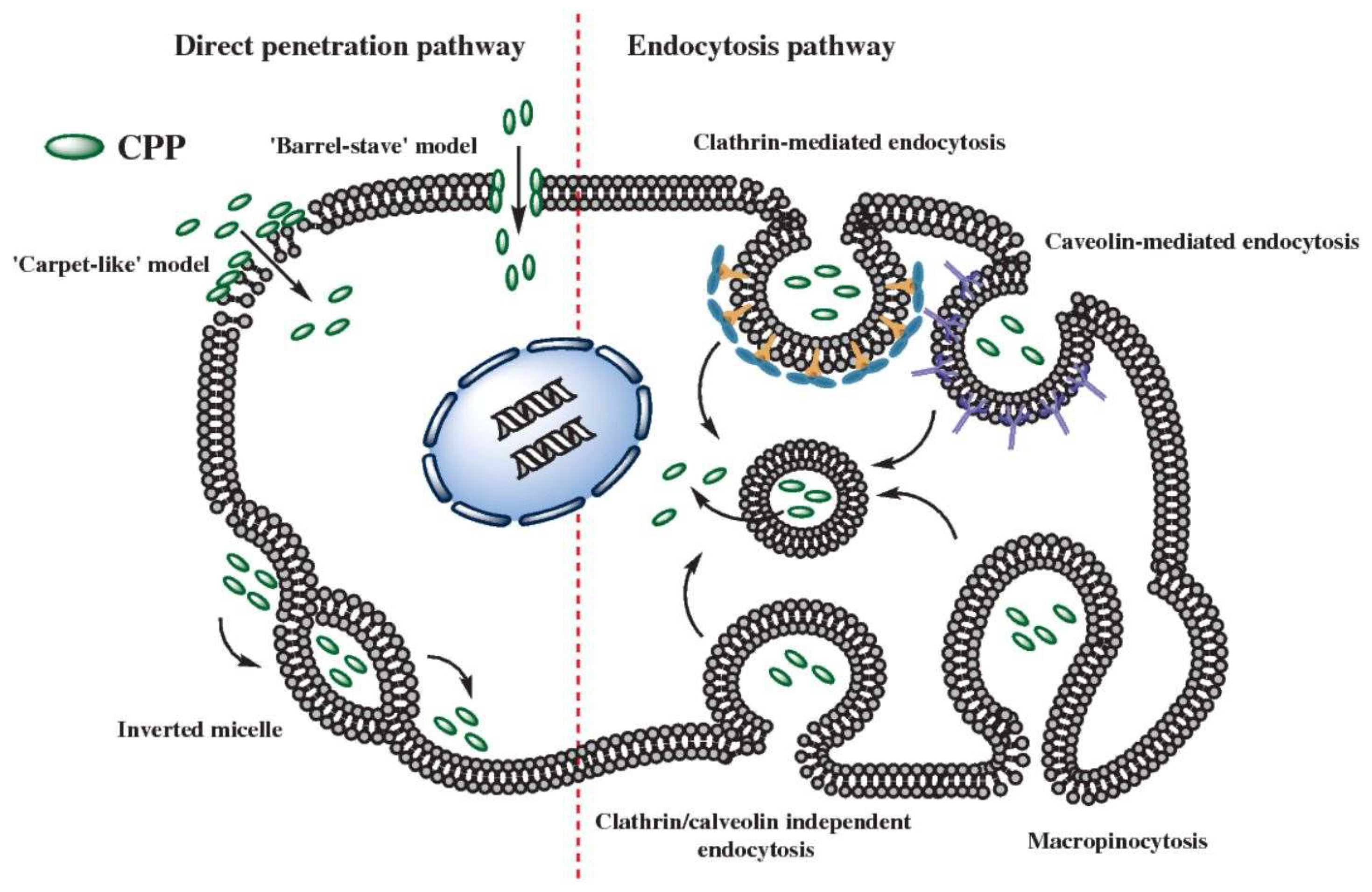
4. Mechanism of CPP Mediated Uptake
5. Future Perspectives of Clinical Application of CPP-ASOs
Acknowledgments
Conflicts of Interest
References
- Nayerossadat, N.; Ali, P.; Maedeh, T. Viral and nonviral delivery systems for gene delivery. Adv. Biomed. Res. 2012, 1, 27. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorth, M.; Narvekar, A. Non viral vectors in gene therapy—An overview. J. Clin. Diagn. Res. 2015, 9, GE01-6. [Google Scholar] [CrossRef] [PubMed]
- Lehto, T.; Ezzat, K.; Wood, M.J.A.; EL Andaloussi, S. Peptides for nucleic acid delivery. Adv. Drug Deliv. Rev. 2016, 106, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, M.; Birch, D.; Nielsen, H.M. Applications and challenges for use of cell-penetrating peptides as delivery vectors for peptide and protein cargos. Int. J. Mol. Sci. 2016, 17, 185. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Derossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar] [PubMed]
- Pooga, M.; Hällbrink, M.; Zorko, M.; Langel, Ü. Cell penetration by transportan. FASEB J. 1998, 12, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Futaki, S.; Suzuki, T.; Ohashi, W.; Yagami, T.; Tanaka, S.; Ueda, K.; Sugiura, Y. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J. Biol. Chem. 2001, 276, 5836–5840. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Moulton, H.M.; Seow, Y.; Boyd, C.; Boutilier, J.; Iverson, P.; Wood, M.J. Cell-penetrating peptide-conjugated antisense oligonucleotides restore systemic muscle and cardiac dystrophin expression and function. Hum. Mol. Genet. 2008, 17, 3909–3918. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Moulton, H.M.; Iversen, P.L.; Jiang, J.; Li, J.; Li, J.; Spurney, C.F.; Sali, A.; Guerron, A.D.; Nagaraju, K.; et al. Effective rescue of dystrophin improves cardiac function in dystrophin-deficient mice by a modified morpholino oligomer. Proc. Natl. Acad. Sci. USA 2008, 105, 14814–14819. [Google Scholar] [CrossRef] [PubMed]
- Leger, A.J.; Mosquea, L.M.; Clayton, N.P.; Wu, I.-H.; Weeden, T.; Nelson, C.A.; Phillips, L.; Roberts, E.; Piepenhagen, P.A.; Cheng, S.H.; et al. Systemic delivery of a Peptide-linked morpholino oligonucleotide neutralizes mutant RNA toxicity in a mouse model of myotonic dystrophy. Nucleic Acid Ther. 2013, 23, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Moulton, H.M.; Betts, C.; Seow, Y.; Boutilier, J.; Iverson, P.L.; Wood, M.J. A fusion peptide directs enhanced systemic dystrophin exon skipping and functional restoration in dystrophin-deficient mdx mice. Hum. Mol. Genet. 2009, 18, 4405–4414. [Google Scholar] [CrossRef] [PubMed]
- Betts, C.A.; Saleh, A.F.; Carr, C.A.; Hammond, S.M.; Coenen-Stass, A.M.L.; Godfrey, C.; McClorey, G.; Varela, M.A.; Roberts, T.C.; Clarke, K.; et al. Prevention of exercised induced cardiomyopathy following Pip-PMO treatment in dystrophic mdx mice. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, C.; Muses, S.; McClorey, G.; Wells, K.E.; Coursindel, T.; Terry, R.L.; Betts, C.; Hammond, S.; O’donovan, L.; Hildyard, J.; et al. How much dystrophin is enough: The physiological consequences of different levels of dystrophin in the mdx mouse. Hum. Mol. Genet. 2015, 24. [Google Scholar] [CrossRef] [PubMed]
- Betts, C.; Saleh, A.F.; Arzumanov, A.A.; Hammond, S.M.; Godfrey, C.; Coursindel, T.; Gait, M.J.; Wood, M.J.A. Pip6-PMO, a new generation of peptide-oligonucleotide conjugates with improved cardiac exon skipping activity for DMD treatment. Mol. Ther. Nucleic Acids 2012, 1. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.M.; Hazell, G.; Shabanpoor, F.; Saleh, A.F.; Bowerman, M.; Sleigh, J.N.; Meijboom, K.E.; Zhou, H.; Muntoni, F.; Talbot, K.; et al. Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 2016, 113, 10962–10967. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhao, J.; Han, G.; Zhang, Y.; Dong, X.; Cao, L.; Wang, Q.; Moulton, H.M.; Yin, H. Effective dystrophin restoration by a novel muscle-homing peptide-morpholino conjugate in dystrophin-deficient mdx mice. Mol. Ther. 2014, 22, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Shabanpoor, F.; Hammond, S.M.; Abendroth, F.; Hazell, G.; Wood, M.J.A.; Gait, M.J. Identification of a Peptide for Systemic Brain Delivery of a Morpholino Oligonucleotide in Mouse Models of Spinal Muscular Atrophy. Nucleic Acid Ther. 2017, 27, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Jirka, S.M.G.; Heemskerk, H.; Tanganyika-de Winter, C.L.; Muilwijk, D.; Pang, K.H.; de Visser, P.C.; Janson, A.; Karnaoukh, T.G.; Vermue, R.; ‘t Hoen, P.A.C.; et al. Peptide Conjugation of 2′-O-methyl Phosphorothioate Antisense Oligonucleotides Enhances Cardiac Uptake and Exon Skipping in mdx Mice. Nucleic Acid Ther. 2014, 24, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Burrer, R.; Neuman, B.W.; Ting, J.P.C.; Stein, D.A.; Moulton, H.M.; Iversen, P.L.; Kuhn, P.; Buchmeier, M.J. Antiviral effects of antisense morpholino oligomers in murine coronavirus infection models. J. Virol. 2007, 81, 5637–5648. [Google Scholar] [CrossRef] [PubMed]
- Geller, B.L.; Marshall-Batty, K.; Schnell, F.J.; McKnight, M.M.; Iversen, P.L.; Greenberg, D.E. Gene-silencing antisense oligomers inhibit acinetobacter growth in vitro and in vivo. J. Infect. Dis. 2013, 208, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Crombez, L.; Morris, M.C.; Dufort, S.; Aldrian-Herrada, G.; Nguyen, Q.; Mc Master, G.; Coll, J.-L.; Heitz, F.; Divita, G. Targeting cyclin B1 through peptide-based delivery of siRNA prevents tumour growth. Nucleic Acids Res. 2009, 37, 4559–4569. [Google Scholar] [CrossRef] [PubMed]
- Crombez, L.; Aldrian-Herrada, G.; Konate, K.; Nguyen, Q.N.; McMaster, G.K.; Brasseur, R.; Heitz, F.; Divita, G. A new potent secondary amphipathic cell-penetrating peptide for siRNA delivery into mammalian cells. Mol. Ther. 2009, 17, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Vaissière, A.; Aldrian, G.; Konate, K.; Lindberg, M.F.; Jourdan, C.; Telmar, A.; Seisel, Q.; Fernandez, F.; Viguier, V.; Genevois, C.; et al. A retro-inverso cell-penetrating peptide for siRNA delivery. J. Nanobiotechnol. 2017, 15. [Google Scholar] [CrossRef] [PubMed]
- Mäe, M.; EL Andaloussi, S.; Lundin, P.; Oskolkov, N.; Johansson, H.J.; Guterstam, P.; Langel, Ü. A stearylated CPP for delivery of splice correcting oligonucleotides using a non-covalent co-incubation strategy. J. Control. Release 2009, 134, 221–227. [Google Scholar] [CrossRef] [PubMed]
- El Andaloussi, S.; Lehto, T.; Mäger, I.; Rosenthal-Aizman, K.; Oprea, I.I.; Simonson, O.E.; Sork, H.; Ezzat, K.; Copolovici, D.M.; Kurrikoff, K.; et al. Design of a peptide-based vector, PepFect6, for efficient delivery of siRNA in cell culture and systemically in vivo. Nucleic Acids Res. 2011, 39, 3972–3987. [Google Scholar] [CrossRef] [PubMed]
- Veiman, K.L.; Mäger, I.; Ezzat, K.; Margus, H.; Lehto, T.; Langel, K.; Kurrikoff, K.; Arukuusk, P.; Suhorutsenko, J.; Padari, K.; et al. PepFect14 peptide vector for efficient gene delivery in cell cultures. Mol. Pharm. 2013, 10, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, K.; El Andaloussi, S.; Zaghloul, E.M.; Lehto, T.; Lindberg, S.; Moreno, P.M.D.; Viola, J.R.; Magdy, T.; Abdo, R.; Guterstam, P.; et al. PepFect 14, a novel cell-penetrating peptide for oligonucleotide delivery in solution and as solid formulation. Nucleic Acids Res. 2011, 39, 5284–5298. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Wu, H.; McBride, J.L.; Jung, K.-E.; Hee Kim, M.; Davidson, B.L.; Lee, S.K.; Shankar, P.; Manjunath, N. Transvascular delivery of small interfering RNA to the central nervous system. Nature 2007, 448, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Cantini, L.; Attaway, C.C.; Butler, B.; Andino, L.M.; Sokolosky, M.L.; Jakymiw, A. Fusogenic-Oligoarginine Peptide-Mediated Delivery of siRNAs Targeting the CIP2A Oncogene into Oral Cancer Cells. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Alexander-Bryant, A.A.; Zhang, H.; Attaway, C.C.; Pugh, W.; Eggart, L.; Sansevere, R.M.; Andino, L.M.; Dinh, L.; Cantini, L.P.; Jakymiw, A. Dual peptide-mediated targeted delivery of bioactive siRNAs to oral cancer cells in vivo. Oral Oncol. 2017, 72, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Leng, Q.; Scaria, P.; Lu, P.; Woodle, M.C.; Mixson, A.J. Systemic delivery of HK Raf-1 siRNA polyplexes inhibits MDA-MB-435 xenografts. Cancer Gene Ther. 2008, 15, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Sazani, P.; Gemignani, F.; Kang, S.H.; Maier, M.A.; Manoharan, M.; Persmark, M.; Bortner, D.; Kole, R. Systemically delivered antisense oligomers upregulate gene expression in mouse tissues. Nat. Biotechnol. 2002, 20, 1228–1233. [Google Scholar] [CrossRef] [PubMed]
- Bendifallah, N.; Rasmussen, F.W.; Zachar, V.; Ebbesen, P.; Nielsen, P.E.; Koppelhus, U. Evaluation of cell-penetrating peptides (CPPs) as vehicles for intracellular delivery of antisense peptide nucleic acid (PNA). Bioconjug. Chem. 2006, 17, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Boffa, L.C.; Cutrona, G.; Cilli, M.; Matis, S.; Damonte, G.; Mariani, M.R.; Millo, E.; Moroni, M.; Roncella, S.; Fedeli, F.; et al. Inhibition of Burkitt’s lymphoma cells growth in SCID mice by a PNA specific for a regulatory sequence of the translocated c-myc. Cancer Gene Ther. 2007, 14, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Fabani, M.M.; Abreu-Goodger, C.; Williams, D.; Lyons, P.A.; Torres, A.G.; Smith, K.G.C.; Enright, A.J.; Gait, M.J.; Vigorito, E. Efficient inhibition of miR-155 function in vivo by peptide nucleic acids. Nucleic Acids Res. 2010, 38, 4466–4475. [Google Scholar] [CrossRef] [PubMed]
- Stirchak, E.P.; Summerton, J.E.; Weller, D.D. Uncharged Stereoregular Nucleic Acid Analogues. 1. Synthesis of a Cytosine-Containing Oligomer with Carbamate Internucleoside Linkages. J. Org. Chem. 1987, 52, 4202–4206. [Google Scholar] [CrossRef]
- Moulton, H.M.; Nelson, M.H.; Hatlevig, S.A.; Reddy, M.T.; Iversen, P.L. Cellular Uptake of Antisense Morpholino Oligomers Conjugated to Arginine-Rich Peptides. Bioconjug. Chem. 2004, 15, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Boisguérin, P.; Deshayes, S.; Gait, M.J.; O’Donovan, L.; Godfrey, C.; Betts, C.A.; Wood, M.J.A.; Lebleu, B. Delivery of therapeutic oligonucleotides with cell penetrating peptides. Adv. Drug Deliv. Rev. 2015, 87, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Amantana, A.; Moulton, H.M.; Cate, M.L.; Reddy, M.T.; Whitehead, T.; Hassinger, J.N.; Youngblood, D.S.; Iversen, P.L. Pharmacokinetics, biodistribution, stability and toxicity of a cell-penetrating peptide—Morpholino oligomer conjugate. Bioconjug. Chem. 2007, 18, 1325–1331. [Google Scholar] [CrossRef] [PubMed]
- Abes, S.; Moulton, H.M.; Clair, P.; Prevot, P.; Youngblood, D.S.; Wu, R.P.; Iversen, P.L.; Lebleu, B. Vectorization of morpholino oligomers by the (R-Ahx-R)4peptide allows efficient splicing correction in the absence of endosomolytic agents. J. Control. Release 2006, 116, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.Q.; Maruyama, R.; Yokota, T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Dev. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Goyenvalle, A.; Griffith, G.; Babbs, A.; El Andaloussi, S.; Ezzat, K.; Avril, A.; Dugovic, B.; Chaussenot, R.; Ferry, A.; Voit, T.; et al. Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers. Nat. Med. 2015, 21, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Samoylova, T.I.; Smith, B.F. Elucidation of muscle-binding peptides by phage display screening. Muscle Nerve 1999, 22, 460–466. [Google Scholar] [CrossRef]
- Echigoya, Y.; Nakamura, A.; Nagata, T.; Urasawa, N.; Lim, K.R.Q.; Trieu, N.; Panesar, D.; Kuraoka, M.; Moulton, H.M.; Saito, T.; et al. Effects of systemic multiexon skipping with peptide-conjugated morpholinos in the heart of a dog model of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2017, 114, 4213–4218. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, G.D.; Arzumanov, A.; Abes, R.; Yin, H.; Wood, M.J.A.; Lebleu, B.; Gait, M.J. Improved cell-penetrating peptide-PNA conjugates for splicing redirection in HeLa cells and exon skipping in mdx mouse muscle. Nucleic Acids Res. 2008, 36, 6418–6428. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Saleh, A.F.; Betts, C.; Camelliti, P.; Seow, Y.; Ashraf, S.; Arzumanov, A.; Hammond, S.; Merritt, T.; Gait, M.J.; et al. Pip5 transduction peptides direct high efficiency oligonucleotide-mediated dystrophin exon skipping in heart and phenotypic correction in mdx mice. Mol. Ther. 2011, 19, 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Kayali, R.; Bertoni, C.; Fike, F.; Hu, H.; Iversen, P.L.; Gatti, R.A. Arginine-rich cell-penetrating peptide dramatically enhances AMO-mediated ATM aberrant splicing correction and enables delivery to brain and cerebellum. Hum. Mol. Genet. 2011, 20, 3151–3160. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Darras, B.T.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2018, 378, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.; Adams, A.M.; Johnsen, R.D.; Greer, K.; Moulton, H.M.; Wilton, S.D. Dystrophin isoform induction in vivo by antisense-mediated alternative splicing. Mol. Ther. 2010, 18, 1218–1223. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.K.; Malerba, A.; Popplewell, L.; Foster, K.; Dickson, G. Antisense-induced myostatin exon skipping leads to muscle hypertrophy in mice following octa guanidine morpholino oligomer treatment. Mol. Ther. 2011, 19, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Malerba, A.; Kang, J.K.; McClorey, G.; Saleh, A.F.; Popplewell, L.; Gait, M.J.; Wood, M.J.A.; Dickson, G. Dual myostatin and dystrophin exon skipping by morpholino nucleic acid oligomers conjugated to a cell-penetrating peptide is a promising therapeutic strategy for the treatment of duchenne muscular dystrophy. Mol. Ther. Nucleic Acids 2012, 1. [Google Scholar] [CrossRef] [PubMed]
- Lu-Nguyen, N.; Malerba, A.; Popplewell, L.; Schnell, F.; Hanson, G.; Dickson, G. Systemic Antisense Therapeutics for Dystrophin and Myostatin Exon Splice Modulation Improve Muscle Pathology of Adult mdx Mice. Mol. Ther. Nucleic Acids 2017, 6, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Shabanpoor, F.; McClorey, G.; Saleh, A.F.; Jarver, P.; Wood, M.J.A.; Gait, M.J. Bi-specific splice-switching PMO oligonucleotides conjugated via a single peptide active in a mouse model of Duchenne muscular dystrophy. Nucleic Acids Res. 2015, 43, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.Y.; Mao, X.G.; Zhou, Y.; Chen, Z.; Hu, Y.; Hou, Z.; Li, M.-K.; Meng, J.-R.; Luo, X.-X. Advances in the delivery of antisense oligonucleotides for combating bacterial infectious diseases. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 745–758. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Vidal, P.; Chaloin, L.; Heitz, F.; Divita, G. A new peptide vector for efficient delivery of oligonucleotides into mammalian cells. Nucleic Acids Res. 1997, 25, 2730–2736. [Google Scholar] [CrossRef] [PubMed]
- Wolfrum, C.; Shi, S.; Jayaprakash, K.N.; Jayaraman, M.; Wang, G.; Pandey, R.K.; Rajeev, K.G.; Nakayama, T.; Charrise, K.; Ndungo, E.M.; et al. Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat. Biotechnol. 2007, 25, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Chorev, M.; Goodman, M. Recent developments in retro peptides and proteins—An ongoing topochemical exploration. Trends Biotechnol. 1995, 13, 438–445. [Google Scholar] [CrossRef]
- Leng, Q.; Scaria, P.; Zhu, J.; Ambulos, N.; Campbell, P.; Mixson, A.J. Highly branched HK peptides are effective carriers of siRNA. J. Gene Med. 2005, 7, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Järver, P.; Zaghloul, E.M.; Arzumanov, A.A.; Saleh, A.F.; McClorey, G.; Hammond, S.M.; Hällbrink, M.; Langel, Ü.; Smith, C.I.; Wood, M.J.; et al. Peptide Nanoparticle Delivery of Charge-Neutral Splice-Switching Morpholino Oligonucleotides. Nucleic Acid Ther. 2015, 25, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Ban, H.S.; Kim, S.S.; Wu, H.; Pearson, T.; Greiner, D.L.; Laouar, A.; Yao, J.; Haridas, V.; Habiro, K.; et al. T Cell-Specific siRNA Delivery Suppresses HIV-1 Infection in Humanized Mice. Cell 2008, 134, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Martín, I.; Teixidó, M.; Giralt, E. Building cell selectivity into CPP-mediated strategies. Pharmaceuticals 2010, 3, 1456–1490. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.S.; Aguilera, T.A.; Jiang, T.; Ellies, L.G.; Nguyen, Q.T.; Wong, E.H.; Gross, L.A.; Tsien, R.Y. In vivo characterization of activatable cell penetrating peptides for targeting protease activity in cancer. Integr. Biol. (Camb.) 2009, 1, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Xiang, B.; Jia, X.L.; Qi, J.L.; Yang, L.P.; Sun, W.H.; Yan, X.; Yang, S.-K.; Cao, D.-Y.; Du, Q.; Qi, X.-R. Enhancing siRNA-based cancer therapy using a new pH-responsive activatable cell-penetrating peptide-modified liposomal system. Int. J. Nanomed. 2017, 12, 2385–2405. [Google Scholar] [CrossRef] [PubMed]
- Luneberg, J.; Martin, I.; Nussler, F.; Ruysschaert, J.M.; Herrmann, A. Structure and topology of the influenza virus fusion peptide in lipid bilayers. J. Biol. Chem. 1995, 270, 27606–27614. [Google Scholar] [CrossRef] [PubMed]
- Vivès, E.; Brodin, P.; Lebleu, B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef] [PubMed]
- Pouny, Y.; Rapaport, D.; Mor, A.; Nicolas, P.; Shai, Y. Interaction of antimicrobial dermaseptin and its fluorescently labeled analogues with phospholipid membranes. Biochemistry 1992, 31, 12416–12423. [Google Scholar] [CrossRef] [PubMed]
- Shai, Y. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by α-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta Biomembr. 1999, 1462, 55–70. [Google Scholar] [CrossRef]
- Derossi, D.; Calvet, S.; Trembleau, A.; Brunissen, A.; Chassaing, G.; Prochiantz, A. Cell internalization of the third helix of the antennapedia homeodomain is receptor-independent. J. Biol. Chem. 1996, 271, 18188–18193. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.P.; Melikov, K.; Vives, E.; Ramos, C.; Verbeure, B.; Gait, M.J.; Chernomordik, L.V.; Lebleu, B. Cell-penetrating peptides: A reevaluation of the mechanism of cellular uptake. J. Biol. Chem. 2003, 278, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Rydström, A.; Deshayes, S.; Konate, K.; Crombez, L.; Padari, K.; Boukhaddaoui, H.; Aldrian, G.; Pooga, M.; Divita, G. Direct translocation as major cellular uptake for CADY self-assembling peptide-based nanoparticles. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Nakase, I.; Tadokoro, A.; Kawabata, N.; Takeuchi, T.; Katoh, H.; Hiramoto, K.; Negishi, M.; Nomizu, M.; Sugiura, Y.; Futaki, S. Interaction of arginine-rich peptides with membrane-associated proteoglycans is crucial for induction of actin organization and macropinocytosis. Biochemistry 2007, 46, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Säälik, P.; Elmquist, A.; Hansen, M.; Padari, K.; Saar, K.; Viht, K.; Langel, U.; Pooga, M. Protein cargo delivery properties of cell-penetrating peptides. A comparative study. Bioconjug. Chem. 2004, 15, 1246–1253. [Google Scholar] [CrossRef] [PubMed]
- Fittipaldi, A.; Ferrari, A.; Zoppé, M.; Arcangeli, C.; Pellegrini, V.; Beltram, F.; Giacca, M. Cell Membrane Lipid Rafts Mediate Caveolar Endocytosis of HIV-1 Tat Fusion Proteins. J. Biol. Chem. 2003, 278, 34141–34149. [Google Scholar] [CrossRef] [PubMed]
- Gestin, M.; Dowaidar, M.; Langel, Ü. Uptake mechanism of cell-penetrating peptides. Adv. Exp. Med. Biol. 2017, 1030, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Soomets, U.; Lindgren, M.; Gallet, X.; Hällbrink, M.; Elmquist, A.; Balaspiri, L.; Zorko, M.; Pooga, M.; Brasseur, R.; Langel, U. Deletion analogues of transportan. Biochim. Biophys. Acta Biomembr. 2000, 1467, 165–176. [Google Scholar] [CrossRef]
- Lehto, T.; Alvarez, A.C.; Gauck, S.; Gait, M.J.; Coursindel, T.; Wood, M.J.A.; Lebleu, B.; Boisguerin, P. Cellular trafficking determines the exon skipping activity of Pip6a-PMO in mdx skeletal and cardiac muscle cells. Nucleic Acids Res. 2014, 42, 3207–3217. [Google Scholar] [CrossRef] [PubMed]
- Pang, H.-B.; Braun, G.B.; Ruoslahti, E. Neuropilin-1 and heparan sulfate proteoglycans cooperate in cellular uptake of nanoparticles functionalized by cationic cell-penetrating peptides. Sci. Adv. 2015, 1, e1500821. [Google Scholar] [CrossRef] [PubMed]
- Helmfors, H.; Lindberg, S.; Langel, Ü. SCARA involvement in the uptake of nanoparticles formed by cell-penetrating peptides. Methods Mol. Biol. 2015, 1324, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Canton, J.; Neculai, D.; Grinstein, S. Scavenger receptors in homeostasis and immunity. Nat. Rev. Immunol. 2013, 13, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, K.; Helmfors, H.; Tudoran, O.; Juks, C.; Lindberg, S.; Padari, K.; El-Andaloussi, S.; Pooga, M.; Langel, U. Scavenger receptor-mediated uptake of cell-penetrating peptide nanocomplexes with oligonucleotides. FASEB J. 2012, 26, 1172–1180. [Google Scholar] [CrossRef] [PubMed]
- Juks, C.; Lorents, A.; Arukuusk, P.; Langel, Ü.; Pooga, M. Cell-penetrating peptides recruit type A scavenger receptors to the plasma membrane for cellular delivery of nucleic acids. FASEB J. 2017, 31, 975–988. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, K.; Aoki, Y.; Koo, T.; McClorey, G.; Benner, L.; Coenen-Stass, A.; O’Donovan, L.; Lehto, T.; Garcia-Guerra, A.; Nordin, J.; et al. Self-Assembly into Nanoparticles Is Essential for Receptor Mediated Uptake of Therapeutic Antisense Oligonucleotides. Nano Lett. 2015, 15, 4364–4373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowaidar, M.; Gestin, M.; Cerrato, C.P.; Jafferali, M.H.; Margus, H.; Kivistik, P.A.; Ezzat, K.; Hallberg, E.; Pooga, M.; Hällbrink, M.; et al. Role of autophagy in cell-penetrating peptide transfection model. Sci. Rep. 2017, 7, 12635. [Google Scholar] [CrossRef] [PubMed]
- Pryor, P.R.; Luzio, J.P. Delivery of endocytosed membrane proteins to the lysosome. Biochim. Biophys. Acta Mol. Cell Res. 2009, 1793, 615–624. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, A.; Futaki, S.; Harashima, H. Delivery of Macromolecules Using Arginine-Rich Cell-Penetrating Peptides: Ways to Overcome Endosomal Entrapment. AAPS J. 2009, 11, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Erazo-Oliveras, A.; Muthukrishnan, N.; Baker, R.; Wang, T.Y.; Pellois, J.P. Improving the endosomal escape of cell-penetrating peptides and their cargos: Strategies and challenges. Pharmaceuticals 2012, 5, 1177–1209. [Google Scholar] [CrossRef] [PubMed]
- Wadia, J.S.; Stan, R.V.; Dowdy, S.F. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat. Med. 2004, 10, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.L.; Wang, S. An endosomolytic Tat peptide produced by incorporation of histidine and cysteine residues as a nonviral vector for DNA transfection. Biomaterials 2008, 29, 2408–2414. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, P.; Bhalla, S.; Usmani, S.S.; Singh, S.; Chaudhary, K.; Raghava, G.P.S.; Gautam, A. CPPsite 2.0: A repository of experimentally validated cell-penetrating peptides. Nucleic Acids Res. 2016, 44, D1098–D1103. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Xing, P.; Su, R.; Shi, G.; Ma, Z.S.; Zou, Q. CPPred-RF: A Sequence-based Predictor for Identifying Cell-Penetrating Peptides and Their Uptake Efficiency. J. Proteome Res. 2017, 16, 2044–2053. [Google Scholar] [CrossRef] [PubMed]
- Margus, H.; Arukuusk, P.; Langel, U.; Pooga, M. Characteristics of Cell-Penetrating Peptide/Nucleic Acid Nanoparticles. Mol. Pharm. 2016, 13, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Carter, E.; Lau, C.Y.; Tosh, D.; Ward, S.G.; Mrsny, R.J. Cell penetrating peptides fail to induce an innate immune response in epithelial cells in vitro: Implications for continued therapeutic use. Eur. J. Pharm. Biopharm. 2013, 85, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Moulton, H.M.; Moulton, J.D. Morpholinos and their peptide conjugates: Therapeutic promise and challenge for Duchenne muscular dystrophy. Biochim. Biophys. Acta Biomembr. 2010, 1798, 2296–2303. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Peptide | Sequence | Application |
|---|---|---|
| Covalent conjugated CPPs | ||
| B | (RXRRBR)2XB | SSO for DMD, DM1 [9,10,11] |
| B-MSP | (RXRRBR)2XBASSLNIA | SSO for DMD [12] |
| Pip6 | RXRRBRRXR YQFLI RXRBRXRB | SSO for DMD, SMA [13,14,15,16] |
| M12 | RRQPPRSISSHP | SSO for DMD [17] |
| Br-ApoE(K→A) | Ac-LRALRARLLRGGAc-LRALRARLLRGGKX-Bpg-G | SSO for SMA [18] |
| P4 | LGAQSNF | SSO (2OMe) for DMD [19] |
| (RXR)4 | RXRRXRRXRRXR | anti-viral anti-bacterial [20,21] |
| Nanoparticle forming CPPs | ||
| MPG-8 | βAFLGWLGAWGTMGWSPKKKRK-Cya | siRNA for xenograft tumor model [22] |
| CADY | Ac-GLWRALWRLLRSLWRLLWRA-Cya | siRNA, cell lines (various) [23] |
| RICK | KWLLRWLSRLLRWLARWLG | siRNA, human glioblastoma cells [24] |
| Pepfect 3 | stearyl-AGYLLGKINLKALAALAKKIL-NH2 | Plasmid DNA, cell lines, intramuscular [25] |
| Pepfect 6 | See reference | siRNA, cell lines (various), systemic IV [26] |
| Pepfect 14 | stearyl-AGYLLGKLLOOLAAAALOOLL-NH2 | Plasmid DNA, SSO [27,28] |
| RVG-9R | YTIWMPENPRPGTPCDIFTNSRGKRASNGGGGRRRRRRRRR | siRNA, brain-targeting disease models [29] |
| 599 | GLFEAIEGFIENGWEGMIDGWYGGGGRRRRRRRRRK | siRNA, oral cancer [30,31] |
| H3K(+H)4b | Branched KHHHKHHHKHHHHKHHHK | siRNA, tumor xenograft [32] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McClorey, G.; Banerjee, S. Cell-Penetrating Peptides to Enhance Delivery of Oligonucleotide-Based Therapeutics. Biomedicines 2018, 6, 51. https://doi.org/10.3390/biomedicines6020051
McClorey G, Banerjee S. Cell-Penetrating Peptides to Enhance Delivery of Oligonucleotide-Based Therapeutics. Biomedicines. 2018; 6(2):51. https://doi.org/10.3390/biomedicines6020051
Chicago/Turabian StyleMcClorey, Graham, and Subhashis Banerjee. 2018. "Cell-Penetrating Peptides to Enhance Delivery of Oligonucleotide-Based Therapeutics" Biomedicines 6, no. 2: 51. https://doi.org/10.3390/biomedicines6020051
APA StyleMcClorey, G., & Banerjee, S. (2018). Cell-Penetrating Peptides to Enhance Delivery of Oligonucleotide-Based Therapeutics. Biomedicines, 6(2), 51. https://doi.org/10.3390/biomedicines6020051