Hypoxia, Metabolism and Immune Cell Function


Abstract
:1. Hypoxia-Induced Transcriptional Machinery
HIFs
2. Hypoxia and Immunometabolism
3. The Effect of Hypoxia on Myeloid Cell Function and Metabolism
3.1. Hypoxia and Granulocytes
3.1.1. Neutrophils
3.1.2. Mast Cells, Basophils and Eosinophils
3.2. Hypoxia and Macrophages
3.3. Hypoxia and Dendritic Cells
4. The Effect of Hypoxia on Adaptive Cell Function and Metabolism
4.1. Hypoxia and T Cells
4.2. Hypoxia and B Cells
5. The Effect of Hypoxia on Innate Lymphoid Cell Function and Metabolism
5.1. Hypoxia and ILC1 Cells
5.2. Hypoxia ILC2 and ILC3.
6. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Semenza, G.L. Life with oxygen. Science 2007, 318, 62–64. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.T.; Colgan, S.P. Regulation of immunity and inflammation by hypoxia in immunological niches. Nat. Rev. Immunol. 2017, 17, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF transcription factors, inflammation, and immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Carreau, A.; El Hafny-Rahbi, B.; Matejuk, A.; Grillon, C.; Kieda, C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J. Cell. Mol. Med. 2011, 15, 1239–1253. [Google Scholar] [CrossRef] [PubMed]
- Fratantonio, D.; Cimino, F.; Speciale, A.; Virgili, F. Need (more than) two to Tango: Multiple tools to adapt to changes in oxygen availability. BioFactors 2018. [Google Scholar] [CrossRef] [PubMed]
- De Santis, V.; Singer, M. Tissue oxygen tension monitoring of organ perfusion: Rationale, methodologies, and literature review. Br. J. Anaesth. 2015, 115, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci. STKE 2007, 2007, cm8. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-Inducible Factors in Physiology and Medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Sen, R.; Baltimore, D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 1986, 46, 705–716. [Google Scholar] [CrossRef]
- Van Uden, P.; Kenneth, N.S.; Webster, R.; Müller, H.A.; Mudie, S.; Rocha, S. Evolutionary conserved regulation of HIF-1β by NF-κB. PLoS Genet. 2011, 7, e1001285. [Google Scholar] [CrossRef] [PubMed]
- Van Uden, P.; Kenneth, N.S.; Rocha, S. Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem. J. 2008, 412, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Zinkernagel, A.S.; Johnson, R.S.; Nizet, V. Hypoxia inducible factor (HIF) function in innate immunity and infection. J. Mol. Med. 2007, 85, 1339–1346. [Google Scholar] [CrossRef] [PubMed]
- Hankinson, O. Why Does ARNT2 Behave Differently from ARNT? Toxicol. Sci. 2008, 103, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Talks, K.L.; Turley, H.; Gatter, K.C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J.; Harris, A.L. The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am. J. Pathol. 2000, 157, 411–421. [Google Scholar] [CrossRef]
- Yang, S.-L.; Wu, C.; Xiong, Z.-F.; Fang, X. Progress on hypoxia-inducible factor-3: Its structure, gene regulation and biological function (Review). Mol. Med. Rep. 2015, 12, 2411–2416. [Google Scholar] [CrossRef] [PubMed]
- Wenger, R.H.; Stiehl, D.P.; Camenisch, G. Integration of oxygen signaling at the consensus HRE. Sci. STKE 2005, 2005, re12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taabazuing, C.Y.; Hangasky, J.A.; Knapp, M.J. Oxygen sensing strategies in mammals and bacteria. J. Inorg. Biochem. 2014, 133, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef] [PubMed]
- D’Ignazio, L.; Batie, M.; Rocha, S. Hypoxia and Inflammation in Cancer, Focus on HIF and NF-κB. Biomedicines 2017, 5, 21. [Google Scholar] [CrossRef] [PubMed]
- Li, S.H.; Chun, Y.-S.; Lim, J.-H.; Huang, L.E.; Park, J.-W. Von Hippel-Lindau protein adjusts oxygen sensing of the FIH asparaginyl hydroxylase. Int. J. Biochem. Cell Biol. 2011, 43, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 2014, 9, 47–71. [Google Scholar] [CrossRef] [PubMed]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, S.E.; O’Neill, L.A.J. HIF1α and metabolic reprogramming in inflammation. J. Clin. Investig. 2016, 126, 3699–3707. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457.e13–470.e13. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Loftus, R.M.; Finlay, D.K. Immunometabolism: Cellular Metabolism Turns Immune Regulator. J. Biol. Chem. 2016, 291, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Roth, P.H.; Fang, H.M.; Wang, G.L. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 1994, 269, 23757–23763. [Google Scholar] [PubMed]
- Bauer, D.E.; Harris, M.H.; Plas, D.R.; Lum, J.J.; Hammerman, P.S.; Rathmell, J.C.; Riley, J.L.; Thompson, C.B. Cytokine stimulation of aerobic glycolysis in hematopoietic cells exceeds proliferative demand. FASEB J. 2004, 18, 1303–1305. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, R.P.; Finlay, D.K. Glucose, glycolysis and lymphocyte responses. Mol. Immunol. 2015, 68, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Halligan, D.N.; Murphy, S.J.E.; Taylor, C.T. The hypoxia-inducible factor (HIF) couples immunity with metabolism. Semin. Immunol. 2016, 28, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Amulic, B.; Cazalet, C.; Hayes, G.L.; Metzler, K.D.; Zychlinsky, A. Neutrophil function: From mechanisms to disease. Annu. Rev. Immunol. 2012, 30, 459–489. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.; Hyun, Y.-M.; Lambert-Emo, K.; Capece, T.; Bae, S.; Miller, R.; Topham, D.J.; Kim, M. Neutrophil trails guide influenza-specific CD8+ T cells in the airways. Science 2015, 349, aaa4352. [Google Scholar] [CrossRef] [PubMed]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.R.; Roy, S.; Leal, S.M.; Sun, Y.; Howell, S.J.; Cobb, B.A.; Li, X.; Pearlman, E. Activation of neutrophils by autocrine IL-17A-IL-17RC interactions during fungal infection is regulated by IL-6, IL-23, RORγt and dectin-2. Nat. Immunol. 2014, 15, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Rosales, C. Neutrophil: A Cell with Many Roles in Inflammation or Several Cell Types? Front. Physiol. 2018, 9, 113. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.L.; Bruyninckx, W.J.; Kelly, C.J.; Glover, L.E.; McNamee, E.N.; Bowers, B.E.; Bayless, A.J.; Scully, M.; Saeedi, B.J.; Golden-Mason, L.; et al. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity 2014, 40, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Majlessi, L.; Deriaud, E.; Leclerc, C.; Lo-Man, R. Coactivation of Syk kinase and MyD88 adaptor protein pathways by bacteria promotes regulatory properties of neutrophils. Immunity 2009, 31, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Wu, W.; Millman, A.; Craft, J.F.; Chen, E.; Patel, N.; Boucher, J.L.; Urban, J.F.; Kim, C.C.; Gause, W.C. Neutrophils prime a long-lived effector macrophage phenotype that mediates accelerated helminth expulsion. Nat. Immunol. 2014, 15, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Sturge, C.R.; Benson, A.; Raetz, M.; Wilhelm, C.L.; Mirpuri, J.; Vitetta, E.S.; Yarovinsky, F. TLR-independent neutrophil-derived IFN-γ is important for host resistance to intracellular pathogens. Proc. Natl. Acad. Sci. USA 2013, 110, 10711–10716. [Google Scholar] [CrossRef] [PubMed]
- Sbarra, A.J.; Karnovsky, M.L. The biochemical basis of phagocytosis. I. Metabolic changes during the ingestion of particles by polymorphonuclear leukocytes. J. Biol. Chem. 1959, 234, 1355–1362. [Google Scholar] [PubMed]
- Walmsley, S.R.; Cadwallader, K.A.; Chilvers, E.R. The role of HIF-1alpha in myeloid cell inflammation. Trends Immunol. 2005, 26, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Cramer, T.; Yamanishi, Y.; Clausen, B.E.; Förster, I.; Pawlinski, R.; Mackman, N.; Haase, V.H.; Jaenisch, R.; Corr, M.; Nizet, V.; et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 2003, 112, 645–657. [Google Scholar] [CrossRef]
- Harris, A.J.; Thompson, A.R.; Whyte, M.K.; Walmsley, S.R. HIF-mediated innate immune responses: Cell signaling and therapeutic implications. Hypoxia 2014, 2, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Kong, T.; Eltzschig, H.K.; Karhausen, J.; Colgan, S.P.; Shelley, C.S. Leukocyte adhesion during hypoxia is mediated by HIF-1-dependent induction of beta2 integrin gene expression. Proc. Natl. Acad. Sci. USA 2004, 101, 10440–10445. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.; Simon, M.C. Hypoxia-inducible factors: Key regulators of myeloid cells during inflammation. J. Clin. Investig. 2016, 126, 3661–3671. [Google Scholar] [CrossRef] [PubMed]
- Peyssonnaux, C.; Datta, V.; Cramer, T.; Doedens, A.; Theodorakis, E.A.; Gallo, R.L.; Hurtado-Ziola, N.; Nizet, V.; Johnson, R.S. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J. Clin. Investig. 2005, 115, 1806–1815. [Google Scholar] [CrossRef] [PubMed]
- Peyssonnaux, C.; Cejudo-Martin, P.; Doedens, A.; Zinkernagel, A.S.; Johnson, R.S.; Nizet, V. Cutting edge: Essential role of hypoxia inducible factor-1alpha in development of lipopolysaccharide-induced sepsis. J. Immunol. 2007, 178, 7516–7519. [Google Scholar] [CrossRef] [PubMed]
- Colgan, S.P.; Campbell, E.L. Oxygen metabolism and innate immune responses in the gut. J. Appl. Physiol. 2017, 123, 1321–1327. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.H.T.; Campbell, E.L.; Colgan, S.P. Neutrophils as Components of Mucosal Homeostasis. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Wéra, O.; Lancellotti, P.; Oury, C. The Dual Role of Neutrophils in Inflammatory Bowel Diseases. J. Clin. Med. 2016, 5, 118. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.A.R.; Elks, P.M.; Marriott, H.M.; Eamsamarng, S.; Higgins, K.R.; Lewis, A.; Williams, L.; Parmar, S.; Shaw, G.; McGrath, E.E.; et al. Hypoxia-inducible factor 2α regulates key neutrophil functions in humans, mice, and zebrafish. Blood 2014, 123, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Sumbayev, V.V.; Yasinska, I.; Oniku, A.E.; Streatfield, C.L.; Gibbs, B.F. Involvement of hypoxia-inducible factor-1 in the inflammatory responses of human LAD2 mast cells and basophils. PLoS ONE 2012, 7, e34259. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.-J.; Chung, H.-S.; Lee, B.-R.; Kim, S.-J.; Yoo, S.-J.; Hong, S.-H.; Kim, H.-M. Expression of proinflammatory cytokines via HIF-1alpha and NF-kappaB activation on desferrioxamine-stimulated HMC-1 cells. Biochem. Biophys. Res. Commun. 2003, 306, 805–811. [Google Scholar] [CrossRef]
- Lee, K.S.; Kim, S.R.; Park, S.J.; Min, K.H.; Lee, K.Y.; Choe, Y.H.; Park, S.Y.; Chai, O.H.; Zhang, X.; Song, C.H.; et al. Mast cells can mediate vascular permeability through regulation of the PI3K-HIF-1alpha-VEGF axis. Am. J. Respir. Crit. Care Med. 2008, 178, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Nissim Ben Efraim, A.H.; Eliashar, R.; Levi-Schaffer, F. Hypoxia modulates human eosinophil function. Clin. Mol. Allergy 2010, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Branitzki-Heinemann, K.; Möllerherm, H.; Völlger, L.; Husein, D.M.; de Buhr, N.; Blodkamp, S.; Reuner, F.; Brogden, G.; Naim, H.Y.; von Köckritz-Blickwede, M. Formation of Neutrophil Extracellular Traps under Low Oxygen Level. Front. Immunol. 2016, 7, 518. [Google Scholar] [CrossRef] [PubMed]
- Möllerherm, H.; von Köckritz-Blickwede, M.; Branitzki-Heinemann, K. Antimicrobial Activity of Mast Cells: Role and Relevance of Extracellular DNA Traps. Front. Immunol. 2016, 7, 265. [Google Scholar] [CrossRef] [PubMed]
- Crotty Alexander, L.E.; Akong-Moore, K.; Feldstein, S.; Johansson, P.; Nguyen, A.; McEachern, E.K.; Nicatia, S.; Cowburn, A.S.; Olson, J.; Cho, J.Y.; et al. Myeloid cell HIF-1α regulates asthma airway resistance and eosinophil function. J. Mol. Med. 2013, 91, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Naldini, A.; Morena, E.; Pucci, A.; Miglietta, D.; Riboldi, E.; Sozzani, S.; Carraro, F. Hypoxia affects dendritic cell survival: Role of the hypoxia-inducible factor-1α and lipopolysaccharide. J. Cell. Physiol. 2012, 227, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Wobben, R.; Hüsecken, Y.; Lodewick, C.; Gibbert, K.; Fandrey, J.; Winning, S. Role of hypoxia inducible factor-1α for interferon synthesis in mouse dendritic cells. Biol. Chem. 2013, 394, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Köhler, T.; Reizis, B.; Johnson, R.S.; Weighardt, H.; Förster, I. Influence of hypoxia-inducible factor 1α on dendritic cell differentiation and migration. Eur. J. Immunol. 2012, 42, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Hammami, A.; Charpentier, T.; Smans, M.; Stäger, S. IRF-5-Mediated Inflammation Limits CD8+ T Cell Expansion by Inducing HIF-1α and Impairing Dendritic Cell Functions during Leishmania Infection. PLoS Pathog. 2015, 11, e1004938. [Google Scholar] [CrossRef] [PubMed]
- Jantsch, J.; Chakravortty, D.; Turza, N.; Prechtel, A.T.; Buchholz, B.; Gerlach, R.G.; Volke, M.; Gläsner, J.; Warnecke, C.; Wiesener, M.S.; et al. Hypoxia and hypoxia-inducible factor-1 alpha modulate lipopolysaccharide-induced dendritic cell activation and function. J. Immunol. 2008, 180, 4697–4705. [Google Scholar] [CrossRef] [PubMed]
- Bosco, M.C.; Varesio, L. Dendritic cell reprogramming by the hypoxic environment. Immunobiology 2012, 217, 1241–1249. [Google Scholar] [CrossRef] [PubMed]
- Flück, K.; Breves, G.; Fandrey, J.; Winning, S. Hypoxia-inducible factor 1 in dendritic cells is crucial for the activation of protective regulatory T cells in murine colitis. Mucosal Immunol. 2016, 9, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, C.M.; Finlay, D.K. What Fuels Natural Killers? Metabolism and NK Cell Responses. Front. Immunol. 2017, 8, 367. [Google Scholar] [CrossRef] [PubMed]
- Krzywinska, E.; Kantari-Mimoun, C.; Kerdiles, Y.; Sobecki, M.; Isagawa, T.; Gotthardt, D.; Castells, M.; Haubold, J.; Millien, C.; Viel, T.; et al. Loss of HIF-1α in natural killer cells inhibits tumour growth by stimulating non-productive angiogenesis. Nat. Commun. 2017, 8, 1597. [Google Scholar] [CrossRef] [PubMed]
- Nizet, V.; Johnson, R.S. Interdependence of hypoxic and innate immune responses. Nat. Rev. Immunol. 2009, 9, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Germeraad, W.T.V.; Rouschop, K.M.A.; Steeghs, E.M.P.; van Gelder, M.; Bos, G.M.J.; Wieten, L. Hypoxia induced impairment of NK cell cytotoxicity against multiple myeloma can be overcome by IL-2 activation of the NK cells. PLoS ONE 2013, 8, e64835. [Google Scholar] [CrossRef] [PubMed]
- Berchem, G.; Noman, M.Z.; Bosseler, M.; Paggetti, J.; Baconnais, S.; Le Cam, E.; Nanbakhsh, A.; Moussay, E.; Mami-Chouaib, F.; Janji, B.; et al. Hypoxic tumor-derived microvesicles negatively regulate NK cell function by a mechanism involving TGF-β and miR23a transfer. Oncoimmunology 2016, 5, e1062968. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Janji, B.; Berchem, G.; Chouaib, S. MiR-210 and hypoxic microvesicles: Two critical components of hypoxia involved in the regulation of killer cells function. Cancer Lett. 2016, 380, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Sceneay, J.; Chow, M.T.; Chen, A.; Halse, H.M.; Wong, C.S.F.; Andrews, D.M.; Sloan, E.K.; Parker, B.S.; Bowtell, D.D.; Smyth, M.J.; et al. Primary Tumor Hypoxia Recruits CD11b+/Ly6Cmed/Ly6G+ Immune Suppressor Cells and Compromises NK Cell Cytotoxicity in the Premetastatic Niche. Cancer Res. 2012, 72, 3906–3911. [Google Scholar] [CrossRef] [PubMed]
- Wolter, F.; Glässner, A.; Krämer, B.; Kokordelis, P.; Finnemann, C.; Kaczmarek, D.J.; Goeser, F.; Lutz, P.; Nischalke, H.D.; Strassburg, C.P.; et al. Hypoxia impairs anti-viral activity of natural killer (NK) cells but has little effect on anti-fibrotic NK cell functions in hepatitis C virus infection. J. Hepatol. 2015, 63, 1334–1344. [Google Scholar] [CrossRef] [PubMed]
- Baginska, J.; Viry, E.; Berchem, G.; Poli, A.; Noman, M.Z.; van Moer, K.; Medves, S.; Zimmer, J.; Oudin, A.; Niclou, S.P.; et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc. Natl. Acad. Sci. USA 2013, 110, 17450–17455. [Google Scholar] [CrossRef] [PubMed]
- Viry, E.; Baginska, J.; Berchem, G.; Noman, M.Z.; Medves, S.; Chouaib, S.; Janji, B. Autophagic degradation of GZMB/granzyme B: A new mechanism of hypoxic tumor cell escape from natural killer cell-mediated lysis. Autophagy 2014, 10, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Li, D.; Zhang, X.; Wan, Q.; Zhang, W.; Zheng, M.; Zou, L.; Elly, C.; Lee, J.H.; Liu, Y.-C. E3 Ligase VHL Promotes Group 2 Innate Lymphoid Cell Maturation and Function via Glycolysis Inhibition and Induction of Interleukin-33 Receptor. Immunity 2018, 48, 258.e5–270.e5. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.-J.; Wang, L.-Y.; Chodosh, L.A.; Keith, B.; Simon, M.C. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol. Cell. Biol. 2003, 23, 9361–9374. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.; O’Neill, L.A.J. Succinate: A metabolic signal in inflammation. Trends Cell Biol. 2014, 24, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Galván-Peña, S.; O’Neill, L.A.J. Metabolic reprograming in macrophage polarization. Front. Immunol. 2014, 5, 420. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Blouin, C.C.; Pagé, E.L.; Soucy, G.M.; Richard, D.E. Hypoxic gene activation by lipopolysaccharide in macrophages: Implication of hypoxia-inducible factor 1alpha. Blood 2004, 103, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J.; Pearce, E.J. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 2016, 213, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Koivunen, P.; Hirsilä, M.; Remes, A.M.; Hassinen, I.E.; Kivirikko, K.I.; Myllyharju, J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: Possible links between cell metabolism and stabilization of HIF. J. Biol. Chem. 2007, 282, 4524–4532. [Google Scholar] [CrossRef] [PubMed]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Takeda, N.; O’Dea, E.L.; Doedens, A.; Kim, J.; Weidemann, A.; Stockmann, C.; Asagiri, M.; Simon, M.C.; Hoffmann, A.; Johnson, R.S. Differential activation and antagonistic function of HIF-{alpha} isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010, 24, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Imtiyaz, H.Z.; Williams, E.P.; Hickey, M.M.; Patel, S.A.; Durham, A.C.; Yuan, L.-J.; Hammond, R.; Gimotty, P.A.; Keith, B.; Simon, M.C. Hypoxia-inducible factor 2alpha regulates macrophage function in mouse models of acute and tumor inflammation. J. Clin. Investig. 2010, 120, 2699–2714. [Google Scholar] [CrossRef] [PubMed]
- Walmsley, S.R.; Print, C.; Farahi, N.; Peyssonnaux, C.; Johnson, R.S.; Cramer, T.; Sobolewski, A.; Condliffe, A.M.; Cowburn, A.S.; Johnson, N.; et al. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J. Exp. Med. 2005, 201, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Green, D.R. The immune diet: Meeting the metabolic demands of lymphocyte activation. F1000 Biol. Rep. 2012, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Gnanaprakasam, J.N.R.; Sherman, J.W.; Wang, R. MYC and HIF in shaping immune response and immune metabolism. Cytokine Growth Factor Rev. 2017, 35, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.Z.; Wang, R.; Huang, G.; Vogel, P.; Neale, G.; Green, D.R.; Chi, H. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 2011, 208, 1367–1376. [Google Scholar] [CrossRef] [PubMed]
- Dang, E.V.; Barbi, J.; Yang, H.-Y.; Jinasena, D.; Yu, H.; Zheng, Y.; Bordman, Z.; Fu, J.; Kim, Y.; Yen, H.-R.; et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 2011, 146, 772–784. [Google Scholar] [CrossRef] [PubMed]
- Clambey, E.T.; McNamee, E.N.; Westrich, J.A.; Glover, L.E.; Campbell, E.L.; Jedlicka, P.; de Zoeten, E.F.; Cambier, J.C.; Stenmark, K.R.; Colgan, S.P.; et al. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc. Natl. Acad. Sci. USA 2012, 109, E2784–E2793. [Google Scholar] [CrossRef] [PubMed]
- Westendorf, A.M.; Skibbe, K.; Adamczyk, A.; Buer, J.; Geffers, R.; Hansen, W.; Pastille, E.; Jendrossek, V. Hypoxia Enhances Immunosuppression by Inhibiting CD4+ Effector T Cell Function and Promoting Treg Activity. Cell. Physiol. Biochem. 2017, 41, 1271–1284. [Google Scholar] [CrossRef] [PubMed]
- Mascanfroni, I.D.; Takenaka, M.C.; Yeste, A.; Patel, B.; Wu, Y.; Kenison, J.E.; Siddiqui, S.; Basso, A.S.; Otterbein, L.E.; Pardoll, D.M.; et al. Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-α. Nat. Med. 2015, 21, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Pot, C.; Apetoh, L.; Awasthi, A.; Kuchroo, V.K. Induction of regulatory Tr1 cells and inhibition of T(H)17 cells by IL-27. Semin. Immunol. 2011, 23, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Tyrakis, P.A.; Palazon, A.; Macias, D.; Lee, K.L.; Phan, A.T.; Veliça, P.; You, J.; Chia, G.S.; Sim, J.; Doedens, A.; et al. S-2-hydroxyglutarate regulates CD8+ T-lymphocyte fate. Nature 2016, 540, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Finlay, D.K.; Rosenzweig, E.; Sinclair, L.V.; Feijoo-Carnero, C.; Hukelmann, J.L.; Rolf, J.; Panteleyev, A.A.; Okkenhaug, K.; Cantrell, D.A. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J. Exp. Med. 2012, 209, 2441–2453. [Google Scholar] [CrossRef] [PubMed]
- Sukumar, M.; Liu, J.; Ji, Y.; Subramanian, M.; Crompton, J.G.; Yu, Z.; Roychoudhuri, R.; Palmer, D.C.; Muranski, P.; Karoly, E.D.; et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Investig. 2013, 123, 4479–4488. [Google Scholar] [CrossRef] [PubMed]
- Biju, M.P.; Neumann, A.K.; Bensinger, S.J.; Johnson, R.S.; Turka, L.A.; Haase, V.H. Vhlh gene deletion induces Hif-1-mediated cell death in thymocytes. Mol. Cell. Biol. 2004, 24, 9038–9047. [Google Scholar] [CrossRef] [PubMed]
- Shehade, H.; Acolty, V.; Moser, M.; Oldenhove, G. Cutting Edge: Hypoxia-Inducible Factor 1 Negatively Regulates Th1 Function. J. Immunol. 2015, 195, 1372–1376. [Google Scholar] [CrossRef] [PubMed]
- Higashiyama, M.; Hokari, R.; Hozumi, H.; Kurihara, C.; Ueda, T.; Watanabe, C.; Tomita, K.; Nakamura, M.; Komoto, S.; Okada, Y.; et al. HIF-1 in T cells ameliorated dextran sodium sulfate-induced murine colitis. J. Leukoc. Biol. 2012, 91, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Raybuck, A.L.; Stengel, K.; Wei, M.; Beck, T.C.; Volanakis, E.; Thomas, J.W.; Hiebert, S.; Haase, V.H.; Boothby, M.R. Germinal centre hypoxia and regulation of antibody qualities by a hypoxia response system. Nature 2016, 537, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Abbott, R.K.; Thayer, M.; Labuda, J.; Silva, M.; Philbrook, P.; Cain, D.W.; Kojima, H.; Hatfield, S.; Sethumadhavan, S.; Ohta, A.; et al. Germinal Center Hypoxia Potentiates Immunoglobulin Class Switch Recombination. J. Immunol. 2016, 197, 4014–4020. [Google Scholar] [CrossRef] [PubMed]
- Jellusova, J.; Cato, M.H.; Apgar, J.R.; Ramezani-Rad, P.; Leung, C.R.; Chen, C.; Richardson, A.D.; Conner, E.M.; Benschop, R.J.; Woodgett, J.R.; et al. Gsk3 is a metabolic checkpoint regulator in B cells. Nat. Immunol. 2017, 18, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Caro-Maldonado, A.; Wang, R.; Nichols, A.G.; Kuraoka, M.; Milasta, S.; Sun, L.D.; Gavin, A.L.; Abel, E.D.; Kelsoe, G.; Green, D.R.; et al. Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed B cells. J. Immunol. 2014, 192, 3626–3636. [Google Scholar] [CrossRef] [PubMed]
- Kojima, H.; Gu, H.; Nomura, S.; Caldwell, C.C.; Kobata, T.; Carmeliet, P.; Semenza, G.L.; Sitkovsky, M.V. Abnormal B lymphocyte development and autoimmunity in hypoxia-inducible factor 1alpha -deficient chimeric mice. Proc. Natl. Acad. Sci. USA 2002, 99, 2170–2174. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.H.; Lin, H.; Zheng, H.; Kim, K.S.; Kim, J.Y.; Chun, Y.S.; Park, J.W.; Nam, J.H.; Kim, W.K.; Zhang, Y.H.; et al. HIF-1α-mediated upregulation of TASK-2 K+ channels augments Ca2+ signaling in mouse B cells under hypoxia. J. Immunol. 2014, 193, 4924–4933. [Google Scholar] [CrossRef] [PubMed]
- Goda, N.; Ryan, H.E.; Khadivi, B.; McNulty, W.; Rickert, R.C.; Johnson, R.S. Hypoxia-inducible factor 1alpha is essential for cell cycle arrest during hypoxia. Mol. Cell. Biol. 2003, 23, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, L.A.; McPhail, L.C.; Henson, P.M.; Johnston, R.B. Priming of neutrophils for enhanced release of oxygen metabolites by bacterial lipopolysaccharide. Evidence for increased activity of the superoxide-producing enzyme. J. Exp. Med. 1984, 160, 1656–1671. [Google Scholar] [CrossRef] [PubMed]
- Rigoni, A.; Colombo, M.P.; Pucillo, C. Mast cells, basophils and eosinophils: From allergy to cancer. Semin. Immunol. 2018, 35, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Bradding, P.; Arthur, G. Mast cells in asthma—State of the art. Clin. Exp. Allergy 2016, 46, 194–263. [Google Scholar] [CrossRef] [PubMed]
- Dahlin, J.S.; Hallgren, J. Mast cell progenitors: Origin, development and migration to tissues. Mol. Immunol. 2015, 63, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Stone, K.D.; Prussin, C.; Metcalfe, D.D. IgE, mast cells, basophils, and eosinophils. J. Allergy Clin. Immunol. 2010, 125, S73–S80. [Google Scholar] [CrossRef] [PubMed]
- Schneider, E.; Thieblemont, N.; De Moraes, M.L.; Dy, M. Basophils: New players in the cytokine network. Eur. Cytokine Netw. 2010, 21, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Voehringer, D. Basophils in allergic immune responses. Curr. Opin. Immunol. 2011, 23, 789–793. [Google Scholar] [CrossRef] [PubMed]
- Muniz, V.S.; Baptista-dos-Reis, R.; Neves, J.S. Functional Extracellular Eosinophil Granules: A Bomb Caught in a Trap. Int. Arch. Allergy Immunol. 2013, 162, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Hogan, S.P.; Rosenberg, H.F.; Moqbel, R.; Phipps, S.; Foster, P.S.; Lacy, P.; Kay, A.B.; Rothenberg, M.E. Eosinophils: Biological Properties and Role in Health and Disease. Clin. Exp. Allergy 2008, 38, 709–750. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, E.A.; Taranova, A.G.; Lee, N.A.; Lee, J.J. Eosinophils: Singularly destructive effector cells or purveyors of immunoregulation? J. Allergy Clin. Immunol. 2007, 119, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Sumbayev, V.V.; Nicholas, S.A.; Streatfield, C.L.; Gibbs, B.F. Involvement of hypoxia-inducible factor-1 HiF(1α) in IgE-mediated primary human basophil responses: Molecular immunology. Eur. J. Immunol. 2009, 39, 3511–3519. [Google Scholar] [CrossRef] [PubMed]
- Labonte, A.C.; Tosello-Trampont, A.-C.; Hahn, Y.S. The role of macrophage polarization in infectious and inflammatory diseases. Mol. Cells 2014, 37, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, S.Z.; Plevy, S.E. The role of the macrophage in sentinel responses in intestinal immunity. Curr. Opin. Gastroenterol. 2010, 26, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Allavena, P.; Mantovani, A. Tumor-associated macrophages: Functional diversity, clinical significance, and open questions. Semin. Immunopathol. 2013, 35, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Dehne, N.; Brüne, B. Nitric oxide causes macrophage migration via the HIF-1-stimulated small GTPases Cdc42 and Rac1. Free Radic. Biol. Med. 2009, 47, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Schioppa, T.; Uranchimeg, B.; Saccani, A.; Biswas, S.K.; Doni, A.; Rapisarda, A.; Bernasconi, S.; Saccani, S.; Nebuloni, M.; Vago, L.; et al. Regulation of the chemokine receptor CXCR4 by hypoxia. J. Exp. Med. 2003, 198, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Bosco, M.C.; Reffo, G.; Puppo, M.; Varesio, L. Hypoxia inhibits the expression of the CCR5 chemokine receptor in macrophages. Cell. Immunol. 2004, 228, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Casazza, A.; Laoui, D.; Wenes, M.; Rizzolio, S.; Bassani, N.; Mambretti, M.; Deschoemaeker, S.; Van Ginderachter, J.A.; Tamagnone, L.; Mazzone, M. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell 2013, 24, 695–709. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Seiki, M. A Membrane Protease Regulates Energy Production in Macrophages by Activating Hypoxia-inducible Factor-1 via a Non-proteolytic Mechanism. J. Biol. Chem. 2010, 285, 29951–29964. [Google Scholar] [CrossRef] [PubMed]
- Mildner, A.; Jung, S. Development and Function of Dendritic Cell Subsets. Immunity 2014, 40, 642–656. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, T.; Nizet, V. Hypoxia-Inducible Factor (HIF) as a Pharmacological Target for Prevention and Treatment of Infectious Diseases. Infect. Dis. Ther. 2014, 3, 159–174. [Google Scholar] [CrossRef] [PubMed]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The Dendritic Cell Lineage: Ontogeny and Function of Dendritic Cells and Their Subsets in the Steady State and the Inflamed Setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef] [PubMed]
- Reizis, B. Regulation of plasmacytoid dendritic cell development. Curr. Opin. Immunol. 2010, 22, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Reizis, B.; Bunin, A.; Ghosh, H.S.; Lewis, K.L.; Sisirak, V. Plasmacytoid dendritic cells: Recent progress and open questions. Annu. Rev. Immunol. 2011, 29, 163–183. [Google Scholar] [CrossRef] [PubMed]
- Villadangos, J.A.; Schnorrer, P. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat. Rev. Immunol. 2007, 7, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Segura, E.; Villadangos, J.A. Antigen presentation by dendritic cells in vivo. Curr. Opin. Immunol. 2009, 21, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Joffre, O.P.; Segura, E.; Savina, A.; Amigorena, S. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 2012, 12, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Förster, R.; Braun, A.; Worbs, T. Lymph node homing of T cells and dendritic cells via afferent lymphatics. Trends Immunol. 2012, 33, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Masopust, D.; Schenkel, J.M. The integration of T cell migration, differentiation and function. Nat. Rev. Immunol. 2013, 13, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Josefowicz, S.Z.; Lu, L.-F.; Rudensky, A.Y. Regulatory T cells: Mechanisms of differentiation and function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, T.; Skepner, J.; Yang, J. Targeting Th17 Effector Cytokines for the Treatment of Autoimmune Diseases. Arch. Immunol. Ther. Exp. 2015, 63, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Tabarkiewicz, J.; Pogoda, K.; Karczmarczyk, A.; Pozarowski, P.; Giannopoulos, K. The Role of IL-17 and Th17 Lymphocytes in Autoimmune Diseases. Arch. Immunol. Ther. Exp. 2015, 63, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Makino, Y.; Nakamura, H.; Ikeda, E.; Ohnuma, K.; Yamauchi, K.; Yabe, Y.; Poellinger, L.; Okada, Y.; Morimoto, C.; Tanaka, H. Hypoxia-inducible factor regulates survival of antigen receptor-driven T cells. J. Immunol. 2003, 171, 6534–6540. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.-Y.; Mauro, C. Similarities in the Metabolic Reprogramming of Immune System and Endothelium. Front. Immunol. 2017, 8, 837. [Google Scholar] [CrossRef] [PubMed]
- Kouidhi, S.; Elgaaied, A.B.; Chouaib, S. Impact of Metabolism on T-Cell Differentiation and Function and Cross Talk with Tumor Microenvironment. Front. Immunol. 2017, 8, 270. [Google Scholar] [CrossRef] [PubMed]
- Talaat, R.M.; Mohamed, S.F.; Bassyouni, I.H.; Raouf, A.A. Th1/Th2/Th17/Treg cytokine imbalance in systemic lupus erythematosus (SLE) patients: Correlation with disease activity. Cytokine 2015, 72, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Mo, H.; Li, D.; Luo, X.; Zhang, L. Th17/Treg imbalance induced by increased incidence of atherosclerosis in patients with systemic lupus erythematosus (SLE). Clin. Rheumatol. 2013, 32, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Shao, S.; Jiao, Z.; Guo, M.; Xu, H.; Wang, S. The Th17/Treg imbalance and cytokine environment in peripheral blood of patients with rheumatoid arthritis. Rheumatol. Int. 2012, 32, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Niu, Q.; Cai, B.; Huang, Z.; Shi, Y.; Wang, L. Disturbed Th17/Treg balance in patients with rheumatoid arthritis. Rheumatol. Int. 2012, 32, 2731–2736. [Google Scholar] [CrossRef] [PubMed]
- Kong, N.; Lan, Q.; Chen, M.; Wang, J.; Shi, W.; Horwitz, D.A.; Quesniaux, V.; Ryffel, B.; Liu, Z.; Brand, D.; et al. Antigen-specific transforming growth factor β-induced Treg cells, but not natural Treg cells, ameliorate autoimmune arthritis in mice by shifting the Th17/Treg cell balance from Th17 predominance to Treg cell predominance. Arthritis Rheum. 2012, 64, 2548–2558. [Google Scholar] [CrossRef] [PubMed]
- Naghavian, R.; Ghaedi, K.; Kiani-Esfahani, A.; Ganjalikhani-Hakemi, M.; Etemadifar, M.; Nasr-Esfahani, M.H. miR-141 and miR-200a, Revelation of New Possible Players in Modulation of Th17/Treg Differentiation and Pathogenesis of Multiple Sclerosis. PLoS ONE 2015, 10, e0124555. [Google Scholar] [CrossRef] [PubMed]
- Jamshidian, A.; Shaygannejad, V.; Pourazar, A.; Zarkesh-Esfahani, S.-H.; Gharagozloo, M. Biased Treg/Th17 balance away from regulatory toward inflammatory phenotype in relapsed multiple sclerosis and its correlation with severity of symptoms. J. Neuroimmunol. 2013, 262, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Eastaff-Leung, N.; Mabarrack, N.; Barbour, A.; Cummins, A.; Barry, S. Foxp3+ regulatory T cells, Th17 effector cells, and cytokine environment in inflammatory bowel disease. J. Clin. Immunol. 2010, 30, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Bai, A.; Lu, N.; Guo, Y.; Liu, Z.; Chen, J.; Peng, Z. All-trans retinoic acid down-regulates inflammatory responses by shifting the Treg/Th17 profile in human ulcerative and murine colitis. J. Leukoc. Biol. 2009, 86, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, Y.; Yang, X.; Wei, J.; Zhou, S.; Zhao, Z.; Cheng, J.; Duan, H.; Jia, T.; Lei, Q.; et al. Characterization of Th17 and FoxP3(+) Treg Cells in Paediatric Psoriasis Patients. Scand. J. Immunol. 2016, 83, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Palazon, A.; Tyrakis, P.A.; Macias, D.; Veliça, P.; Rundqvist, H.; Fitzpatrick, S.; Vojnovic, N.; Phan, A.T.; Loman, N.; Hedenfalk, I.; et al. An HIF-1α/VEGF-A Axis in Cytotoxic T Cells Regulates Tumor Progression. Cancer Cell 2017, 32, 669.e5–683.e5. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.L.; Poffenberger, M.C.; Chang, C.-H.; Jones, R.G. Fueling immunity: Insights into metabolism and lymphocyte function. Science 2013, 342, 1242454. [Google Scholar] [CrossRef] [PubMed]
- Phan, A.T.; Goldrath, A.W. Hypoxia-inducible factors regulate T cell metabolism and function. Mol. Immunol. 2015, 68, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.D.; O’Sullivan, D.; Pearce, E.L. T cell metabolism drives immunity. J. Exp. Med. 2015, 212, 1345–1360. [Google Scholar] [CrossRef] [PubMed]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [PubMed]
- Phan, A.T.; Doedens, A.L.; Palazon, A.; Tyrakis, P.A.; Cheung, K.P.; Johnson, R.S.; Goldrath, A.W. Constitutive Glycolytic Metabolism Supports CD8+ T Cell Effector Memory Differentiation during Viral Infection. Immunity 2016, 45, 1024–1037. [Google Scholar] [CrossRef] [PubMed]
- Pieper, K.; Grimbacher, B.; Eibel, H. B-cell biology and development. J. Allergy Clin. Immunol. 2013, 131, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, W.; Lakkis, F.G.; Chalasani, G. B Cells, Antibodies, and More. Clin. J. Am. Soc. Nephrol. 2016, 11, 137–154. [Google Scholar] [CrossRef] [PubMed]
- Kalampokis, I.; Yoshizaki, A.; Tedder, T.F. IL-10-producing regulatory B cells (B10 cells) in autoimmune disease. Arthritis Res. Ther. 2013, 15, S1. [Google Scholar] [CrossRef] [PubMed]
- Shen, P.; Roch, T.; Lampropoulou, V.; O’Connor, R.A.; Stervbo, U.; Hilgenberg, E.; Ries, S.; Dang, V.D.; Jaimes, Y.; Daridon, C.; et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature 2014, 507, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Stott, R.T.; Zhao, G.; SooHoo, J.; Xiong, W.; Lian, M.M.; Fitzgerald, L.; Shi, S.; Akrawi, E.; Lei, J.; et al. TGF-β-producing regulatory B cells induce regulatory T cells and promote transplantation tolerance: Immunomodulation. Eur. J. Immunol. 2014, 44, 1728–1736. [Google Scholar] [CrossRef] [PubMed]
- Mauri, C.; Bosma, A. Immune Regulatory Function of B Cells. Annu. Rev. Immunol. 2012, 30, 221–241. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Grötsch, B.; Luo, Y.; Knaup, K.X.; Wiesener, M.S.; Chen, X.-X.; Jantsch, J.; Fillatreau, S.; Schett, G.; Bozec, A. Hypoxia-inducible factor-1α is a critical transcription factor for IL-10-producing B cells in autoimmune disease. Nat. Commun. 2018, 9, 251. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, C.; Kharabi Masouleh, S.; Kazakov, A. Metabolic Regulation of Innate Lymphoid Cell-Mediated Tissue Protection-Linking the Nutritional State to Barrier Immunity. Front. Immunol. 2017, 8, 1742. [Google Scholar] [CrossRef] [PubMed]
- Marçais, A.; Cherfils-Vicini, J.; Viant, C.; Degouve, S.; Viel, S.; Fenis, A.; Rabilloud, J.; Mayol, K.; Tavares, A.; Bienvenu, J.; et al. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat. Immunol. 2014, 15, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Keppel, M.P.; Saucier, N.; Mah, A.Y.; Vogel, T.P.; Cooper, M.A. Activation-specific metabolic requirements for NK Cell IFN-γ production. J. Immunol. 2015, 194, 1954–1962. [Google Scholar] [CrossRef] [PubMed]
- Velásquez, S.Y.; Killian, D.; Schulte, J.; Sticht, C.; Thiel, M.; Lindner, H.A. Short Term Hypoxia Synergizes with Interleukin 15 Priming in Driving Glycolytic Gene Transcription and Supports Human Natural Killer Cell Activities. J. Biol. Chem. 2016, 291, 12960–12977. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, R.P.; Loftus, R.M.; Keating, S.E.; Liou, K.T.; Biron, C.A.; Gardiner, C.M.; Finlay, D.K. mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. J. Immunol. 2014, 193, 4477–4484. [Google Scholar] [CrossRef] [PubMed]
- Keating, S.E.; Zaiatz-Bittencourt, V.; Loftus, R.M.; Keane, C.; Brennan, K.; Finlay, D.K.; Gardiner, C.M. Metabolic Reprogramming Supports IFN-γ Production by CD56bright NK Cells. J. Immunol. 2016, 196, 2552–2560. [Google Scholar] [CrossRef] [PubMed]
- Viel, S.; Marçais, A.; Guimaraes, F.S.-F.; Loftus, R.; Rabilloud, J.; Grau, M.; Degouve, S.; Djebali, S.; Sanlaville, A.; Charrier, E.; et al. TGF-β inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci. Signal. 2016, 9, ra19. [Google Scholar] [CrossRef] [PubMed]
- Moro, K.; Yamada, T.; Tanabe, M.; Takeuchi, T.; Ikawa, T.; Kawamoto, H.; Furusawa, J.-I.; Ohtani, M.; Fujii, H.; Koyasu, S. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 2010, 463, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Saenz, S.A.; Siracusa, M.C.; Perrigoue, J.G.; Spencer, S.P.; Urban, J.F.; Tocker, J.E.; Budelsky, A.L.; Kleinschek, M.A.; Kastelein, R.A.; Kambayashi, T.; et al. IL25 elicits a multipotent progenitor cell population that promotes T(H)2 cytokine responses. Nature 2010, 464, 1362–1366. [Google Scholar] [CrossRef] [PubMed]
- Artis, D.; Spits, H. The biology of innate lymphoid cells. Nature 2015, 517, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Licona-Limón, P.; Kim, L.K.; Palm, N.W.; Flavell, R.A. TH2, allergy and group 2 innate lymphoid cells. Nat. Immunol. 2013, 14, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Everaere, L.; Ait Yahia, S.; Bouté, M.; Audousset, C.; Chenivesse, C.; Tsicopoulos, A. Innate lymphoid cells at the interface between obesity and asthma. Immunology 2018, 153, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Ebbo, M.; Crinier, A.; Vély, F.; Vivier, E. Innate lymphoid cells: Major players in inflammatory diseases. Nat. Rev. Immunol. 2017, 17, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Cella, M.; Fuchs, A.; Vermi, W.; Facchetti, F.; Otero, K.; Lennerz, J.K.M.; Doherty, J.M.; Mills, J.C.; Colonna, M. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature 2009, 457, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Satoh-Takayama, N.; Vosshenrich, C.A.J.; Lesjean-Pottier, S.; Sawa, S.; Lochner, M.; Rattis, F.; Mention, J.-J.; Thiam, K.; Cerf-Bensussan, N.; Mandelboim, O.; et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity 2008, 29, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Satoh-Takayama, N. Heterogeneity and diversity of group 3 innate lymphoid cells: New cells on the block. Int. Immunol. 2016, 28, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Louis, C.A.; Reichner, J.S.; Henry, W.L.; Mastrofrancesco, B.; Gotoh, T.; Mori, M.; Albina, J.E. Distinct arginase isoforms expressed in primary and transformed macrophages: Regulation by oxygen tension. Am. J. Physiol. 1998, 274, R775–R782. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
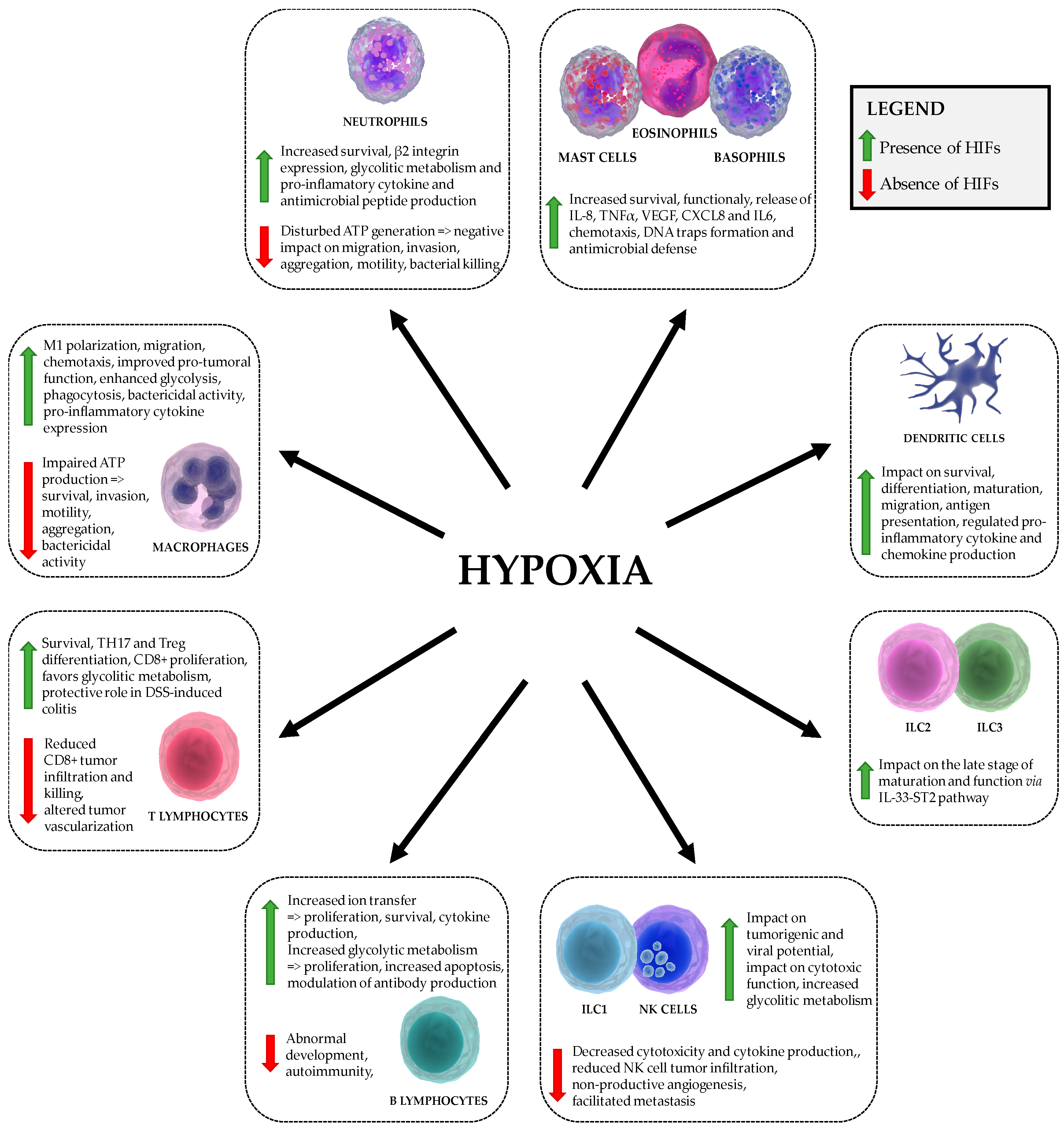{kind=link}
| Cell Type | HIFα-Mediated Effects |
|---|---|
| basophils, eosinophils, mast cells | HIF1: Survival and function, chemotaxis, IL-8 and TNF-α production, stimulation of VEGF, CXCL8 and IL-6 production, formation of DNA traps [55,56,57,58,59,60,61] HIF2: Chemotaxis [61] |
| dendritic cells | HIF1: Survival, migration, pro-inflammatory cytokine (INF, IL-22, IL-10) production, differentiation, activation, T cell stimulation, antigen presentation [62,63,64,65,66,67,68] |
| innate lymphoid cells ILC1 and NK cells, ILC2, ILC3 | HIF1 (NK cells): Metabolic reprogramming, impact on tumorigenic and viral potential, impact on number of tumor infiltrating NK cells expressing sVEGFR1, impact on cytotoxic function [28,69,70,71,72,73,74,75,76,77,78] HIF1 (ILC2): impact on the late stage of maturation and function via IL33-ST2 pathway [79] |
| macrophages Classic M1 macrophages, Regulatory M2 macrophages | HIF1: M1 polarization, motility, aggregation, invasion, metabolism, phagocytosis, chemotaxis, bactericidal activity, tumorigenic potential, expression of pro-inflammatory cytokines, increased TLR4 expression [45,48,49,50,80,81,82,83,84,85,86,87] HIF2: M1 polarization, motility, metabolism, bactericidal activity, tumorigenic potential [88,89] |
| neutrophils | HIF1: Survival, migration, invasion, bactericidal activity, promoted the on-state, increased pro-inflammatory cytokine production and nitric oxide [45,46,47,48,49,50,90] HIF2: Survival, increased resistance to nitrosative stress (Catalase) [46,54] |
| T cells T helper CD4+, Cytotoxic CD8+ T cells | HIF1: TH17 and Treg differentiation, survival, proliferation, migration, metabolic reprogramming; CD4+: increased IL17A production; CD8+: increased cytolitic activity, granzyme and perforin production and expression of costimulatory/inhibitory molecules (CTLA-4, GITR, 4-1BB) [91,92,93,94,95,96,97,98,99,100,101,102,103,104] HIF2: T cell suppression (Arginase), impact on thymocyte development [46] |
| B cells | HIF1: Abnormal B-cell development, impact on proliferation and cell death, autoimmunity, ion transfer, enhanced IgG2c production [105,106,107,108,109,110,111] HIF2: impact on proliferation and cell death, enhanced IgG2c production [105,109,111] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krzywinska, E.; Stockmann, C. Hypoxia, Metabolism and Immune Cell Function. Biomedicines 2018, 6, 56. https://doi.org/10.3390/biomedicines6020056
Krzywinska E, Stockmann C. Hypoxia, Metabolism and Immune Cell Function. Biomedicines. 2018; 6(2):56. https://doi.org/10.3390/biomedicines6020056
Chicago/Turabian StyleKrzywinska, Ewelina, and Christian Stockmann. 2018. "Hypoxia, Metabolism and Immune Cell Function" Biomedicines 6, no. 2: 56. https://doi.org/10.3390/biomedicines6020056
APA StyleKrzywinska, E., & Stockmann, C. (2018). Hypoxia, Metabolism and Immune Cell Function. Biomedicines, 6(2), 56. https://doi.org/10.3390/biomedicines6020056