Human Granzyme B Based Targeted Cytolytic Fusion Proteins

 ,
, 
Abstract
:1. Introduction to Targeted Therapy and Humanization of Immunotoxins
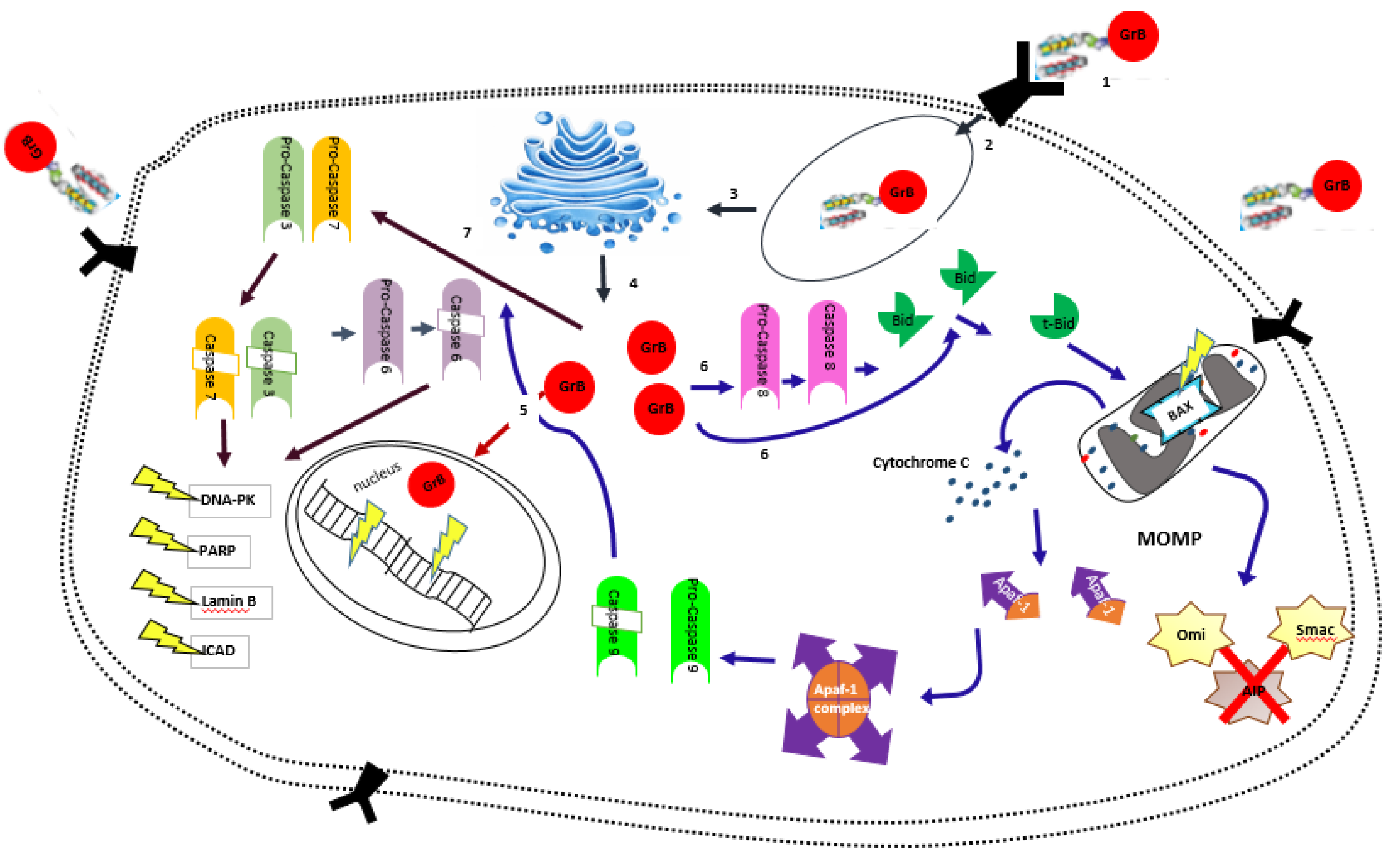
2. Granzyme B and Its Anti-Tumor Activity
Granzyme B in Targeted Therapy
3. Reducing the Off-Target Toxicity of Granzyme B for Future Targeted Therapies
4. Overcoming Natural Granzyme Inhibitors
5. Effective Cytosolic Delivery of Granzyme B to Target Cells: A Major Bottleneck
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Akbari, B.; Farajnia, S.; Ahdi Khosroshahi, S.; Safari, F.; Yousefi, M.; Dariushnejad, H.; Rahbarnia, L. Immunotoxins in cancer therapy: Review and update. Int. Rev. Immunol. 2017, 36, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Onda, M.; Lee, B.; Kreitman, R.J.; Hassan, R.; Xiang, L.; Pastan, I. Recombinant immunotoxin engineered for low immunogenicity and antigenicity by identifying and silencing human B-cell epitopes. Proc. Natl. Acad. Sci. USA 2012, 109, 11782–11787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Marks, J.D.; Huang, Q.; Rudnick, S.I.; Xiong, C.; Hittelman, W.N.; Wen, X.; Marks, J.W.; Cheung, L.H.; Boland, K.; et al. Single-Chain Antibody-Based Immunotoxins Targeting Her2/neu: Design Optimization and Impact of Affinity on Antitumor Efficacy and Off-Target Toxicity. Mol. Cancer Ther. 2012, 11, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, H.; Weng, A.; Gilabert-Oriol, R. Augmenting the efficacy of immunotoxins and other targeted protein toxins by endosomal escape enhancers. Toxins (Basel) 2016, 8, 200. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-F.; Xiang, L.; FitzGerald, D.J.; Pastan, I. Antitumor Effects of Immunotoxins Are Enhanced by Lowering HCK or Treatment with Src Kinase Inhibitors. Mol. Cancer Ther. 2014, 13, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Kessler, C.; Pardo, A.; Tur, M.K.; Gattenlöhner, S.; Fischer, R.; Kolberg, K.; Barth, S. Novel PSCA targeting scFv-fusion proteins for diagnosis and immunotherapy of prostate cancer. J. Cancer Res. Clin. Oncol. 2017, 143, 2025–2038. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Hu, Y.; Tan, Y.; Shen, Z.; Jiang, S.; Qian, H.; Liang, B.; Shan, D. Immunotherapy of lymphomas with T cells modified by anti-CD20 scFv/CD28/ CD3ζ recombinant gene. Leuk. Lymphoma 2008, 49, 1368–1373. [Google Scholar] [CrossRef] [PubMed]
- Bochicchio, A.; Jordaan, S.; Losasso, V.; Chetty, S.; Perera, R.; Ippoliti, E.; Barth, S.; Carloni, P. Designing the Sniper: Improving Targeted Human Cytolytic Fusion Proteins for Anti-Cancer Therapy via Molecular Simulation. Biomedicines 2017, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, S.; Hansen, H.P.; Hehmann-Titt, G.; Huhn, M.; Fischer, R.; Barth, S.; Thepen, T. Efficacy of an adapted granzyme B-based anti-CD30 cytolytic fusion protein against PI-9-positive classical Hodgkin lymphoma cells in a murine model. Blood Cancer J. 2013, 3, e106-7. [Google Scholar] [CrossRef] [PubMed]
- Akinrinmade, O.; Jordaan, S.; Hristodorov, D.; Mladenov, R.; Mungra, N.; Chetty, S.; Barth, S. Human MAP Tau Based Targeted Cytolytic Fusion Proteins. Biomedicines 2017, 5, 36. [Google Scholar] [CrossRef] [PubMed]
- Jordaan, S.; Chetty, S.; Mungra, N.; Koopmans, I.; van Bommel, P.; Helfrich, W.; Barth, S. CSPG4: A Target for Selective Delivery of Human Cytolytic Fusion Proteins and TRAIL. Biomedicines 2017, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Mathew, M.; Verma, R.S. Humanized immunotoxins: A new generation of immunotoxins for targeted cancer therapy. Cancer Sci. 2009, 100, 1359–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krauss, J.; Arndt, M.A.E.; Vu, B.K.; Newton, D.L.; Seeber, S.; Rybak, S.M. Efficient killing of CD22+ tumor cells by a humanized diabody-RNase fusion protein. Biochem. Biophys. Res. Commun. 2005, 331, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Arndt, M.A.E.; Vu, B.K.; Newton, D.L.; Rybak, S.M. A Dimeric Angiogenin Immunofusion Protein Mediates Selective Toxicity Toward CD22+ Tumor Cells. J. Immunother. 2005, 28, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Tomita, Y.; Yuno, A.; Yoshitake, Y.; Shinohara, M. Cancer immunotherapy using novel tumor-associated antigenic peptides identified by genome-wide cDNA microarray analyses. Cancer Sci. 2015, 106, 505–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krzewski, K.; Coligan, J.E. Human NK cell lytic granules and regulation of their exocytosis. Front. Immunol. 2012, 3, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Veugelers, K.; Motyka, B.; Frantz, C.; Shostak, I.; Sawchuk, T.; Bleackley, R.C. The granzyme B-serglycin complex from cytotoxic granules requires dynamin for endocytosis. Blood 2004, 103, 3845–3853. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.J.; Amarante-Mendes, G.P.; Shi, L.; Chuang, T.H.; Casiano, C.A.; O’Brien, G.A.; Fitzgerald, P.; Tan, E.M.; Bokoch, G.M.; Greenberg, A.H.; et al. The cytotoxic cell protease granzyme B initiates apoptosis in a cell-free system by proteolytic processing and activation of the ICE/CED-3 family protease, CPP32, via a novel two-step mechanism. EMBO J. 1996, 15, 2407–2416. [Google Scholar] [PubMed]
- Li, P.; Zheng, G.; Yang, Y.; Zhang, C.; Xiong, P.; Xu, Y.; Fang, M.; Tan, Z.; Zheng, F.; Gong, F. Granzyme B is recovered by natural killer cells via clathrin-dependent endocytosis. Cell. Mol. Life Sci. 2010, 67, 3197–3208. [Google Scholar] [CrossRef] [PubMed]
- Afonina, I.S.; Tynan, G.A.; Logue, S.E.; Cullen, S.P.; Bots, M.; Lüthi, A.U.; Reeves, E.P.; McElvaney, N.G.; Medema, J.P.; Lavelle, E.C.; et al. Granzyme B-dependent proteolysis acts as a switch to enhance the proinflammatory activity of IL-1α. Mol. Cell 2011, 44, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Kurschus, F.C.; Fellows, E.; Stegmann, E.; Jenne, D.E. Granzyme B delivery via perforin is restricted by size, but not by heparan sulfate-dependent endocytosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13799–13804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-sanz, J.A.; Macdonald, H.R.; Jenne, D.E.; Nabholz, M.; Tschopp, J. Cell specificity of granzyme gene expression. J. Immunol. 1990, 145, 3111–3118. [Google Scholar] [PubMed]
- Cremer, C.; Hehmann-Titt, G.; Schiffer, S.; Melmer, G.; Carloni, P.; Barth, S.; Nachreiner, T. Engineered Versions of Granzyme B and Angiogenin Overcome Intrinsic Resistance to Apoptosis Mediated by Human Cytolytic Fusion Proteins. In Resistance to Immunotoxins in Cancer Therapy; Verma, R.S., Bonavida, B., Eds.; Springer: Cham, Switzerland, 2015; pp. 185–219. ISBN 978-3-319-17275-0. [Google Scholar]
- Beseničar, M.P.; Metkar, S.; Wang, B.; Froelich, C.J.; Anderluh, G. Granzyme B translocates across the lipid membrane only in the presence of lytic agents. Biochem. Biophys. Res. Commun. 2008, 371, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Bots, M.; Medema, J.P. Granzymes at a glance. J. Cell Sci. 2006, 119, 5011–5014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, T.; Opferman, J.T.; Shah, R.; Liu, N.; Froelich, C.J.; Ashton-Rickardt, P.G. A role for the granzyme B inhibitor serine protease inhibitor 6 in CD8+ memory cell homeostasis. J. Immunol. (Baltimore, Md. 1950) 2004, 173, 3801–3809. [Google Scholar] [CrossRef]
- Bird, C.H.; Sun, J.; Ung, K.; Karambalis, D.; Whisstock, J.C.; Trapani, J.A.; Bird, P.I. Cationic Sites on Granzyme B Contribute to Cytotoxicity by Promoting Its Uptake into Target Cells. Mol. Cell. Biol. 2005, 25, 7854–7867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buzza, M.S.; Hirst, C.E.; Bird, C.H.; Hosking, P.; McKendrick, J.; Bird, P.I. The granzyme B inhibitor, PI-9, is present in endothelial and mesothelial cells, suggesting that it protects bystander cells during immune responses. Cell. Immunol. 2001, 210, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, M.G.; Barth, S. Development of novel, highly cytotoxic fusion constructs containing granzyme B: Unique mechanisms and functions. Curr. Pharm. Des. 2009, 15, 2676–2692. [Google Scholar] [CrossRef] [PubMed]
- Stahnke, B.; Thepen, T.; Stocker, M.; Rosinke, R.; Jost, E.; Fischer, R.; Tur, M.K.; Barth, S. Granzyme B-H22(scFv), a human immunotoxin targeting CD64 in acute myeloid leukemia of monocytic subtypes. Mol. Cancer Ther. 2008, 7, 2924–2932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Cheung, L.H.; Thorpe, P.; Rosenblum, M.G. Mechanistic studies of a novel human fusion toxin composed of vascular endothelial growth factor (VEGF)121 and the serine protease granzyme B: Directed apoptotic events in vascular endothelial cells. Mol. Cancer Ther. 2003, 2, 949–959. [Google Scholar] [PubMed]
- Dälken, B.; Giesübel, U.; Knauer, S.K.; Wels, W.S. Targeted induction of apoptosis by chimeric granzyme B fusion proteins carrying antibody and growth factor domains for cell recognition. Cell Death Differ. 2006, 13, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Kurschus, F.C.; Kleinschmidt, M.; Fellows, E.; Dornmair, K.; Rudolph, R.; Lilie, H.; Jenne, D.E. Killing of target cells by redirected granzyme B in the absence of perforin. FEBS Lett. 2004, 562, 87–92. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Zhao, J.; Ren, J.L.; Zhang, L.; Wen, W.H.; Zhang, R.; Qin, W.W.; Jia, L.T.; Yao, L.B.; Zhang, Y.Q.; et al. Recombinant immunoproapoptotic proteins with furin site can translocate and kill HER2-positive cancer cells. Cancer Res. 2007, 67, 11830–11839. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, S.; Rosinke, R.; Jost, E.; Hehmann-Titt, G.; Huhn, M.; Melmer, G.; Barth, S.; Thepen, T. Targeted ex vivo reduction of CD64-positive monocytes in chronic myelomonocytic leukemia and acute myelomonocytic leukemia using human granzyme B-based cytolytic fusion proteins. Int. J. Cancer 2014, 135, 1497–1508. [Google Scholar] [CrossRef] [PubMed]
- Niesen, J.; Hehmann-Titt, G.; Woitok, M.; Fendel, R.; Barth, S.; Fischer, R.; Stein, C. A novel fully-human cytolytic fusion protein based on granzyme B shows in vitro cytotoxicity and ex vivo binding to solid tumors overexpressing the epidermal growth factor receptor. Cancer Lett. 2016, 374, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Amoury, M.; Kolberg, K.; Pham, A.T.; Hristodorov, D.; Mladenov, R.; Di Fiore, S.; Helfrich, W.; Kiessling, F.; Fischer, R.; Pardo, A.; et al. Granzyme B-based cytolytic fusion protein targeting EpCAM specifically kills triple negative breast cancer cells in vitro and inhibits tumor growth in a subcutaneous mouse tumor model. Cancer Lett. 2016, 372, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Lee, M.H.; Choi, E.; Pardo-Villamizar, C.A.; Lee, S.B.; Yang, I.H.; Calabresi, P.A.; Nath, A. Granzyme B-Induced Neurotoxicity Is Mediated via Activation of PAR-1 Receptor and Kv1.3 Channel. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Jabulowsky, R.A.; Oberoi, P.; Bähr-Mahmud, H.; Dälken, B.; Wels, W.S. Surface charge-modification prevents sequestration and enhances tumor-cell specificity of a recombinant granzyme B-TGFα fusion protein. Bioconjug. Chem. 2012, 23, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Raja, S.M.; Metkar, S.S.; Höning, S.; Wang, B.; Russin, W.A.; Pipalia, N.H.; Menaa, C.; Belting, M.; Cao, X.; Dressel, R.; et al. A novel mechanism for protein delivery: Granzyme B undergoes electrostatic exchange from serglycin to target cells. J. Biol. Chem. 2005, 280, 20752–20761. [Google Scholar] [CrossRef] [PubMed]
- Mohamedali, K.A.; Cheung, L.H.; Rosenblum, M.G. Tumor-targeted fusion constructs containing engineered granzyme B variants with optimized stability and potency. Cancer Res. 2015, 75, 632 LP-632. [Google Scholar] [CrossRef]
- Ho, P.; Ede, C.; Chen, Y.Y. Modularly Constructed Synthetic Granzyme B Molecule Enables Interrogation of Intracellular Proteases for Targeted Cytotoxicity. ACS Synth. Biol. 2017, 6, 1484–1495. [Google Scholar] [CrossRef] [PubMed]
- Bird, C.H.; Christensen, M.E.; Mangan, M.S.J.; Prakash, M.D.; Sedelies, K.A.; Smyth, M.J.; Harper, I.; Waterhouse, N.J.; Bird, P.I. The granzyme B-Serpinb9 axis controls the fate of lymphocytes after lysosomal stress. Cell Death Differ. 2014, 21, 876–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullen, S.P.; Martin, S.J. Mechanisms of granule-dependent killing. Cell Death Differ. 2008, 15, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Kaiserman, D.; Bird, P.I. Control of granzymes by serpins. Cell Death Differ. 2010, 17, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Azzi, J.; Ohori, S.; Ting, C.; Uehara, M.; Abdoli, R.; Smith, B.D.; Safa, K.; Solhjou, Z.; Lukyanchykov, P.; Patel, J.; et al. Serine protease inhibitor-6 differentially affects the survival of effector and memory alloreactive CD8-T cells. Am. J. Transplant. 2015, 15, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Ashton-Rickardt, P.G. An emerging role for Serine Protease Inhibitors in T lymphocyte immunity and beyond. Immunol. Lett. 2013, 152, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Soriano, C.; Mukaro, V.; Hodge, G.; Ahern, J.; Holmes, M.; Jersmann, H.; Moffat, D.; Meredith, D.; Jurisevic, C.; Reynolds, P.N.; et al. Increased proteinase inhibitor-9 (PI-9) and reduced granzyme B in lung cancer: Mechanism for immune evasion? Lung Cancer 2012, 77, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Ellison, S.J.; Alarid, E.T.; Shapiro, D.J. Interplay between the levels of estrogen and estrogen receptor controls the level of the granzyme inhibitor, proteinase inhibitor 9 and susceptibility to immune surveillance by natural killer cells. Oncogene 2007, 26, 4106–4114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, M.; Hostetter, D.R.; Loeb, C.R.K.; Simko, J.; Craik, C.S. Inhibition of Granzyme B by PI-9 protects prostate cancer cells from apoptosis. Prostate 2012, 72, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Mohamedali, K.A.; Marks, J.W.; Cheung, L.H.; Hittelman, W.N.; Rosenblum, M.G. Construction and characterization of novel, completely human serine protease therapeutics targeting Her2/neu. Mol. Cancer Ther. 2013, 12, 979–991. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Mohamedali, K.A.; Gonzalez-Angulo, A.M.; Cao, Y.; Migliorini, M.; Cheung, L.H.; LoBello, J.; Lei, X.; Qi, Y.; Hittelman, W.N.; et al. Development of human serine protease-based therapeutics targeting Fn14 and identification of Fn14 as a new target overexpressed in TNBC. Mol. Cancer Ther. 2014, 13, 2688–2705. [Google Scholar] [CrossRef] [PubMed]
- Baginska, J.; Viry, E.; Berchem, G.; Poli, A.; Noman, M.Z.; van Moer, K.; Medves, S.; Zimmer, J.; Oudin, A.; Niclou, S.P.; et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc. Natl. Acad. Sci. USA 2013, 110, 17450–17455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Losasso, V.; Schiffer, S.; Barth, S.; Carloni, P. Design of human granzyme B variants resistant to serpin B9. Proteins Struct. Funct. Bioinform. 2012, 80, 2514–2522. [Google Scholar] [CrossRef] [PubMed]
- Froelich, C.J.; Orth, K.; Turbov, J.; Seth, P.; Gottlieb, R.; Babior, B.; Shah, G.M.; Bleackley, R.C.; Dixit, V.M.; Hanna, W. New Paradigm for Lymphocyte Granule-mediated Cytotoxicity. J. Biol. Chem. 1996, 271, 29073–29079. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, L.H.; Jia, L.T.; Zhang, L.; Xu, Y.M.; Wang, Z.; Yu, C.J.; Peng, W.D.; Wen, W.H.; Wang, C.J.; et al. Secreted antibody/granzyme B fusion protein stimulates selective killing of HER2-overexpressing tumor cells. J. Biol. Chem. 2004, 279, 21343–21348. [Google Scholar] [CrossRef] [PubMed]
- Kurschus, F.C.; Jenne, D.E. Delivery and therapeutic potential of human granzyme B. Immunol. Rev. 2010, 235, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Hetzel, C.; Bachran, C.; Fischer, R.; Fuchs, H.; Barth, S.; Stöcker, M. Small cleavable adapters enhance the specific cytotoxicity of a humanized immunotoxin directed against CD64-positive cells. J. Immunother. 2008, 31, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, H.; Bachran, C.; Li, T.; Heisler, I.; Dürkop, H.; Sutherland, M. A cleavable molecular adapter reduces side effects and concomitantly enhances efficacy in tumor treatment by targeted toxins in mice. J. Control. Release 2007, 117, 342–350. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Construct | Disease * | Target | Cell Line | P19 Expression in Cell Line | Cytotoxicity | Reference |
|---|---|---|---|---|---|---|
| GrB (wt)-H22(scFv) and GrBR201K-H22(scFv) | CMML | CD64+ | Cells from AMML and CMML patients. | Yes | Not specified | [35] |
| CD64+ HL60 | No | 4–7 nM | ||||
| GrB (wt)-ki4(scFv) and GrBR201K-Ki4(scFv) | cHL | CD30+ | L428 | Yes | [12] | |
| L540cy | No | |||||
| GrBR201K-scFv1711 | Epidermoid cancer cells | EGFR+ | A431 | Yes | 133.3 nM | [36] |
| RD target cells | Yes | 21.1 nM | ||||
| GrBR201K-αEpCAM(scFv) | TNBC | EpCAM+ | MDA-MB-231 | Yes | N/A | [37] |
| MDA-MB-468 | yes | 221 nM | ||||
| MDA-MB-453 | No | 307 nM |
| Granzyme B Variant | Mutation | Implication of Mutation | Result | Reference |
|---|---|---|---|---|
| GzmBFacD | The kktmrkry sequence at the C-terminus was replaced with the acidic peptide DSVLA derived from human complement factor D | This sequence motif is not positively charged and should have little immunogenic potential because complement factor D occurs at relatively high levels in human plasma | Binding to HL60 cells was completely abolished | [21] |
| GzmBKD | The region around K127 and K131 is known to function as a heparin binding site in thrombin. To stabilize this, both lysines were replaced with aspartate residues | Reduced HS binding | Reduced binding to HL60 cells compared to wild type GrB | [21] |
| GzmBKD-FacD | Double mutant consisting with aspartate replacement at position K127 and K131 and the acidic C-terminal peptide DSVLA | Combined effect of mutation | The binding and internalization efficiency was completely abolished | [21] |
| cs1 | Arginine in position 110, 114 and 116 (R110, R114, and R116) replaced with alanine. Constitutes an altered classical GAG-binding motif | Most proteins bind GAG. This is dependent on electrostatic interaction between the positively and negatively charged cells. Mutation in this region alters binding of GrB to negatively charged cells | Reduced cytotoxic activity. 20-fold less cytotoxic compared to wild type GrB. Abolished binding to Heparin region | [27] |
| cs2 | Lysine in position 239, 240, 243 and 244 (K239, K240, K243, and K244) replaced with alanine. Constitutes an altered C-terminal helix | Amphipathic C-terminal helix that has paired basic residues that bind GAGs. Mutation in this region alters binding of GrB to negatively charged cells. | Reduced cytotoxic activity. 2.5-fold less cytotoxic compared to wild type GrB. Reduced binding to Heparin region | [27] |
| cs1+2 | Combined mutation of cs1 and cs2 | Combined mutation of cs1 and cs3 | Reduced cytotoxic activity. 20-fold less cytotoxic compared to wild type GrB. Abolished binding to Heparin region | [27] |
| Granzyme B Variant | Mutation | Implication of Mutation | References |
|---|---|---|---|
| R28A | Substitution of Arginine residue with Alanine (constitutes a neutral charge at position 28) | In the presence of PI-9 the GrBR28A mutant contains 54% activity | [54] |
| R28E | Substitution of Arginine residue with Glutamate (constitutes an opposite charge at position 28) | In the presence of PI-9 the GrBR28E mutant contains 25% activity | [54] |
| R28K | Substitution of Arginine residue with Lysine (constitutes an identical charge at position 28) | In the presence of PI-9, the GrBR28K and mutants retained 76% of their original activity | [54] |
| R201A | Substitution of Arginine residue with Alanine (constitutes a neutral charge at position 201) | In the presence of PI-9, the GrBR201A mutants retained 46% of their original activity | [54] |
| R201E | Substitution of Arginine residue with Glutamate (constitutes an opposite charge at position 201) | No activity in the presence of PI-9 | [54] |
| R201K | Substitution of Arginine residue with Lysine (constitutes an identical charge at position 28) | In the presence of PI-9, the GrBR201K mutant retained 94% of its activity | [54] |
| K27A | Substitution of Lysine residue with Alanine (constitutes a neutral charge at position 27) | Insensitive to P1-9 activity and K27A mutant showed a marked decrease in the ability to bind and cleave a substrate (substrate 3) containing P9 residues | [54] |
| R28A & R201A | Double mutant; Arginine replaced with Alanine at position 28 and 201 | In the presence of PI-9 the double mutant contains 0.5% activity | [54] |
| K27E & R28A (EA) | Double mutant; Lysine replaced with Glutamate at position 27 and Arginine replaced with Alanine at position 28 | In the presence of 50% human serum, the enzymatic activity of EA remained over 40% over 24 h | [41] |
| K27L & R28A (LA) | Double mutant; Lysine replaced with Leucine at position 27 and Arginine replaced with Alanine at position 28 | LA double mutant appeared to behave intermediate to the wild-type protein (GrB/VEGF121) and the EA construct | [41] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hlongwane, P.; Mungra, N.; Madheswaran, S.; Akinrinmade, O.A.; Chetty, S.; Barth, S. Human Granzyme B Based Targeted Cytolytic Fusion Proteins. Biomedicines 2018, 6, 72. https://doi.org/10.3390/biomedicines6020072
Hlongwane P, Mungra N, Madheswaran S, Akinrinmade OA, Chetty S, Barth S. Human Granzyme B Based Targeted Cytolytic Fusion Proteins. Biomedicines. 2018; 6(2):72. https://doi.org/10.3390/biomedicines6020072
Chicago/Turabian StyleHlongwane, Precious, Neelakshi Mungra, Suresh Madheswaran, Olusiji A. Akinrinmade, Shivan Chetty, and Stefan Barth. 2018. "Human Granzyme B Based Targeted Cytolytic Fusion Proteins" Biomedicines 6, no. 2: 72. https://doi.org/10.3390/biomedicines6020072
APA StyleHlongwane, P., Mungra, N., Madheswaran, S., Akinrinmade, O. A., Chetty, S., & Barth, S. (2018). Human Granzyme B Based Targeted Cytolytic Fusion Proteins. Biomedicines, 6(2), 72. https://doi.org/10.3390/biomedicines6020072