Breast Cancer Stem Cells



Abstract
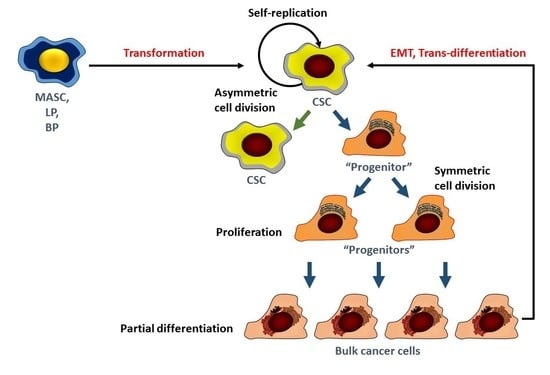
:
{kind=link}
1. Introduction
2. The Study of Breast Cancer Stem Cells
3. Origins of Breast Cancer Stem Cells
4. Signaling Pathways in BCSC
5. Notch
6. Hedgehog (Hh)
7. BMI1
8. Wnt/β-Catenin
9. Hippo
10. Estrogen Receptor α/β
11. Other Pathways
12. Conclusions and Future Directions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NCI. Surveillance, Epidemiology and End Results (SEER) Program. Available online: https://seer.cancer.gov/statfacts/html/breast.html (accessed on 27 May 2018).
- Alferez, D.G.; Simoes, B.M.; Howell, S.J.; Clarke, R.B. The Role of Steroid Hormones in Breast and Effects on Cancer Stem Cells. Curr. Stem Cell Rep. 2018, 4, 81–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sin, W.C.; Lim, C.L. Breast cancer stem cells-from origins to targeted therapy. Stem Cell Investig. 2017, 4, 96. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Wicha, M.S.; Liu, S.; Dontu, G. Cancer stem cells: An old idea—A paradigm shift. Cancer Res. 2006, 66, 1883–1890. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, Y.; de Giorgio, A.; Coombes, C.R.; Stebbing, J.; Castellano, L. Mammosphere formation assay from human breast cancer tissues and cell lines. J. Vis. Exp. 2015, 97. [Google Scholar] [CrossRef] [PubMed]
- Dontu, G.; Jackson, K.W.; McNicholas, E.; Kawamura, M.J.; Abdallah, W.M.; Wicha, M.S. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 2004, 6, R605–R615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dontu, G.; Abdallah, W.M.; Foley, J.M.; Jackson, K.W.; Clarke, M.F.; Kawamura, M.J.; Wicha, M.S. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003, 17, 1253–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimshaw, M.J.; Cooper, L.; Papazisis, K.; Coleman, J.A.; Bohnenkamp, H.R.; Chiapero-Stanke, L.; Taylor-Papadimitriou, J.; Burchell, J.M. Mammosphere culture of metastatic breast cancer cells enriches for tumorigenic breast cancer cells. Breast Cancer Res. 2008, 10, R52. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Hirsch, H.A.; Wang, G.; Struhl, K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc. Natl. Acad. Sci. USA 2011, 108, 1397–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponti, D.; Costa, A.; Zaffaroni, N.; Pratesi, G.; Petrangolini, G.; Coradini, D.; Pilotti, S.; Pierotti, M.A.; Daidone, M.G. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005, 65, 5506–5511. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ablett, M.P.; Singh, J.K.; Clarke, R.B. Stem cells in breast tumours: Are they ready for the clinic? Eur. J. Cancer 2012, 48, 2104–2116. [Google Scholar] [CrossRef] [PubMed]
- Croker, A.K.; Goodale, D.; Chu, J.; Postenka, C.; Hedley, B.D.; Hess, D.A.; Allan, A.L. High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. J. Cell Mol. Med. 2009, 13, 2236–2252. [Google Scholar] [CrossRef] [PubMed]
- Honeth, G.; Bendahl, P.O.; Ringner, M.; Saal, L.H.; Gruvberger-Saal, S.K.; Lovgren, K.; Grabau, D.; Ferno, M.; Borg, A.; Hegardt, C. The CD44+/CD24− phenotype is enriched in basal-like breast tumors. Breast Cancer Res. 2008, 10, R53. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Wang, X.; Chen, G.Y.; Dalerba, P.; Gurney, A.; Hoey, T.; Sherlock, G.; Lewicki, J.; Shedden, K.; Clarke, M.F. The prognostic role of a gene signature from tumorigenic breast-cancer cells. N. Engl. J. Med. 2007, 356, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Pham, P.V.; Phan, N.L.; Nguyen, N.T.; Truong, N.H.; Duong, T.T.; Le, D.V.; Truong, K.D.; Phan, N.K. Differentiation of breast cancer stem cells by knockdown of CD44: Promising differentiation therapy. J. Transl. Med. 2011, 9, 209. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, N.; Ghahremani, M.H.; Amini, M.; Atyabi, F.; Ostad, S.N.; Shabani Ravari, N.; Nateghian, N.; Dinarvand, R. CD44-targeted docetaxel conjugate for cancer cells and cancer stem-like cells: A novel hyaluronic acid-based drug delivery system. Chem. Biol. Drug Des. 2014, 83, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Muntimadugu, E.; Kumar, R.; Saladi, S.; Rafeeqi, T.A.; Khan, W. CD44 targeted chemotherapy for co-eradication of breast cancer stem cells and cancer cells using polymeric nanoparticles of salinomycin and paclitaxel. Colloids Surf. B Biointerfaces 2016, 143, 532–546. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T.M.; McBride, W.H.; Pajonk, F. The response of CD24−/low/CD44+ breast cancer-initiating cells to radiation. J. Natl. Cancer Inst. 2006, 98, 1777–1785. [Google Scholar] [CrossRef] [PubMed]
- Schabath, H.; Runz, S.; Joumaa, S.; Altevogt, P. CD24 affects CXCR4 function in pre-B lymphocytes and breast carcinoma cells. J. Cell Sci. 2006, 119, 314–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bensimon, J.; Altmeyer-Morel, S.; Benjelloun, H.; Chevillard, S.; Lebeau, J. CD24(−/low) stem-like breast cancer marker defines the radiation-resistant cells involved in memorization and transmission of radiation-induced genomic instability. Oncogene 2013, 32, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Charafe-Jauffret, E.; Ginestier, C.; Iovino, F.; Tarpin, C.; Diebel, M.; Esterni, B.; Houvenaeghel, G.; Extra, J.M.; Bertucci, F.; Jacquemier, J.; et al. Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clin. Cancer Res. 2010, 16, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Moreb, J.S.; Ucar, D.; Han, S.; Amory, J.K.; Goldstein, A.S.; Ostmark, B.; Chang, L.J. The enzymatic activity of human aldehyde dehydrogenases 1A2 and 2 (ALDH1A2 and ALDH2) is detected by Aldefluor, inhibited by diethylaminobenzaldehyde and has significant effects on cell proliferation and drug resistance. Chem. Biol. Interact. 2012, 195, 52–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcato, P.; Dean, C.A.; Giacomantonio, C.A.; Lee, P.W. Aldehyde dehydrogenase: Its role as a cancer stem cell marker comes down to the specific isoform. Cell Cycle 2011, 10, 1378–1384. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.J.; Sun, B.C.; Zhao, X.L.; Zhao, X.M.; Sun, T.; Gu, Q.; Yao, Z.; Dong, X.Y.; Zhao, N.; Liu, N. CD133+ cells with cancer stem cell characteristics associates with vasculogenic mimicry in triple-negative breast cancer. Oncogene 2013, 32, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Friedrichs, K.; Ruiz, P.; Franke, F.; Gille, I.; Terpe, H.J.; Imhof, B.A. High expression level of alpha 6 integrin in human breast carcinoma is correlated with reduced survival. Cancer Res. 1995, 55, 901–906. [Google Scholar] [PubMed]
- Lu, H.; Clauser, K.R.; Tam, W.L.; Frose, J.; Ye, X.; Eaton, E.N.; Reinhardt, F.; Donnenberg, V.S.; Bhargava, R.; Carr, S.A.; et al. Addendum: A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat. Cell Biol. 2015, 17, 1607. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Clauser, K.R.; Tam, W.L.; Frose, J.; Ye, X.; Eaton, E.N.; Reinhardt, F.; Donnenberg, V.S.; Bhargava, R.; Carr, S.A.; et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat. Cell Biol. 2014, 16, 1105–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Cao, L.; Sun, Z.; Jin, J.; Fang, H.; Zhang, W.; Guan, X. Evaluation of Breast Cancer Stem Cells and Intratumor Stemness Heterogeneity in Triple-negative Breast Cancer as Prognostic Factors. Int. J. Biol. Sci. 2016, 12, 1568–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passegue, E.; Jamieson, C.H.; Ailles, L.E.; Weissman, I.L. Normal and leukemic hematopoiesis: Are leukemias a stem cell disorder or a reacquisition of stem cell characteristics? Proc. Natl. Acad. Sci. USA 2003, 100 (Suppl. 1), 11842–11849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herschkowitz, J.I.; Simin, K.; Weigman, V.J.; Mikaelian, I.; Usary, J.; Hu, Z.; Rasmussen, K.E.; Jones, L.P.; Assefnia, S.; Chandrasekharan, S.; et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007, 8, R76. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Jovanovic, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef] [PubMed]
- Dontu, G.; Al-Hajj, M.; Abdallah, W.M.; Clarke, M.F.; Wicha, M.S. Stem cells in normal breast development and breast cancer. Cell Prolif. 2003, 36 (Suppl. 1), 59–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dontu, G.; El-Ashry, D.; Wicha, M.S. Breast cancer, stem/progenitor cells and the estrogen receptor. Trends Endocrinol. Metab. 2004, 15, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Hwang-Verslues, W.W.; Kuo, W.H.; Chang, P.H.; Pan, C.C.; Wang, H.H.; Tsai, S.T.; Jeng, Y.M.; Shew, J.Y.; Kung, J.T.; Chen, C.H.; et al. Multiple lineages of human breast cancer stem/progenitor cells identified by profiling with stem cell markers. PLoS ONE 2009, 4, e8377. [Google Scholar] [CrossRef] [PubMed]
- Tsang, J.Y.; Huang, Y.H.; Luo, M.H.; Ni, Y.B.; Chan, S.K.; Lui, P.C.; Yu, A.M.; Tan, P.H.; Tse, G.M. Cancer stem cell markers are associated with adverse biomarker profiles and molecular subtypes of breast cancer. Breast Cancer Res. Treat. 2012, 136, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.; Vaillant, F.; Wu, D.; Forrest, N.C.; Pal, B.; Hart, A.H.; Asselin-Labat, M.L.; Gyorki, D.E.; Ward, T.; Partanen, A.; et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat. Med. 2009, 15, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visvader, J.E.; Stingl, J. Mammary stem cells and the differentiation hierarchy: Current status and perspectives. Genes Dev. 2014, 28, 1143–1158. [Google Scholar] [CrossRef] [PubMed]
- Molyneux, G.; Geyer, F.C.; Magnay, F.A.; McCarthy, A.; Kendrick, H.; Natrajan, R.; Mackay, A.; Grigoriadis, A.; Tutt, A.; Ashworth, A.; et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell 2010, 7, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Rios, A.C.; Fu, N.Y.; Lindeman, G.J.; Visvader, J.E. In situ identification of bipotent stem cells in the mammary gland. Nature 2014, 506, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Van Keymeulen, A.; Rocha, A.S.; Ousset, M.; Beck, B.; Bouvencourt, G.; Rock, J.; Sharma, N.; Dekoninck, S.; Blanpain, C. Distinct stem cells contribute to mammary gland development and maintenance. Nature 2011, 479, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Lagadec, C.; Vlashi, E.; Della Donna, L.; Dekmezian, C.; Pajonk, F. Radiation-induced reprogramming of breast cancer cells. Stem Cells 2012, 30, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Marjanovic, N.D.; Lee, T.; Bell, G.; Kleer, C.G.; Reinhardt, F.; D’Alessio, A.C.; Young, R.A.; Weinberg, R.A. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 2013, 154, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Reavie, L.; Couto, J.P.; De Silva, D.; Stadler, M.B.; Roloff, T.; Britschgi, A.; Eichlisberger, T.; Kohler, H.; Aina, O.; et al. PIK3CA(H1047R) induces multipotency and multi-lineage mammary tumours. Nature 2015, 525, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Poli, V.; Fagnocchi, L.; Fasciani, A.; Cherubini, A.; Mazzoleni, S.; Ferrillo, S.; Miluzio, A.; Gaudioso, G.; Vaira, V.; Turdo, A.; et al. MYC-driven epigenetic reprogramming favors the onset of tumorigenesis by inducing a stem cell-like state. Nat. Commun. 2018, 9, 1024. [Google Scholar] [CrossRef] [PubMed]
- Bhola, N.E.; Jansen, V.M.; Koch, J.P.; Li, H.; Formisano, L.; Williams, J.A.; Grandis, J.R.; Arteaga, C.L. Treatment of Triple-Negative Breast Cancer with TORC1/2 Inhibitors Sustains a Drug-Resistant and Notch-Dependent Cancer Stem Cell Population. Cancer Res. 2016, 76, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Eirew, P.; Steif, A.; Khattra, J.; Ha, G.; Yap, D.; Farahani, H.; Gelmon, K.; Chia, S.; Mar, C.; Wan, A.; et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature 2015, 518, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Liu, Y.; Deng, L.; Wang, D.; He, X.; Zhou, L.; Wicha, M.S.; Bai, F.; Liu, S. Transcriptional profiles of different states of cancer stem cells in triple-negative breast cancer. Mol. Cancer 2018, 17, 65. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: Clinical update. Nat. Rev. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef] [PubMed]
- Pannuti, A.; Foreman, K.; Rizzo, P.; Osipo, C.; Golde, T.; Osborne, B.; Miele, L. Targeting Notch to target cancer stem cells. Clin. Cancer Res. 2010, 16, 3141–3152. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.S.; Vadgama, J.V. Curcumin and epigallocatechin gallate inhibit the cancer stem cell phenotype via down-regulation of STAT3-NFkappaB signaling. Anticancer Res. 2015, 35, 39–46. [Google Scholar] [PubMed]
- Hori, K.; Sen, A.; Artavanis-Tsakonas, S. Notch signaling at a glance. J. Cell Sci. 2013, 126, 2135–2140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz-Martinez, F.; Gutierrez-Avino, F.J.; Sanmartin, E.; Pomares-Navarro, E.; Villalba-Riquelme, C.; Garcia-Martinez, A.; Lerma, E.; Peiro, G. Association of Notch pathway down-regulation with Triple Negative/Basal-like breast carcinomas and high tumor-infiltrating FOXP3+ Tregs. Exp. Mol. Pathol. 2016, 100, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Perumalsamy, L.R.; Marcel, N.; Kulkarni, S.; Radtke, F.; Sarin, A. Distinct spatial and molecular features of notch pathway assembly in regulatory T cells. Sci. Signal 2012, 5, ra53. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.H.; Gallahan, D.; Diella, F.; Jhappan, C.; Merlino, G.; Callahan, R. Constitutive expression of a truncated INT3 gene in mouse mammary epithelium impairs differentiation and functional development. Cell Growth Differ. 1995, 6, 563–577. [Google Scholar] [PubMed]
- Soriano, J.V.; Uyttendaele, H.; Kitajewski, J.; Montesano, R. Expression of an activated Notch4(int-3) oncoprotein disrupts morphogenesis and induces an invasive phenotype in mammary epithelial cells in vitro. Int. J. Cancer 2000, 86, 652–659. [Google Scholar] [CrossRef]
- Uyttendaele, H.; Soriano, J.V.; Montesano, R.; Kitajewski, J. Notch4 and Wnt-1 proteins function to regulate branching morphogenesis of mammary epithelial cells in an opposing fashion. Dev. Biol. 1998, 196, 204–217. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, R.C.; Ouzounova, M.; Davis, A.; Choi, D.; Tchuenkam, S.M.; Kim, G.; Luther, T.; Quraishi, A.A.; Senbabaoglu, Y.; Conley, S.J.; et al. Notch reporter activity in breast cancer cell lines identifies a subset of cells with stem cell activity. Mol. Cancer Ther. 2015, 14, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Grudzien, P.; Lo, S.; Albain, K.S.; Robinson, P.; Rajan, P.; Strack, P.R.; Golde, T.E.; Miele, L.; Foreman, K.E. Inhibition of Notch signaling reduces the stem-like population of breast cancer cells and prevents mammosphere formation. Anticancer Res. 2010, 30, 3853–3867. [Google Scholar] [CrossRef] [PubMed]
- Simmons, M.J.; Serra, R.; Hermance, N.; Kelliher, M.A. NOTCH1 inhibition in vivo results in mammary tumor regression and reduced mammary tumorsphere-forming activity in vitro. Breast Cancer Res. 2012, 14, R126. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Shen, S.; Zhou, Y.; Mao, F.; Lin, Y.; Guan, J.; Xu, Y.; Zhang, S.; Liu, X.; Sun, Q. NOTCH1 is a poor prognostic factor for breast cancer and is associated with breast cancer stem cells. Onco Targets Ther. 2016, 9, 6865–6871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simoes, B.M.; O’Brien, C.S.; Eyre, R.; Silva, A.; Yu, L.; Sarmiento-Castro, A.; Alferez, D.G.; Spence, K.; Santiago-Gomez, A.; Chemi, F.; et al. Anti-estrogen Resistance in Human Breast Tumors Is Driven by JAG1-NOTCH4-Dependent Cancer Stem Cell Activity. Cell Rep. 2015, 12, 1968–1977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sansone, P.; Ceccarelli, C.; Berishaj, M.; Chang, Q.; Rajasekhar, V.K.; Perna, F.; Bowman, R.L.; Vidone, M.; Daly, L.; Nnoli, J.; et al. Self-renewal of CD133(hi) cells by IL6/Notch3 signalling regulates endocrine resistance in metastatic breast cancer. Nat. Commun. 2016, 7, 10442. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, I.; Pochampally, R.; Xing, F.; Watabe, K.; Miele, L. Notch signaling: Targeting cancer stem cells and epithelial-to-mesenchymal transition. Onco Targets Ther. 2013, 6, 1249–1259. [Google Scholar] [PubMed]
- Chen, M.H.; Wilson, C.W.; Chuang, P.T. SnapShot: Hedgehog signaling pathway. Cell 2007, 130, 386. [Google Scholar] [CrossRef] [PubMed]
- Kubo, M.; Nakamura, M.; Tasaki, A.; Yamanaka, N.; Nakashima, H.; Nomura, M.; Kuroki, S.; Katano, M. Hedgehog signaling pathway is a new therapeutic target for patients with breast cancer. Cancer Res. 2004, 64, 6071–6074. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Dontu, G.; Mantle, I.D.; Patel, S.; Ahn, N.S.; Jackson, K.W.; Suri, P.; Wicha, M.S. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006, 66, 6063–6071. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wicha, M.S. Targeting breast cancer stem cells. J. Clin. Oncol. 2010, 28, 4006–4012. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Nakamura, M.; Kameda, C.; Kubo, M.; Sato, N.; Kuroki, S.; Tanaka, M.; Katano, M. The Hedgehog signaling pathway plays an essential role in maintaining the CD44+CD24−/low subpopulation and the side population of breast cancer cells. Anticancer Res. 2009, 29, 2147–2157. [Google Scholar] [PubMed]
- Tao, Y.; Mao, J.; Zhang, Q.; Li, L. Overexpression of Hedgehog signaling molecules and its involvement in triple-negative breast cancer. Oncol. Lett. 2011, 2, 995–1001. [Google Scholar] [PubMed] [Green Version]
- Fu, Y.Z.; Yan, Y.Y.; He, M.; Xiao, Q.H.; Yao, W.F.; Zhao, L.; Wu, H.Z.; Yu, Z.J.; Zhou, M.Y.; Lv, M.T.; et al. Salinomycin induces selective cytotoxicity to MCF-7 mammosphere cells through targeting the Hedgehog signaling pathway. Oncol. Rep. 2016, 35, 912–922. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Fu, Y.; Yan, Y.; Xiao, Q.; Wu, H.; Yao, W.; Zhao, H.; Zhao, L.; Jiang, Q.; Yu, Z.; et al. The Hedgehog signalling pathway mediates drug response of MCF-7 mammosphere cells in breast cancer patients. Clin. Sci. 2015, 129, 809–822. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, S.A.; Machalek, D.A.; Shearer, R.F.; Millar, E.K.; Nair, R.; Schofield, P.; McLeod, D.; Cooper, C.L.; McNeil, C.M.; McFarland, A.; et al. Hedgehog overexpression is associated with stromal interactions and predicts for poor outcome in breast cancer. Cancer Res. 2011, 71, 4002–4014. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Tang, H.; Xiao, Q.; He, M.; Zhao, L.; Fu, Y.; Wu, H.; Yu, Z.; Jiang, Q.; Yan, Y.; et al. The Hedgehog signaling pathway is associated with poor prognosis in breast cancer patients with the CD44+/CD24 phenotype. Mol. Med. Rep. 2016, 14, 5261–5270. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, A.; Binju, M.; Arfuso, F.; Newsholme, P.; Dharmarajan, A. Role of epigenetic modulation in cancer stem cell fate. Int. J. Biochem. Cell Biol. 2017, 90, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.J.; Kieboom, K.; Marino, S.; DePinho, R.A.; van Lohuizen, M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 1999, 397, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Van der Lugt, N.M.; Domen, J.; Linders, K.; van Roon, M.; Robanus-Maandag, E.; te Riele, H.; van der Valk, M.; Deschamps, J.; Sofroniew, M.; van Lohuizen, M.; et al. Posterior transformation, neurological abnormalities, and severe hematopoietic defects in mice with a targeted deletion of the bmi-1 proto-oncogene. Genes Dev. 1994, 8, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhe, H.; Ding, Z.; Gao, P.; Zhang, N.; Li, G. Cancer stem cell marker Bmi-1 expression is associated with basal-like phenotype and poor survival in breast cancer. World J. Surg. 2012, 36, 1189–1194. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, M.; Bharali, D.J.; Sudha, T.; Khedr, M.; Guest, I.; Sell, S.; Glinsky, G.V.; Mousa, S.A. Downregulation of Bmi1 in breast cancer stem cells suppresses tumor growth and proliferation. Oncotarget 2017, 8, 38731–38742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojo, D.; Lin, X.; Wu, Y.; Cockburn, J.; Bane, A.; Tang, D. Polycomb complex protein BMI1 confers resistance to tamoxifen in estrogen receptor positive breast cancer. Cancer Lett. 2018, 426, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Wang, Y.; Zhao, P.; Ma, W.; Hu, Z.; Zhang, K. BMI-1 Promotes Self-Renewal of Radio- and Temozolomide (TMZ)-Resistant Breast Cancer Cells. Reprod. Sci. 2017, 24, 1620–1629. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Chang, W.W.; Chen, Y.Y.; Tsai, Y.H.; Chou, Y.H.; Tseng, H.C.; Chen, H.L.; Wu, C.C.; Chang-Chien, J.; Lee, H.T.; et al. Hsp90alpha Mediates BMI1 Expression in Breast Cancer Stem/Progenitor Cells through Facilitating Nuclear Translocation of c-Myc and EZH2. Int. J. Mol. Sci. 2017, 18, 1986. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Li, S.; Song, E.; Liu, S. The roles of ncRNAs and histone-modifiers in regulating breast cancer stem cells. Protein Cell 2016, 7, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.M.; Wang, B.Y.; Lee, C.H.; Lee, H.T.; Li, J.J.; Hong, G.C.; Hung, Y.C.; Chien, P.J.; Chang, C.Y.; Hsu, L.S.; et al. Hinokitiol up-regulates miR-494-3p to suppress BMI1 expression and inhibits self-renewal of breast cancer stem/progenitor cells. Oncotarget 2017, 8, 76057–76068. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Kipps, T.J.; Zhang, S. Corrigendum to “Wnt5a Signaling in Normal and Cancer Stem Cells”. Stem Cells Int. 2017, 2017, 3467360. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Kipps, T.J.; Zhang, S. Wnt5a Signaling in Normal and Cancer Stem Cells. Stem Cells Int. 2017, 2017, 5295286. [Google Scholar] [PubMed]
- Veeck, J.; Niederacher, D.; An, H.; Klopocki, E.; Wiesmann, F.; Betz, B.; Galm, O.; Camara, O.; Durst, M.; Kristiansen, G.; et al. Aberrant methylation of the Wnt antagonist SFRP1 in breast cancer is associated with unfavourable prognosis. Oncogene 2006, 25, 3479–3488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klopocki, E.; Kristiansen, G.; Wild, P.J.; Klaman, I.; Castanos-Velez, E.; Singer, G.; Stohr, R.; Simon, R.; Sauter, G.; Leibiger, H.; et al. Loss of SFRP1 is associated with breast cancer progression and poor prognosis in early stage tumors. Int. J. Oncol. 2004, 25, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R. Wnt signaling and stem cell control. Cell Res. 2008, 18, 523–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteiro, J.; Gaspar, C.; Richer, W.; Franken, P.F.; Sacchetti, A.; Joosten, R.; Idali, A.; Brandao, J.; Decraene, C.; Fodde, R. Cancer stemness in Wnt-driven mammary tumorigenesis. Carcinogenesis 2014, 35, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Jang, G.B.; Kim, J.Y.; Cho, S.D.; Park, K.S.; Jung, J.Y.; Lee, H.Y.; Hong, I.S.; Nam, J.S. Blockade of Wnt/beta-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Sci. Rep. 2015, 5, 12465. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, A.; Kumar, S.; Arfuso, F.; Newsholme, P.; Dharmarajan, A. Secreted Frizzled-related protein 4 (sFRP4) chemo-sensitizes cancer stem cells derived from human breast, prostate, and ovary tumor cell lines. Sci. Rep. 2017, 7, 2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, N.; Barwick, B.G.; Moreno, C.S.; Ordanic-Kodani, M.; Chen, Z.; Oprea-Ilies, G.; Tang, W.; Catzavelos, C.; Kerstann, K.F.; Sledge, G.W., Jr.; et al. Wnt signaling in triple negative breast cancer is associated with metastasis. BMC Cancer 2013, 13, 537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pohl, S.G.; Brook, N.; Agostino, M.; Arfuso, F.; Kumar, A.P.; Dharmarajan, A. Wnt signaling in triple-negative breast cancer. Oncogenesis 2017, 6, e310. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Yuan, Z.; Zeng, L.; Wang, Y.; Lai, L.; Li, J.; Sun, P.; Xue, X.; Qi, J.; Yang, Z.; et al. LGR4 modulates breast cancer initiation, metastasis, and cancer stem cells. FASEB J. 2018, 32, 2422–2437. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.; Camargo, F.D. The Hippo signaling pathway and stem cell biology. Trends Cell Biol. 2012, 22, 339–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staley, B.K.; Irvine, K.D. Hippo signaling in Drosophila: Recent advances and insights. Dev. Dyn. 2012, 241, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Means-Powell, J.; Minton, S.E.; Mayer, I.A.; Abramson, V.; Ismail-Khan, R.; Arteaga, C.; Ayers, D.; Sanders, M.; Lush, R.; Miele, L. A Phase 1b Dose Escalation Trial of RO4929097 (a g-secretase inhibitor) in Combination with Exemestane in Patients with ER+ Metastatic Breast Cancer. Cancer Res. 2012, 72, 280. [Google Scholar] [CrossRef]
- St John, M.A.; Tao, W.; Fei, X.; Fukumoto, R.; Carcangiu, M.L.; Brownstein, D.G.; Parlow, A.F.; McGrath, J.; Xu, T. Mice deficient of Lats1 develop soft-tissue sarcomas, ovarian tumours and pituitary dysfunction. Nat. Genet. 1999, 21, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhang, N.; Gray, R.S.; Li, H.; Ewald, A.J.; Zahnow, C.A.; Pan, D. A temporal requirement for Hippo signaling in mammary gland differentiation, growth, and tumorigenesis. Genes Dev. 2014, 28, 432–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartucci, M.; Dattilo, R.; Moriconi, C.; Pagliuca, A.; Mottolese, M.; Federici, G.; Benedetto, A.D.; Todaro, M.; Stassi, G.; Sperati, F.; et al. TAZ is required for metastatic activity and chemoresistance of breast cancer stem cells. Oncogene 2015, 34, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Martin, J.; Lopez-Garcia, M.A.; Romero-Perez, L.; Atienza-Amores, M.R.; Pecero, M.L.; Castilla, M.A.; Biscuola, M.; Santon, A.; Palacios, J. Nuclear TAZ expression associates with the triple-negative phenotype in breast cancer. Endocr. Relat. Cancer 2015, 22, 443–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.K.; Jung, W.H.; Koo, J.S. Yes-associated protein (YAP) is differentially expressed in tumor and stroma according to the molecular subtype of breast cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 3224–3234. [Google Scholar] [PubMed]
- Lehn, S.; Tobin, N.P.; Sims, A.H.; Stal, O.; Jirstrom, K.; Axelson, H.; Landberg, G. Decreased expression of Yes-associated protein is associated with outcome in the luminal A breast cancer subgroup and with an impaired tamoxifen response. BMC Cancer 2014, 14, 119. [Google Scholar] [CrossRef] [PubMed]
- Min Kim, H.; Kim, S.K.; Jung, W.H.; Koo, J.S. Metaplastic carcinoma show different expression pattern of YAP compared to triple-negative breast cancer. Tumour Biol. 2015, 36, 1207–1212. [Google Scholar] [CrossRef] [PubMed]
- Vici, P.; Mottolese, M.; Pizzuti, L.; Barba, M.; Sperati, F.; Terrenato, I.; Di Benedetto, A.; Natoli, C.; Gamucci, T.; Angelucci, D.; et al. The Hippo transducer TAZ as a biomarker of pathological complete response in HER2-positive breast cancer patients treated with trastuzumab-based neoadjuvant therapy. Oncotarget 2014, 5, 9619–9625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendinelli, P.; Maroni, P.; Matteucci, E.; Luzzati, A.; Perrucchini, G.; Desiderio, M.A. Hypoxia inducible factor-1 is activated by transcriptional co-activator with PDZ-binding motif (TAZ) versus WWdomain-containing oxidoreductase (WWOX) in hypoxic microenvironment of bone metastasis from breast cancer. Eur. J. Cancer 2013, 49, 2608–2618. [Google Scholar] [CrossRef] [PubMed]
- Hiemer, S.E.; Szymaniak, A.D.; Varelas, X. The transcriptional regulators TAZ and YAP direct transforming growth factor beta-induced tumorigenic phenotypes in breast cancer cells. J. Biol. Chem. 2014, 289, 13461–13474. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Morrison, C.D.; Liu, P.; Miecznikowski, J.; Bshara, W.; Han, S.; Zhu, Q.; Omilian, A.R.; Li, X.; Zhang, J. TAZ induces growth factor-independent proliferation through activation of EGFR ligand amphiregulin. Cell Cycle 2012, 11, 2922–2930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordenonsi, M.; Zanconato, F.; Azzolin, L.; Forcato, M.; Rosato, A.; Frasson, C.; Inui, M.; Montagner, M.; Parenti, A.R.; Poletti, A.; et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 2011, 147, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.W.; Shen, H.; Frangou, C.; Yang, N.; Guo, J.; Xu, B.; Bshara, W.; Shepherd, L.; Zhu, Q.; Wang, J.; et al. Characterization of TAZ domains important for the induction of breast cancer stem cell properties and tumorigenesis. Cell Cycle 2015, 14, 146–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asselin-Labat, M.L.; Vaillant, F.; Sheridan, J.M.; Pal, B.; Wu, D.; Simpson, E.R.; Yasuda, H.; Smyth, G.K.; Martin, T.J.; Lindeman, G.J.; et al. Control of mammary stem cell function by steroid hormone signalling. Nature 2010, 465, 798–802. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Karthik, G.M.; Lovrot, J.; Haglund, F.; Rosin, G.; Katchy, A.; Zhang, X.; Viberg, L.; Frisell, J.; Williams, C.; et al. Estrogen Receptor beta as a Therapeutic Target in Breast Cancer Stem Cells. J. Natl. Cancer Inst. 2017, 109, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Zhang, X.T.; Wang, M.L.; Zheng, H.Y.; Liu, L.J.; Wang, Z.Y. ER-alpha36-mediated rapid estrogen signaling positively regulates ER-positive breast cancer stem/progenitor cells. PLoS ONE 2014, 9, e88034. [Google Scholar]
- Gelsomino, L.; Panza, S.; Giordano, C.; Barone, I.; Gu, G.; Spina, E.; Catalano, S.; Fuqua, S.; Ando, S. Mutations in the estrogen receptor alpha hormone binding domain promote stem cell phenotype through notch activation in breast cancer cell lines. Cancer Lett. 2018, 428, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44+CD24− stem cell-like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, G.; Zhao, Y.; Liu, X.; Ding, Q.; Shi, J.; Ding, Y.; Wang, S. STAT3 mediates resistance of CD44+CD24−/low breast cancer stem cells to tamoxifen in vitro. J. Biomed. Res. 2012, 26, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kang, J.W.; Song, X.; Kim, B.K.; Yoo, Y.D.; Kwon, Y.T.; Lee, Y.J. Role of the IL-6-JAK1-STAT3-Oct-4 pathway in the conversion of non-stem cancer cells into cancer stem-like cells. Cell Signal 2013, 25, 961–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kordon, E.C.; McKnight, R.A.; Jhappan, C.; Hennighausen, L.; Merlino, G.; Smith, G.H. Ectopic TGF beta 1 expression in the secretory mammary epithelium induces early senescence of the epithelial stem cell population. Dev. Biol. 1995, 168, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Moses, H.; Barcellos-Hoff, M.H. TGF-beta biology in mammary development and breast cancer. Cold Spring Harb. Perspect. Biol. 2011, 3, a003277. [Google Scholar] [CrossRef] [PubMed]
- Sakaki-Yumoto, M.; Katsuno, Y.; Derynck, R. TGF-beta family signaling in stem cells. Biochim. Biophys. Acta 2013, 1830, 2280–2296. [Google Scholar] [CrossRef] [PubMed]
- Scheel, C.; Eaton, E.N.; Li, S.H.; Chaffer, C.L.; Reinhardt, F.; Kah, K.J.; Bell, G.; Guo, W.; Rubin, J.; Richardson, A.L.; et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 2011, 145, 926–940. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Bandyopadhyay, A.; Nichols, R.W.; Wang, L.; Hinck, A.P.; Wang, S.; Sun, L.Z. Blockade of Autocrine TGF-beta Signaling Inhibits Stem Cell Phenotype, Survival, and Metastasis of Murine Breast Cancer Cells. J. Stem Cell Res. Ther. 2012, 2, 1–8. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crabtree, J.S.; Miele, L. Breast Cancer Stem Cells. Biomedicines 2018, 6, 77. https://doi.org/10.3390/biomedicines6030077
Crabtree JS, Miele L. Breast Cancer Stem Cells. Biomedicines. 2018; 6(3):77. https://doi.org/10.3390/biomedicines6030077
Chicago/Turabian StyleCrabtree, Judy S., and Lucio Miele. 2018. "Breast Cancer Stem Cells" Biomedicines 6, no. 3: 77. https://doi.org/10.3390/biomedicines6030077
APA StyleCrabtree, J. S., & Miele, L. (2018). Breast Cancer Stem Cells. Biomedicines, 6(3), 77. https://doi.org/10.3390/biomedicines6030077