CRISPR/Cas9 for Cancer Therapy: Hopes and Challenges

 and
and {kind=link}
{kind=link}
Abstract
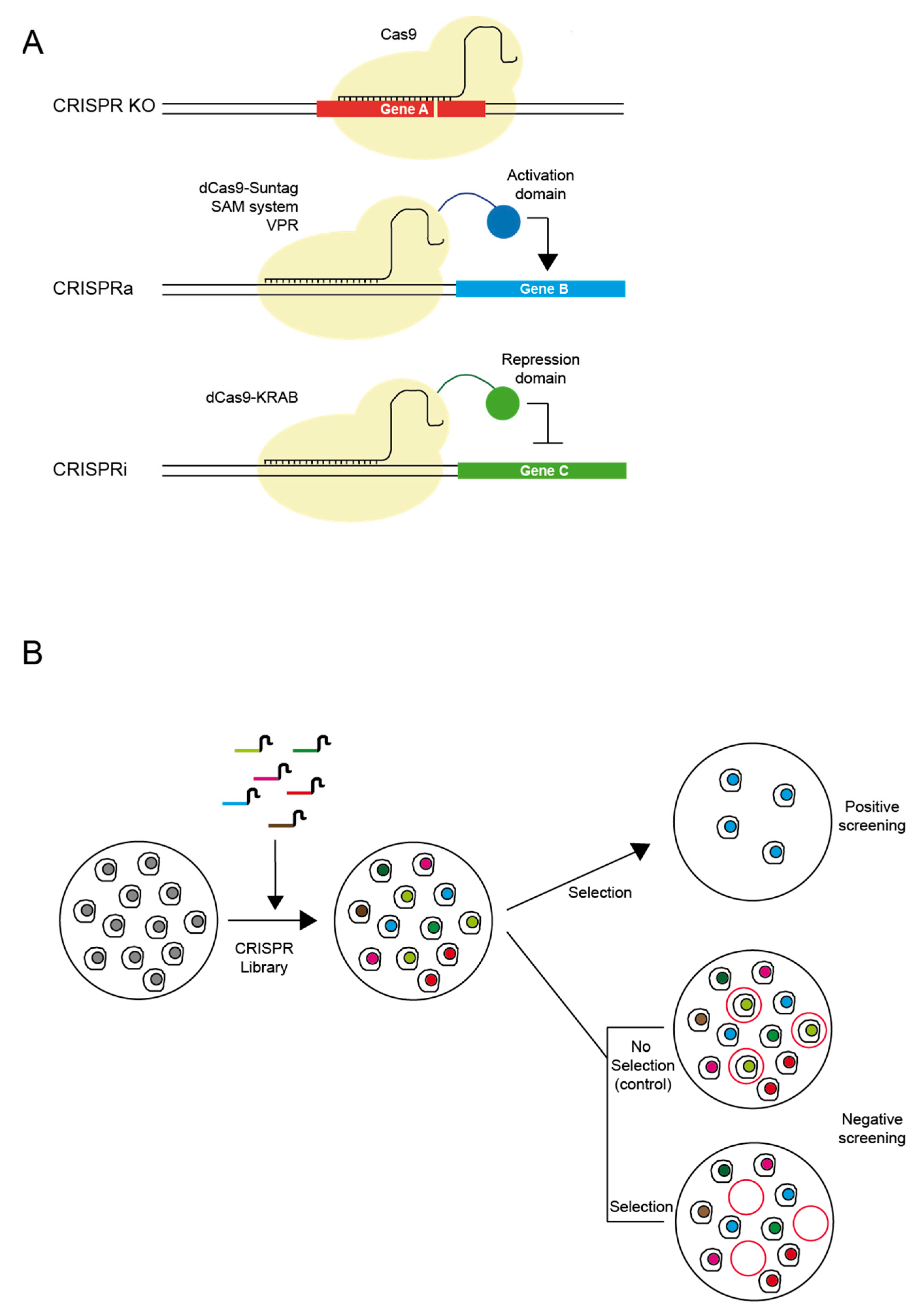
:1. Mechanism and Advantages of CRISPR Genome Editing
2. CRISPR in Drug Discovery
3. CRISPR/Cas9 Library Screens for Drug Target Discovery
4. CRISPR/Cas9 in Drug Resistance
5. Disease Models for Drug Efficacy
6. CRISPR in Cancer Therapy
7. In Vivo Delivery Technologies for Gene Editing
8. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef] [PubMed]
- Mojica, F.J.; Juez, G.; Rodríguez-Valera, F. Transcription at different salinities of Haloferax mediterranei sequences adjacent to partially modified PstI sites. Mol. Microbiol. 1993, 9, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Van Soolingen, D.; de Haas, P.E.; Hermans, P.W.; Groenen, P.M.; van Embden, J.D. Comparison of various repetitive DNA elements as genetic markers for strain differentiation and epidemiology of Mycobacterium tuberculosis. J. Clin. Microbiol. 1993, 31, 1987–1995. [Google Scholar] [PubMed]
- Bolotin, A.; Quinquis, B.; Sorokin, A.; Ehrlich, S.D. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 2005, 151, 2551–2561. [Google Scholar] [PubMed] [Green Version]
- Mojica, F.J.M.; Díez-Villaseñor, C.; García-Martínez, J.; Soria, E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 2005, 60, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Pourcel, C.; Salvignol, G.; Vergnaud, G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 2005, 151, 653–663. [Google Scholar] [PubMed]
- Van der Oost, J.; Jore, M.M.; Westra, E.R.; Lundgren, M.; Brouns, S.J.J. CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem. Sci. 2009, 34, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Kim, S.; Kim, J.M.; Kim, J.-S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Esvelt, K.M.; Church, G.M. Cas9 as a versatile tool for engineering biology. Nat. Methods 2013, 10, 957–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Haft, D.H.; Barrangou, R.; Brouns, S.J.J.; Charpentier, E.; Horvath, P.; Moineau, S.; Mojica, F.J.M.; Wolf, Y.I.; Yakunin, A.F.; et al. Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 2011, 9, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef] [PubMed]
- Mojica, F.J.M.; Díez-Villaseñor, C.; García-Martínez, J.; Almendros, C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 2009, 155, 733–740. [Google Scholar] [PubMed]
- Liang, F.; Han, M.; Romanienko, P.J.; Jasin, M. Homology-directed repair is a major double-strand break repair pathway in mammalian cells. Proc. Natl. Acad. Sci. USA 1998, 95, 5172–5177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bibikova, M.; Carroll, D.; Segal, D.J.; Trautman, J.K.; Smith, J.; Kim, Y.G.; Chandrasegaran, S. Stimulation of homologous recombination through targeted cleavage by chimeric nucleases. Mol. Cell. Biol. 2001, 21, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Fishman-Lobell, J.; Haber, J.E. Removal of nonhomologous DNA ends in double-strand break recombination: The role of the yeast ultraviolet repair gene RAD1. Science 1992, 258, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Jakočiūnas, T.; Bonde, I.; Herrgård, M.; Harrison, S.J.; Kristensen, M.; Pedersen, L.E.; Jensen, M.K.; Keasling, J.D. Multiplex metabolic pathway engineering using CRISPR/Cas9 in Saccharomyces cerevisiae. Metab. Eng. 2015, 28, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, H.; Shivalila, C.S.; Dawlaty, M.M.; Cheng, A.W.; Zhang, F.; Jaenisch, R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013, 153, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Teng, F.; Li, T.; Zhou, Q. Simultaneous generation and germline transmission of multiple gene mutations in rat using CRISPR-Cas systems. Nat. Biotechnol. 2013, 31, 684–686. [Google Scholar] [CrossRef] [PubMed]
- Torres, R.; Martin, M.C.; Garcia, A.; Cigudosa, J.C.; Ramirez, J.C.; Rodriguez-Perales, S. Engineering human tumour-associated chromosomal translocations with the RNA-guided CRISPR-Cas9 system. Nat. Commun. 2014, 5, 3964. [Google Scholar] [CrossRef] [PubMed]
- Choi, P.S.; Meyerson, M. Targeted genomic rearrangements using CRISPR/Cas technology. Nat. Commun. 2014, 5, 3728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adikusuma, F.; Williams, N.; Grutzner, F.; Hughes, J.; Thomas, P. Targeted Deletion of an Entire Chromosome Using CRISPR/Cas9. Mol. Ther. 2017, 25, 1736–1738. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, A.A.; Lim, W.A.; Qi, L.S. Beyond editing: Repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat. Rev. Mol. Cell Biol. 2016, 17, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Q.; Joung, J.K. Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nat. Rev. Genet. 2016, 17, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.-S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Staahl, B.T.; Alla, R.K.; Doudna, J.A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife 2014, 3, e04766. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishna, S.; Kwaku Dad, A.-B.; Beloor, J.; Gopalappa, R.; Lee, S.-K.; Kim, H. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Res. 2014, 24, 1020–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuris, J.A.; Thompson, D.B.; Shu, Y.; Guilinger, J.P.; Bessen, J.L.; Hu, J.H.; Maeder, M.L.; Joung, J.K.; Chen, Z.-Y.; Liu, D.R. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 2015, 33, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Torres-Ruiz, R.; Martinez-Lage, M.; Martin, M.C.; Garcia, A.; Bueno, C.; Castaño, J.; Ramirez, J.C.; Menendez, P.; Cigudosa, J.C.; Rodriguez-Perales, S. Efficient Recreation of t(11;22) EWSR1-FLI1+ in Human Stem Cells Using CRISPR/Cas9. Stem Cell Rep. 2017, 8, 1408–1420. [Google Scholar] [CrossRef] [PubMed]
- Nihongaki, Y.; Kawano, F.; Nakajima, T.; Sato, M. Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat. Biotechnol. 2015, 33, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Dow, L.E.; Fisher, J.; O’Rourke, K.P.; Muley, A.; Kastenhuber, E.R.; Livshits, G.; Tschaharganeh, D.F.; Socci, N.D.; Lowe, S.W. Inducible in vivo genome editing with CRISPR-Cas9. Nat. Biotechnol. 2015, 33, 390–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, K.M.; Pattanayak, V.; Thompson, D.B.; Zuris, J.A.; Liu, D.R. Small molecule-triggered Cas9 protein with improved genome-editing specificity. Nat. Chem. Biol. 2015, 11, 316–318. [Google Scholar] [CrossRef] [PubMed]
- Truong, D.-J.J.; Kühner, K.; Kühn, R.; Werfel, S.; Engelhardt, S.; Wurst, W.; Ortiz, O. Development of an intein-mediated split-Cas9 system for gene therapy. Nucleic Acids Res. 2015, 43, 6450–6458. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.V.; Sternberg, S.H.; Taylor, D.W.; Staahl, B.T.; Bardales, J.A.; Kornfeld, J.E.; Doudna, J.A. Rational design of a split-Cas9 enzyme complex. Proc. Natl. Acad. Sci. USA 2015, 112, 2984–2989. [Google Scholar] [CrossRef] [PubMed]
- Oakes, B.L.; Nadler, D.C.; Flamholz, A.; Fellmann, C.; Staahl, B.T.; Doudna, J.A.; Savage, D.F. Profiling of engineering hotspots identifies an allosteric CRISPR-Cas9 switch. Nat. Biotechnol. 2016, 34, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Lin, C.-Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Guilinger, J.P.; Thompson, D.B.; Liu, D.R. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat. Biotechnol. 2014, 32, 577–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Fleuren, E.D.G.; Zhang, L.; Wu, J.; Daly, R.J. The kinome “at large” in cancer. Nat. Rev. Cancer 2016, 16, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, G.; Amiji, M. Use of CRISPR/Cas9 gene-editing tools for developing models in drug discovery. Drug Discov. Today 2018, 23, 519–533. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Joung, J.; Konermann, S.; Gootenberg, J.S.; Abudayyeh, O.O.; Platt, R.J.; Brigham, M.D.; Sanjana, N.E.; Zhang, F. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 2017, 12, 828–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J. CRISPR/Cas9: From Genome Engineering to Cancer Drug Discovery. Trends Cancer 2016, 2, 313–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeder, M.L.; Linder, S.J.; Cascio, V.M.; Fu, Y.; Ho, Q.H.; Joung, J.K. CRISPR RNA-guided activation of endogenous human genes. Nat. Methods 2013, 10, 977–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavez, A.; Tuttle, M.; Pruitt, B.W.; Ewen-Campen, B.; Chari, R.; Ter-Ovanesyan, D.; Haque, S.J.; Cecchi, R.J.; Kowal, E.J.K.; Buchthal, J.; et al. Comparison of Cas9 activators in multiple species. Nat. Methods 2016, 13, 563–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Kurata, M.; Yamamoto, K.; Moriarity, B.S.; Kitagawa, M.; Largaespada, D.A. CRISPR/Cas9 library screening for drug target discovery. J. Hum. Genet. 2018, 63, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Guichard, S.M. CRISPR–Cas9 for Drug Discovery in Oncology. In Platform Technologies in Drug Discovery and Validation; Elsevier: Amsterdam, The Netherlands, 2017; pp. 61–85. [Google Scholar]
- Scott, A. How CRISPR is transforming drug discovery. Nature 2018, 555, S10–S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smurnyy, Y.; Cai, M.; Wu, H.; McWhinnie, E.; Tallarico, J.A.; Yang, Y.; Feng, Y. DNA sequencing and CRISPR-Cas9 gene editing for target validation in mammalian cells. Nat. Chem. Biol. 2014, 10, 623–625. [Google Scholar] [CrossRef] [PubMed]
- Neggers, J.E.; Vercruysse, T.; Jacquemyn, M.; Vanstreels, E.; Baloglu, E.; Shacham, S.; Crochiere, M.; Landesman, Y.; Daelemans, D. Identifying drug-target selectivity of small-molecule CRM1/XPO1 inhibitors by CRISPR/Cas9 genome editing. Chem. Biol. 2015, 22, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Walton, J.; Blagih, J.; Ennis, D.; Leung, E.; Dowson, S.; Farquharson, M.; Tookman, L.A.; Orange, C.; Athineos, D.; Mason, S.; et al. CRISPR/Cas9-Mediated Trp53 and Brca2 Knockout to Generate Improved Murine Models of Ovarian High-Grade Serous Carcinoma. Cancer Res. 2016, 76, 6118–6129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedland, A.E.; Tzur, Y.B.; Esvelt, K.M.; Colaiácovo, M.P.; Church, G.M.; Calarco, J.A. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat. Methods 2013, 10, 741–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Gilbert, L.A.; Cimini, B.A.; Schnitzbauer, J.; Zhang, W.; Li, G.-W.; Park, J.; Blackburn, E.H.; Weissman, J.S.; Qi, L.S.; et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 2013, 155, 1479–1491. [Google Scholar] [CrossRef] [PubMed]
- Klann, T.S.; Black, J.B.; Chellappan, M.; Safi, A.; Song, L.; Hilton, I.B.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. CRISPR-Cas9 epigenome editing enables high-throughput screening for functional regulatory elements in the human genome. Nat. Biotechnol. 2017, 35, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, E.R.; Thiele, C.J. Epigenetic changes in pediatric solid tumors: Promising new targets. Clin. Cancer Res. 2012, 18, 2768–2779. [Google Scholar] [CrossRef] [PubMed]
- Shachaf, C.M.; Kopelman, A.M.; Arvanitis, C.; Karlsson, A.; Beer, S.; Mandl, S.; Bachmann, M.H.; Borowsky, A.D.; Ruebner, B.; Cardiff, R.D.; et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 2004, 431, 1112–1117. [Google Scholar] [CrossRef] [PubMed]
- Oiseth, S.J.; Aziz, M.S. Cancer immunotherapy: A brief review of the history, possibilities, and challenges ahead. J. Cancer Metastasis Treat. 2017, 3, 250. [Google Scholar] [CrossRef]
- Choi, A.H.; O’Leary, M.P.; Fong, Y.; Chen, N.G. From Benchtop to Bedside: A Review of Oncolytic Virotherapy. Biomedicines 2016, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, K.; Chen, W.; Johnson, D.C.; Hendricks, R.L. Infected cell protein (ICP)47 enhances herpes simplex virus neurovirulence by blocking the CD8+ T cell response. J. Exp. Med. 1998, 187, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Aghi, M.; Visted, T.; Depinho, R.A.; Chiocca, E.A. Oncolytic herpes virus with defective ICP6 specifically replicates in quiescent cells with homozygous genetic mutations in p16. Oncogene 2008, 27, 4249–4254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandara, L.R.; La Thangue, N.B. Adenovirus E1a prevents the retinoblastoma gene product from complexing with a cellular transcription factor. Nature 1991, 351, 494–497. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, M.; Raychaudhuri, P. Retinoblastoma-repression of E2F-dependent transcription depends on the ability of the retinoblastoma protein to interact with E2F and is abrogated by the adenovirus E1A oncoprotein. Nucleic Acids Res. 1992, 20, 5947–5954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyson, N.; Harlow, E. Adenovirus E1A targets key regulators of cell proliferation. Cancer Surv. 1992, 12, 161–195. [Google Scholar] [PubMed]
- Su, S.; Hu, B.; Shao, J.; Shen, B.; Du, J.; Du, Y.; Zhou, J.; Yu, L.; Zhang, L.; Chen, F.; et al. CRISPR-Cas9 mediated efficient PD-1 disruption on human primary T cells from cancer patients. Sci. Rep. 2016, 6, 20070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, S.Y.; Otsuji, M.; Gorski, K.; Huang, X.; Slansky, J.E.; Pai, S.I.; Shalabi, A.; Shin, T.; Pardoll, D.M.; Tsuchiya, H. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J. Exp. Med. 2001, 193, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsushima, F.; Yao, S.; Shin, T.; Flies, A.; Flies, S.; Xu, H.; Tamada, K.; Pardoll, D.M.; Chen, L. Interaction between B7-H1 and PD-1 determines initiation and reversal of T-cell anergy. Blood 2007, 110, 180–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fellmann, C.; Gowen, B.G.; Lin, P.-C.; Doudna, J.A.; Corn, J.E. Cornerstones of CRISPR-Cas in drug discovery and therapy. Nat. Rev. Drug Discov. 2017, 16, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Maus, M.V.; Grupp, S.A.; Porter, D.L.; June, C.H. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood 2014, 123, 2625–2635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cyranoski, D. CRISPR gene-editing tested in a person for the first time. Nature 2016, 539, 479. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, J.; Turtle, C.J. Insights into cytokine release syndrome and neurotoxicity after CD19-specific CAR-T cell therapy. Curr. Res. Transl. Med. 2018, 66, 50–52. [Google Scholar] [CrossRef] [PubMed]
- Muffly, L.S.; Reizine, N.; Stock, W. Management of acute lymphoblastic leukemia in young adults. Clin. Adv. Hematol. Oncol. 2018, 16, 138–146. [Google Scholar] [PubMed]
- Kay, M.A. State-of-the-art gene-based therapies: The road ahead. Nat. Rev. Genet. 2011, 12, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Song, C.-Q.; Dorkin, J.R.; Zhu, L.J.; Li, Y.; Wu, Q.; Park, A.; Yang, J.; Suresh, S.; Bizhanova, A.; et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat. Biotechnol. 2016, 34, 328–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, R.; Garcia, A.; Jimenez, M.; Rodriguez, S.; Ramirez, J.C. An integration-defective lentivirus-based resource for site-specific targeting of an edited safe-harbour locus in the human genome. Gene Ther. 2014, 21, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Kauffman, K.J.; Anderson, D.G. Delivery technologies for genome editing. Nat. Rev. Drug Discov. 2017, 16, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Daya, S.; Berns, K.I. Gene therapy using adeno-associated virus vectors. Clin. Microbiol. Rev. 2008, 21, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Chang, R.; Yang, H.; Zhao, T.; Hong, Y.; Kong, H.E.; Sun, X.; Qin, Z.; Jin, P.; Li, S.; et al. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J. Clin. Investig. 2017, 127, 2719–2724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaj, T.; Ojala, D.S.; Ekman, F.K.; Byrne, L.C.; Limsirichai, P.; Schaffer, D.V. In vivo genome editing improves motor function and extends survival in a mouse model of ALS. Sci Adv. 2017, 3, eaar3952. [Google Scholar] [CrossRef] [PubMed]
- Kotterman, M.A.; Schaffer, D.V. Engineering adeno-associated viruses for clinical gene therapy. Nat. Rev. Genet. 2014, 15, 445–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Follenzi, A.; Santambrogio, L.; Annoni, A. Immune responses to lentiviral vectors. Curr. Gene Ther. 2007, 7, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Ahi, Y.S.; Bangari, D.S.; Mittal, S.K. Adenoviral vector immunity: Its implications and circumvention strategies. Curr. Gene Ther. 2011, 11, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, J.C.; Kowalski, P.S.; Anderson, D.G. Advances in the delivery of RNA therapeutics: From concept to clinical reality. Genome Med. 2017, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Ji, W.; Hall, J.M.; Hu, Q.; Wang, C.; Beisel, C.L.; Gu, Z. Self-assembled DNA nanoclews for the efficient delivery of CRISPR-Cas9 for genome editing. Angew. Chem. Int. Ed. Engl. 2015, 54, 12029–12033. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Conboy, M.; Park, H.M.; Jiang, F.; Kim, H.J.; Dewitt, M.A.; Mackley, V.A.; Chang, K.; Rao, A.; Skinner, C.; et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat. Biomed. Eng. 2017, 1, 889–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.P.; Zeng, Q.H.; Lu, G.Q.; Yu, A.B. Inorganic nanoparticles as carriers for efficient cellular delivery. Chem. Eng. Sci. 2006, 61, 1027–1040. [Google Scholar] [CrossRef]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinez-Lage, M.; Puig-Serra, P.; Menendez, P.; Torres-Ruiz, R.; Rodriguez-Perales, S. CRISPR/Cas9 for Cancer Therapy: Hopes and Challenges. Biomedicines 2018, 6, 105. https://doi.org/10.3390/biomedicines6040105
Martinez-Lage M, Puig-Serra P, Menendez P, Torres-Ruiz R, Rodriguez-Perales S. CRISPR/Cas9 for Cancer Therapy: Hopes and Challenges. Biomedicines. 2018; 6(4):105. https://doi.org/10.3390/biomedicines6040105
Chicago/Turabian StyleMartinez-Lage, Marta, Pilar Puig-Serra, Pablo Menendez, Raul Torres-Ruiz, and Sandra Rodriguez-Perales. 2018. "CRISPR/Cas9 for Cancer Therapy: Hopes and Challenges" Biomedicines 6, no. 4: 105. https://doi.org/10.3390/biomedicines6040105
APA StyleMartinez-Lage, M., Puig-Serra, P., Menendez, P., Torres-Ruiz, R., & Rodriguez-Perales, S. (2018). CRISPR/Cas9 for Cancer Therapy: Hopes and Challenges. Biomedicines, 6(4), 105. https://doi.org/10.3390/biomedicines6040105