Dissecting the Immune Landscape of Acute Myeloid Leukemia


Abstract
:1. Introduction
2. The Tumor Immunological Microenvironment
3. High-Resolution Platforms to Decipher the Complexity of the TME
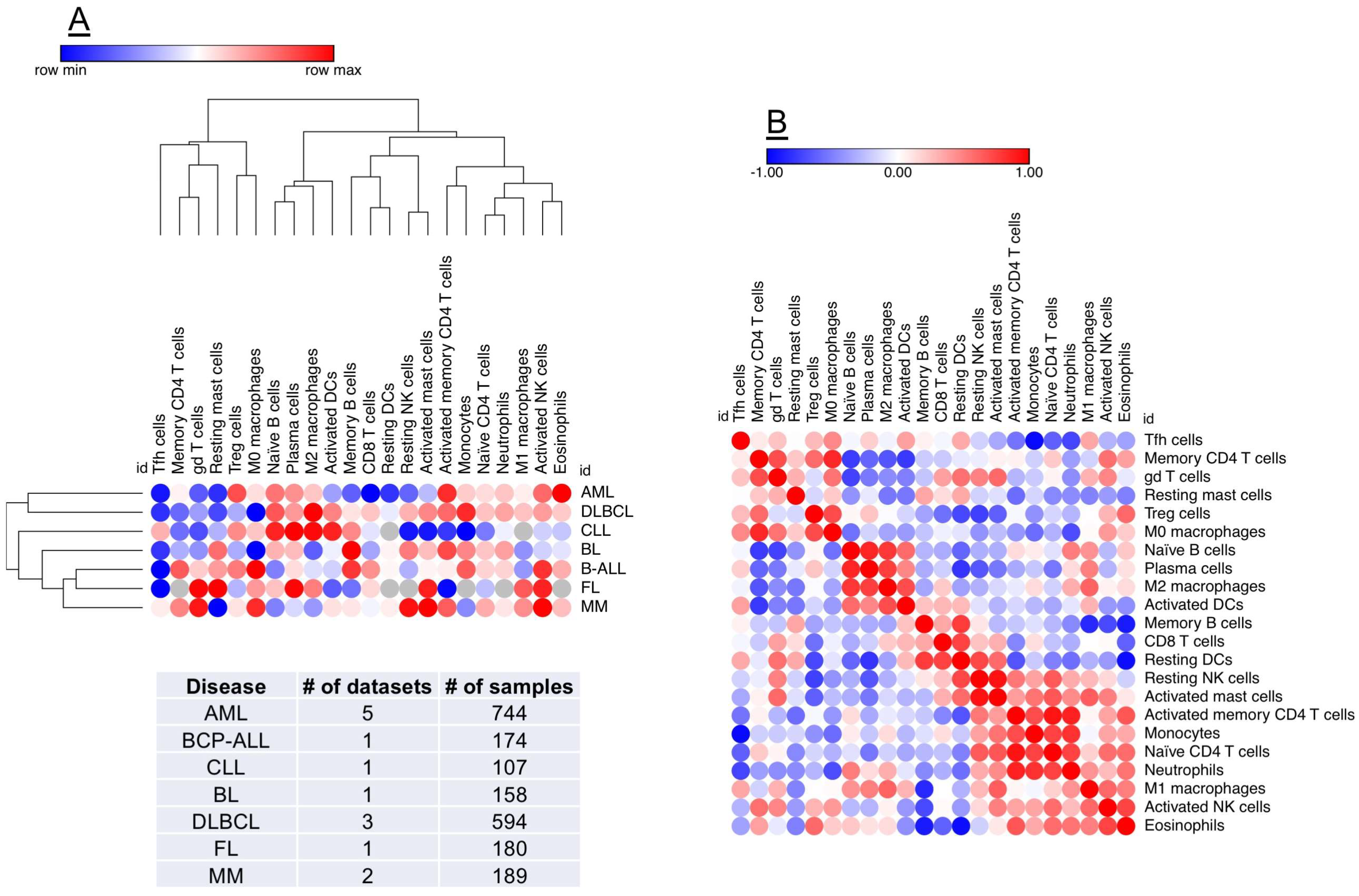
4. Composition of the AML TME
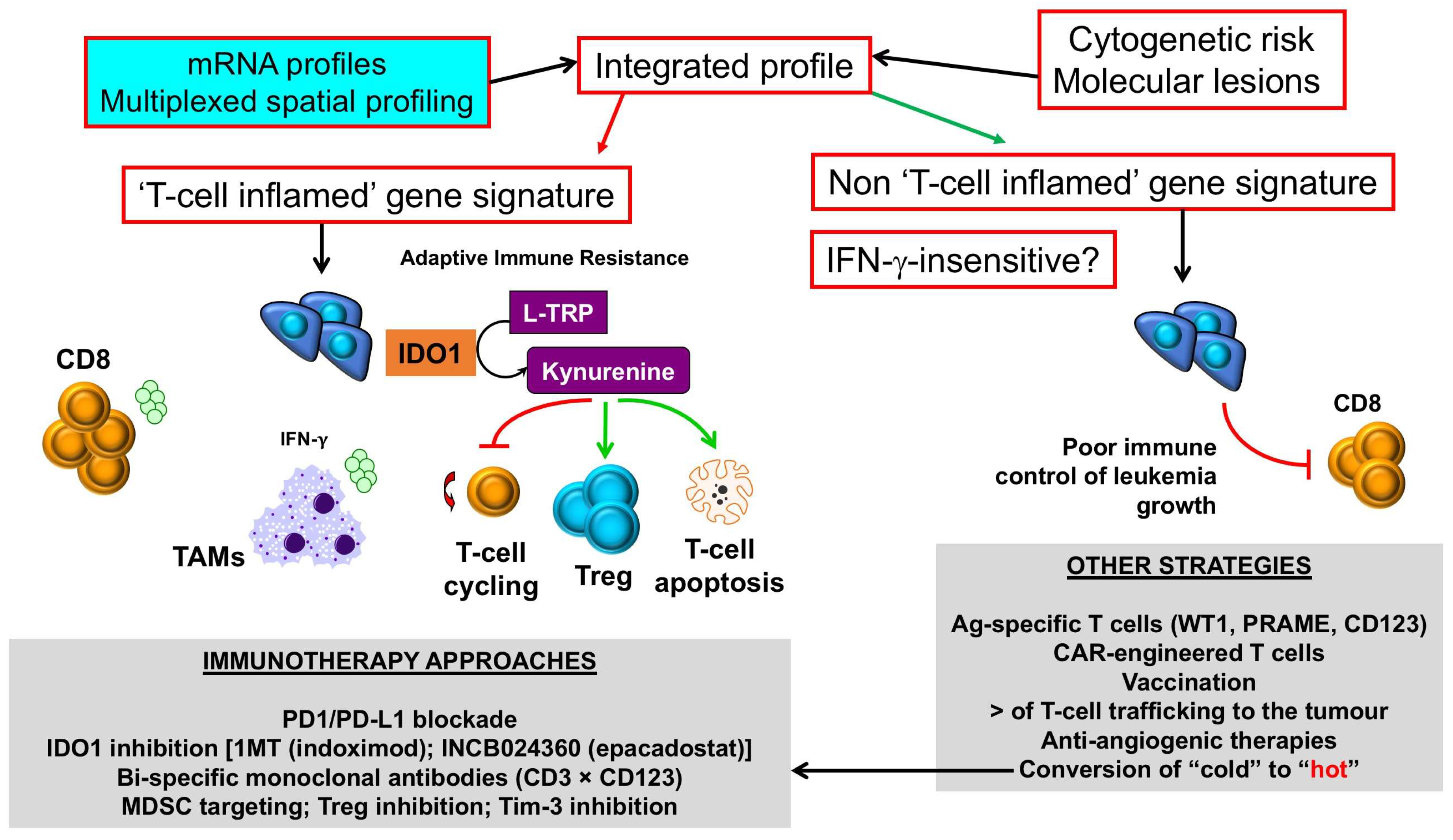
5. Prognostic and Predictive Immune Biomarkers in the AML TME
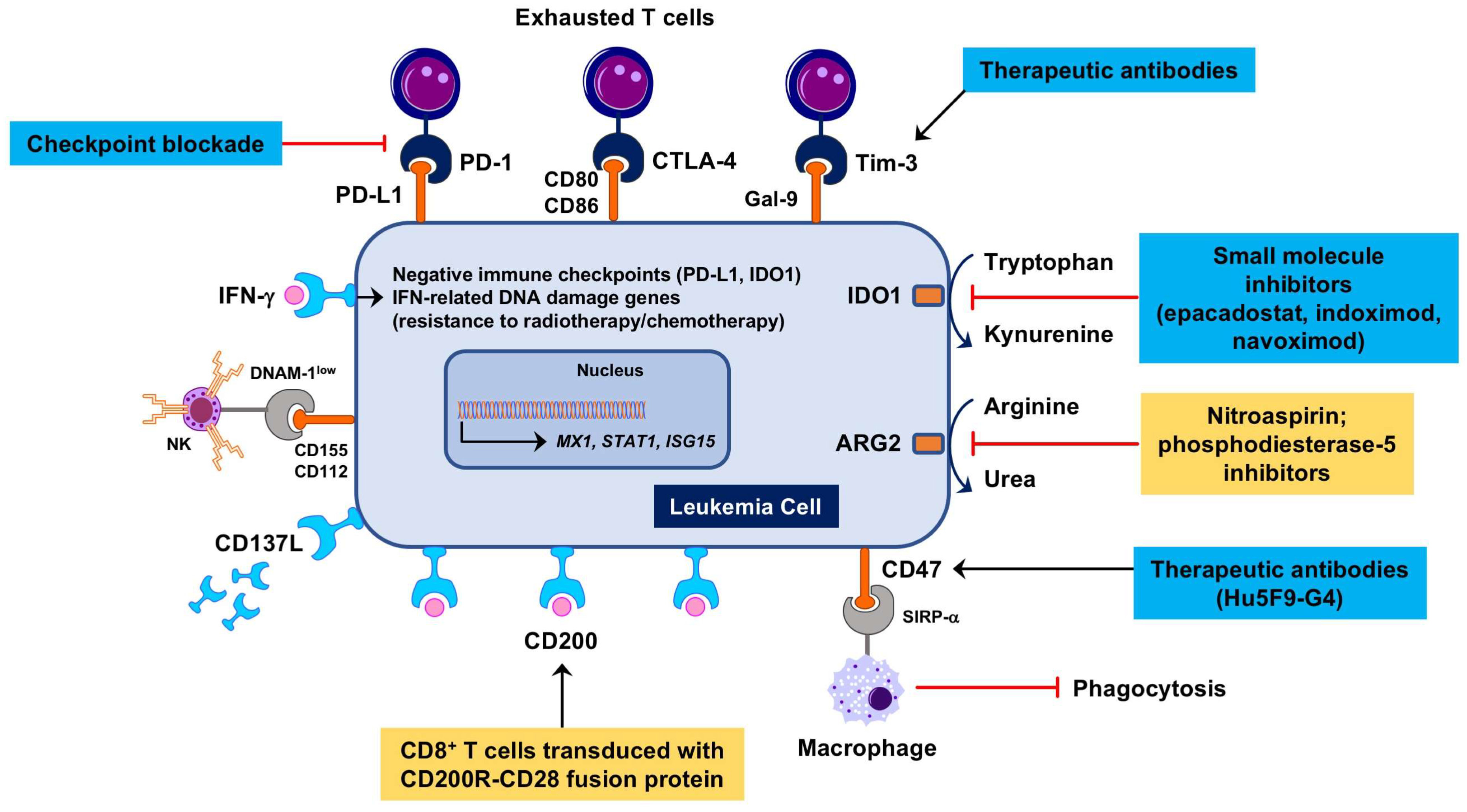
6. Immune Checkpoint Blockade and Novel Immunotherapies for “Inflamed” AMLs
7. Conclusions and Translational Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Bullinger, L.; Dohner, K.; Bair, E.; Frohling, S.; Schlenk, R.F.; Tibshirani, R.; Dohner, H.; Pollack, J.R. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N. Engl. J. Med. 2004, 350, 1605–1616. [Google Scholar] [CrossRef] [PubMed]
- Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; Baty, J.D.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute myeloid leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Valk, P.J.; Verhaak, R.G.; Beijen, M.A.; Erpelinck, C.A.; Barjesteh van Waalwijk van Doorn-Khosrovani, S.; Boer, J.M.; Beverloo, H.B.; Moorhouse, M.J.; van der Spek, P.J.; Lowenberg, B.; et al. Prognostically useful gene-expression profiles in acute myeloid leukemia. N. Engl. J. Med. 2004, 350, 1617–1628. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed]
- Gerstung, M.; Papaemmanuil, E.; Martincorena, I.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Heuser, M.; Thol, F.; Bolli, N.; Ganly, P.; et al. Precision oncology for acute myeloid leukemia using a knowledge bank approach. Nat. Genet. 2017, 49, 332–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coombs, C.C.; Tallman, M.S.; Levine, R.L. Molecular therapy for acute myeloid leukaemia. Nat. Rev. Clin. Oncol. 2016, 13, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Uy, G.L.; Godwin, J.; Rettig, M.P.; Vey, N.; Foster, M.; Arellano, M.L.; Rizzieri, D.A.; Topp, M.S.; Huls, G.; Lowenberg, B.; et al. Preliminary results of a phase 1 study of flotetuzumab, a CD123 × CD3 bispecific DART® protein, in patients with relapsed/refractory acute myeloid leukemia and myelodysplastic syndrome. Blood 2017, 130, 637. [Google Scholar]
- Ng, S.W.; Mitchell, A.; Kennedy, J.A.; Chen, W.C.; McLeod, J.; Ibrahimova, N.; Arruda, A.; Popescu, A.; Gupta, V.; Schimmer, A.D.; et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 2016, 540, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjostedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The immune landscape of cancer. Immunity 2018, 48, 812–830. [Google Scholar] [CrossRef] [PubMed]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P.N. The cancer immunotherapy revolution. Science 2018, 359, 1344–1345. [Google Scholar] [CrossRef] [PubMed]
- Heymach, J.; Krilov, L.; Alberg, A.; Baxter, N.; Chang, S.M.; Corcoran, R.; Dale, W.; DeMichele, A.; Magid Diefenbach, C.S.; Dreicer, R.; et al. Clinical Cancer Advances 2018: Annual report on progress against cancer from the American Society of Clinical Oncology. J. Clin. Oncol. 2018, 36, 1020–1044. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.G.; Stromnes, I.M.; Greenberg, P.D. Obstacles posed by the tumor microenvironment to T cell activity: A case for synergistic therapies. Cancer Cell 2017, 31, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Haanen, J.B.; Ribas, A.; Schumacher, T.N. The cancer immunogram. Science 2016, 352, 658–660. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taube, J.M.; Anders, R.A.; Young, G.D.; Xu, H.; Sharma, R.; McMiller, T.L.; Chen, S.; Klein, A.P.; Pardoll, D.M.; Topalian, S.L.; et al. Colocalization of inflammatory response with B7-H1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci. Transl. Med. 2012, 4, 127ra137. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syn, N.L.; Teng, M.W.L.; Mok, T.S.K.; Soo, R.A. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017, 18, e731–e741. [Google Scholar] [CrossRef]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell 2016, 167, 1540–1554. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Gajewski, T.F. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 2018, 18, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Iglesia, M.D.; Parker, J.S.; Hoadley, K.A.; Serody, J.S.; Perou, C.M.; Vincent, B.G. Genomic analysis of immune cell infiltrates across 11 tumor types. J. Natl. Cancer Inst. 2016, 108, djw144. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Luke, J.J.; Bao, R.; Zha, Y.; Hernandez, K.M.; Li, Y.; Gajewski, A.P.; Andrade, J.; Gajewski, T.F. Density of immunogenic antigens does not explain the presence or absence of the T-cell-inflamed tumor microenvironment in melanoma. Proc. Natl. Acad. Sci. USA 2016, 113, E7759–E7768. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.K.; Herbst, R.S.; Chen, L. Defining and understanding adaptive resistance in cancer immunotherapy. Trends Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B. IFNgamma: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Weichselbaum, R.R.; Ishwaran, H.; Yoon, T.; Nuyten, D.S.; Baker, S.W.; Khodarev, N.; Su, A.W.; Shaikh, A.Y.; Roach, P.; Kreike, B.; et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 18490–18495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khodarev, N.N.; Beckett, M.; Labay, E.; Darga, T.; Roizman, B.; Weichselbaum, R.R. STAT1 is overexpressed in tumors selected for radioresistance and confers protection from radiation in transduced sensitive cells. Proc. Natl. Acad. Sci. USA 2004, 101, 1714–1719. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Yang, S.W.; Yu, K.R.; Ka, S.H.; Lee, S.W.; Seol, J.H.; Jeon, Y.J.; Chung, C.H. Modification of PCNA by ISG15 plays a crucial role in termination of error-prone translesion DNA synthesis. Mol. Cell 2014, 54, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Danaher, P.; Warren, S.; Lu, R.; Samayoa, J.; Sullivan, A.; Pekker, I.; Wallden, B.; Marincola, F.M.; Cesano, A. Pan-cancer adaptive immune resistance as defined by the Tumor Inflammation Signature (TIS): Results from The Cancer Genome Atlas (TCGA). J. Immunother. Cancer 2018, 6, 63. [Google Scholar] [CrossRef] [PubMed]
- Wargo, J.A.; Reddy, S.M.; Reuben, A.; Sharma, P. Monitoring immune responses in the tumor microenvironment. Curr. Opin. Immunol. 2016, 41, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stroncek, D.F.; Butterfield, L.H.; Cannarile, M.A.; Dhodapkar, M.V.; Greten, T.F.; Grivel, J.C.; Kaufman, D.R.; Kong, H.H.; Korangy, F.; Lee, P.P.; et al. Systematic evaluation of immune regulation and modulation. J. Immunother. Cancer 2017, 5, 21. [Google Scholar] [CrossRef] [PubMed]
- Mlecnik, B.; Bindea, G.; Angell, H.K.; Sasso, M.S.; Obenauf, A.C.; Fredriksen, T.; Lafontaine, L.; Bilocq, A.M.; Kirilovsky, A.; Tosolini, M.; et al. Functional network pipeline reveals genetic determinants associated with in situ lymphocyte proliferation and survival of cancer patients. Sci. Transl. Med. 2014, 6, 228ra237. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef] [PubMed]
- Wellenstein, M.D.; de Visser, K.E. Cancer-cell-intrinsic mechanisms shaping the tumor immune landscape. Immunity 2018, 48, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Gajewski, T.F. Tumor-intrinsic oncogene pathways mediating immune avoidance. Oncoimmunology 2016, 5, e1086862. [Google Scholar] [CrossRef] [PubMed]
- Bezzi, M.; Seitzer, N.; Ishikawa, T.; Reschke, M.; Chen, M.; Wang, G.; Mitchell, C.; Ng, C.; Katon, J.; Lunardi, A.; et al. Diverse genetic-driven immune landscapes dictate tumor progression through distinct mechanisms. Nat. Med. 2018, 24, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Kohanbash, G.; Carrera, D.A.; Shrivastav, S.; Ahn, B.J.; Jahan, N.; Mazor, T.; Chheda, Z.S.; Downey, K.M.; Watchmaker, P.B.; Beppler, C.; et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+T cell accumulation in gliomas. J. Clin. Investig. 2017, 127, 1425–1437. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, A.H.; Tiong, I.S. Midostaurin, enasidenib, CPX-351, gemtuzumab ozogamicin, and venetoclax bring new hope to AML. Blood 2017, 130, 2469–2474. [Google Scholar] [CrossRef] [PubMed]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A.; Robert, C.; Hodi, F.S.; Wolchok, J.D.; Joshua, A.M.; Hwu, W.-J.; Weber, J.S.; Zarour, H.M.; Kefford, R.; Loboda, A.; et al. Association of response to programmed death receptor 1 (PD-1) blockade with pembrolizumab (MK-3475) with an interferon-inflammatory immune gene signature. J. Clin. Oncol. 2015, 33, 3001. [Google Scholar]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Damotte, D.; Arrondeau, J.; Boudou-Rouquette, P.; Lupo, A.; Biton, J.; Ouakrim, H.; Alifano, M.; Claire, G.; Bellesoeur, A.; Kramkimel, N.; et al. The tumor inflammation signature is predictive of anti-PD1 treatment benefıt in the CERTIM pan-cancer cohort. Proc. Am. Assoc. Cancer Res. 2018, 59, 1163. [Google Scholar]
- Warren, S.; Vallabhaneni, P.; El-Sawada, J.; White, A.; Ren, X.; Cesano, A.; Beechem, J.; Tarhini, A. Immunological profiling of baseline and resected biopsies from locally/regionally advanced/recurrent melanoma treated with neoadjuvant combination ipilimumab (3 mg/kg or 10 mg/kg) and high dose IFN-α2B. J. Immunother. Cancer 2017, 5, 61. [Google Scholar]
- Rozeman, L.; Franchi, L.; Kuilman, T.; Krijgsman, O.; van Akkooi, A.; Kvistborg, P.; van Thienen, H.; Stegenga, B.; Cullen, D.; Lamon, B.; et al. Biomarker analysis from the OpACIN trial (Neo-/adjuvant ipilimumab + nivolumab (IPI + NIVO) in palpable stage 3 melanoma). J. Immunother. Cancer 2017, 5, 61. [Google Scholar]
- Danaher, P.; Warren, S.; Dennis, L.; D’Amico, L.; White, A.; Disis, M.L.; Geller, M.A.; Odunsi, K.; Beechem, J.; Fling, S.P. Gene expression markers of tumor infiltrating leukocytes. J. Immunother. Cancer 2017, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.L.; Roh, W.; Reuben, A.; Cooper, Z.A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Bassett, R.L.; Gopalakrishnan, V.; Wani, K.; et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. 2016, 6, 827–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stack, E.C.; Foukas, P.G.; Lee, P.P. Multiplexed tissue biomarker imaging. J. Immunother. Cancer 2016, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.Y.; Bhattacharya, N.; Mu, J.; Setiadi, A.F.; Carcamo-Cavazos, V.; Lee, G.H.; Simons, D.L.; Yadegarynia, S.; Hemati, K.; Kapelner, A.; et al. Spatial organization of dendritic cells within tumor draining lymph nodes impacts clinical outcome in breast cancer patients. J. Transl. Med. 2013, 11, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesano, A. nCounter(R) PanCancer Immune Profiling Panel (NanoString Technologies, Inc., Seattle, WA). J. Immunother. Cancer 2015, 3, 42. [Google Scholar] [CrossRef] [PubMed]
- White, A.; Elliott, N.; Liang, Y.; Warren, S.E.; Beechem, J.M. Spatially resolved, multiplexed digital characterization of protein distribution and abundance in Formalin-Fixed, Paraffin-Embedded (FFPE) Diffuse Large B Cell Lymphoma tissue sections based on the Nanostring® Digital Spatial Profiling (DSP) Technology. Blood 2017, 130, 1196. [Google Scholar]
- Rutella, S.; Vadakekolathu, J.; Altmann, H.; Patel, T.; Reeder, S.; Liang, Y.; Schmitz, M.; Hood, T.; Danaher, P.; Warren, S.; et al. Capturing the complexity of the immune microenvironment of acute myeloid leukemia with 3D biology technology. J. Clin. Oncol. 2018, 36, 50. [Google Scholar] [CrossRef]
- Simoni, Y.; Becht, E.; Fehlings, M.; Loh, C.Y.; Koo, S.L.; Teng, K.W.W.; Yeong, J.P.S.; Nahar, R.; Zhang, T.; Kared, H.; et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 2018, 557, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Krieg, C.; Nowicka, M.; Guglietta, S.; Schindler, S.; Hartmann, F.J.; Weber, L.M.; Dummer, R.; Robinson, M.D.; Levesque, M.P.; Becher, B. Author Correction: High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Krieg, C.; Nowicka, M.; Guglietta, S.; Schindler, S.; Hartmann, F.J.; Weber, L.M.; Dummer, R.; Robinson, M.D.; Levesque, M.P.; Becher, B. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat. Med. 2018, 24, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Mercier, F.E.; Ragu, C.; Scadden, D.T. The bone marrow at the crossroads of blood and immunity. Nat. Rev. Immunol. 2011, 12, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, M.H.; Gherardini, P.F.; Fragiadakis, G.K.; Bhattacharya, N.; Yuan, R.T.; Hotson, A.N.; Finck, R.; Carmi, Y.; Zunder, E.R.; Fantl, W.J.; et al. An interactive reference framework for modeling a dynamic immune system. Science 2015, 349, 1259425. [Google Scholar] [CrossRef] [PubMed]
- Sapoznikov, A.; Pewzner-Jung, Y.; Kalchenko, V.; Krauthgamer, R.; Shachar, I.; Jung, S. Perivascular clusters of dendritic cells provide critical survival signals to B cells in bone marrow niches. Nat. Immunol. 2008, 9, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Tokoyoda, K.; Egawa, T.; Sugiyama, T.; Choi, B.I.; Nagasawa, T. Cellular niches controlling B lymphocyte behavior within bone marrow during development. Immunity 2004, 20, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Tokoyoda, K.; Zehentmeier, S.; Hegazy, A.N.; Albrecht, I.; Grun, J.R.; Lohning, M.; Radbruch, A. Professional memory CD4+ T lymphocytes preferentially reside and rest in the bone marrow. Immunity 2009, 30, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Okhrimenko, A.; Grun, J.R.; Westendorf, K.; Fang, Z.; Reinke, S.; von Roth, P.; Wassilew, G.; Kuhl, A.A.; Kudernatsch, R.; Demski, S.; et al. Human memory T cells from the bone marrow are resting and maintain long-lasting systemic memory. Proc. Natl. Acad. Sci. USA 2014, 111, 9229–9234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, T.C.; Coley, S.M.; Wherry, E.J.; Ahmed, R. Bone marrow is a preferred site for homeostatic proliferation of memory CD8 T cells. J. Immunol. 2005, 174, 1269–1273. [Google Scholar] [CrossRef] [PubMed]
- Mazo, I.B.; Honczarenko, M.; Leung, H.; Cavanagh, L.L.; Bonasio, R.; Weninger, W.; Engelke, K.; Xia, L.; McEver, R.P.; Koni, P.A.; et al. Bone marrow is a major reservoir and site of recruitment for central memory CD8+T cells. Immunity 2005, 22, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.D.; Tokoyoda, K.; Radbruch, A. Immunological memories of the bone marrow. Immunol. Rev. 2018, 283, 86–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prins, R.M.; Shu, C.J.; Radu, C.G.; Vo, D.D.; Khan-Farooqi, H.; Soto, H.; Yang, M.Y.; Lin, M.S.; Shelly, S.; Witte, O.N.; et al. Anti-tumor activity and trafficking of self, tumor-specific T cells against tumors located in the brain. Cancer Immunol. Immunother. 2008, 57, 1279–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curran, E.K.; Godfrey, J.; Kline, J. Mechanisms of immune tolerance in leukemia and lymphoma. Trends Immunol. 2017, 38, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Mussai, F.; De Santo, C.; Abu-Dayyeh, I.; Booth, S.; Quek, L.; McEwen-Smith, R.M.; Qureshi, A.; Dazzi, F.; Vyas, P.; Cerundolo, V. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood 2013, 122, 749–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folgiero, V.; Goffredo, B.M.; Filippini, P.; Masetti, R.; Bonanno, G.; Caruso, R.; Bertaina, V.; Mastronuzzi, A.; Gaspari, S.; Zecca, M.; et al. Indoleamine 2,3-dioxygenase 1 (IDO1) activity in leukemia blasts correlates with poor outcome in childhood acute myeloid leukemia. Oncotarget 2014, 5, 2052–2064. [Google Scholar] [CrossRef] [PubMed]
- Curti, A.; Pandolfi, S.; Valzasina, B.; Aluigi, M.; Isidori, A.; Ferri, E.; Salvestrini, V.; Bonanno, G.; Rutella, S.; Durelli, I.; et al. Modulation of tryptophan catabolism by human leukemic cells results in the conversion of CD25- into CD25+ T regulatory cells. Blood 2007, 109, 2871–2877. [Google Scholar] [CrossRef] [PubMed]
- Zafeiris, D.; Vadakekolathu, J.; Wagner, S.; Pockley, A.G.; Ball, G.R.; Rutella, S. Discovery and application of immune biomarkers for hematological malignancies. Expert Rev. Mol. Diagn. 2017, 17, 983–1000. [Google Scholar] [CrossRef] [PubMed]
- Austin, R.; Smyth, M.J.; Lane, S.W. Harnessing the immune system in acute myeloid leukaemia. Crit. Rev. Oncol. Hematol. 2016, 103, 62–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, T.; Matsumoto, T.; Shibata, Y.; Nakamura, N.; Nakamura, H.; Ninomiya, S.; Kitagawa, J.; Nannya, Y.; Shimizu, M.; Ito, H.; et al. Prognostic value of the combination of serum L-kynurenine level and indoleamine 2,3-dioxygenase mRNA expression in acute myeloid leukemia. Leuk. Lymphoma 2016, 57, 2208–2211. [Google Scholar] [CrossRef] [PubMed]
- Vadakekolathu, J.; Meredith, R.; Ross, P.M.; Wagner, S.; Reeder, S.; Demirkan, G.; Kuhar, J.R.; Ball, G.R.; Pockley, A.G.; Rutella, S. Interferon-γ induces distinct mRNA and protein profiles in acute and chronic myeloid leukemia. Blood 2017, 130, 3945. [Google Scholar]
- Vadakekolathu, J.; Patel, T.; Reeder, S.; Schaarschmidt, H.; Schmitz, M.; Bornhäuser, M.; Warren, S.E.; Hood, T.; Danaher, P.; Cesano, A.; et al. Immune gene expression profiling in children and adults with acute myeloid leukemia identifies distinct phenotypic patterns. Blood 2017, 130, 3942. [Google Scholar]
- Schurch, C.; Riether, C.; Amrein, M.A.; Ochsenbein, A.F. Cytotoxic T cells induce proliferation of chronic myeloid leukemia stem cells by secreting interferon-gamma. J. Exp. Med. 2013, 210, 605–621. [Google Scholar] [CrossRef] [PubMed]
- Nayak-Kapoor, A.; Hao, Z.; Sadek, R.; Dobbins, R.; Marshall, L.; Vahanian, N.N.; Jay Ramsey, W.; Kennedy, E.; Mautino, M.R.; Link, C.J.; et al. Phase Ia study of the indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor navoximod (GDC-0919) in patients with recurrent advanced solid tumors. J. Immunother. Cancer 2018, 6, 61. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; O’Dwyer, P.J.; Clark, J.; Shi, J.G.; Bowman, K.J.; Scherle, P.A.; Newton, R.C.; Schaub, R.; Maleski, J.; Leopold, L.; et al. First-in-human phase I study of the oral inhibitor of indoleamine 2,3-dioxygenase-1 epacadostat (INCB024360) in patients with advanced solid malignancies. Clin. Cancer Res. 2017, 23, 3269–3276. [Google Scholar] [CrossRef] [PubMed]
- De Santo, C.; Serafini, P.; Marigo, I.; Dolcetti, L.; Bolla, M.; Del Soldato, P.; Melani, C.; Guiducci, C.; Colombo, M.P.; Iezzi, M.; et al. Nitroaspirin corrects immune dysfunction in tumor-bearing hosts and promotes tumor eradication by cancer vaccination. Proc. Natl. Acad. Sci. USA 2005, 102, 4185–4190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pende, D.; Spaggiari, G.M.; Marcenaro, S.; Martini, S.; Rivera, P.; Capobianco, A.; Falco, M.; Lanino, E.; Pierri, I.; Zambello, R.; et al. Analysis of the receptor-ligand interactions in the natural killer-mediated lysis of freshly isolated myeloid or lymphoblastic leukemias: Evidence for the involvement of the Poliovirus receptor (CD155) and Nectin-2 (CD112). Blood 2005, 105, 2066–2073. [Google Scholar] [CrossRef] [PubMed]
- Hentschel, N.; Krusch, M.; Kiener, P.A.; Kolb, H.J.; Salih, H.R.; Schmetzer, H.M. Serum levels of sCD137 (4-1BB) ligand are prognostic factors for progression in acute myeloid leukemia but not in non-Hodgkin’s lymphoma. Eur. J. Haematol. 2006, 77, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Coles, S.J.; Gilmour, M.N.; Reid, R.; Knapper, S.; Burnett, A.K.; Man, S.; Tonks, A.; Darley, R.L. The immunosuppressive ligands PD-L1 and CD200 are linked in AML T-cell immunosuppression: Identification of a new immunotherapeutic synapse. Leukemia 2015, 29, 1952–1954. [Google Scholar] [CrossRef] [PubMed]
- Oda, S.K.; Daman, A.W.; Garcia, N.M.; Wagener, F.; Schmitt, T.M.; Tan, X.; Chapuis, A.G.; Greenberg, P.D. A CD200R-CD28 fusion protein appropriates an inhibitory signal to enhance T-cell function and therapy of murine leukemia. Blood 2017, 130, 2410–2419. [Google Scholar] [CrossRef] [PubMed]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D., Jr.; van Rooijen, N.; Weissman, I.L. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Kikushige, Y.; Shima, T.; Takayanagi, S.; Urata, S.; Miyamoto, T.; Iwasaki, H.; Takenaka, K.; Teshima, T.; Tanaka, T.; Inagaki, Y.; et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell 2010, 7, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Armand, P.; Shipp, M.A.; Ribrag, V.; Michot, J.M.; Zinzani, P.L.; Kuruvilla, J.; Snyder, E.S.; Ricart, A.D.; Balakumaran, A.; Rose, S.; et al. Programmed Death-1 blockade with pembrolizumab in patients with classical Hodgkin lymphoma after brentuximab vedotin failure. J. Clin. Oncol. 2016, 34, 3733–3739. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Melero, I.; Berman, D.M.; Aznar, M.A.; Korman, A.J.; Perez Gracia, J.L.; Haanen, J. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat. Rev. Cancer 2015, 15, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Farsaci, B.; Donahue, R.N.; Grenga, I.; Lepone, L.M.; Kim, P.S.; Dempsey, B.; Siebert, J.C.; Ibrahim, N.K.; Madan, R.A.; Heery, C.R.; et al. Analyses of pretherapy peripheral immunoscore and response to vaccine therapy. Cancer Immunol. Res. 2016, 4, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Gnjatic, S.; Bronte, V.; Brunet, L.R.; Butler, M.O.; Disis, M.L.; Galon, J.; Hakansson, L.G.; Hanks, B.A.; Karanikas, V.; Khleif, S.N.; et al. Identifying baseline immune-related biomarkers to predict clinical outcome of immunotherapy. J. Immunother. Cancer 2017, 5, 44. [Google Scholar] [CrossRef] [PubMed]
- Church, S.E.; Galon, J. Tumor microenvironment and immunotherapy: The whole picture is better than a glimpse. Immunity 2015, 43, 631–633. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, H.; Kitada, K.; Nakai, K.; Sarai, A. PrognoScan: A new database for meta-analysis of the prognostic value of genes. BMC Med. Genom. 2009, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goswami, C.P.; Nakshatri, H. PROGgeneV2: Enhancements on the existing database. BMC Cancer 2014, 14, 970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, J. Too much of a good thing? Science 2018, 359, 1346–1347. [Google Scholar] [CrossRef] [PubMed]
- Armand, P. Immune checkpoint blockade in hematologic malignancies. Blood 2015, 125, 3393–3400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeidner, J.F.; Vincent, B.G.; Ivanova, A.; Foster, M.; Coombs, C.C.; Jamieson, K.; Van Deventer, H.; Scibilia, R.; Blanchard, L.; Frank, C.; et al. Phase II study of high dose cytarabine followed by pembrolizumab in relapsed/refractory acute myeloid leukemia (AML). Blood 2017, 130, 1349. [Google Scholar]
- Kubasch, A.S.; Wehner, R.; Bazzurri, S.; Tunger, A.; Stasik, S.; Garzarolli, M.; Meinel, J.; Baretton, G.; Meier, F.; Thiede, C.; et al. Clinical, molecular, and immunological responses to pembrolizumab treatment of synchronous melanoma and acute myeloid leukemia. Blood Adv. 2018, 2, 1187–1190. [Google Scholar] [CrossRef] [PubMed]
- Chichili, G.R.; Huang, L.; Li, H.; Burke, S.; He, L.; Tang, Q.; Jin, L.; Gorlatov, S.; Ciccarone, V.; Chen, F.; et al. A CD3 × CD123 bispecific DART for redirecting host T cells to myelogenous leukemia: Preclinical activity and safety in nonhuman primates. Sci. Transl. Med. 2015, 7, 289ra282. [Google Scholar] [CrossRef] [PubMed]
- Rutella, S.; Church, S.E.; Vadakekolathu, J.; Viboch, E.; Sullivan, A.H.; Hood, T.; Warren, S.E.; Cesano, A.; La Motte-Mohs, R.; Muth, J.; et al. Adaptive immune gene signatures correlate with response to Flotetuzumab, a CD123 × CD3 bispecific DART® molecule, in patients with relapsed/refractory acute myeloid leukemia. Blood 2018, in press. [Google Scholar]
- Uy, G.L.; Rettig, M.P.; Vey, N.; Godwin, J.; Foster, M.C.; Rizzieri, D.A.; Arellano, M.L.; Topp, M.S.; Huls, G.; Jongen-Lavrencic, M.; et al. Phase 1 cohort expansion of Flotetuzumab, a CD123 × CD3 bispecific DART® protein in patients with relapsed/refractory acute myeloid leukemia (AML). Blood 2018, in press. [Google Scholar]
- Godwin, J.; Ballesteros-Merino, C.; Bifulco, C.B.; Lelievre, H.; Davidson-Moncada, J.; Wigginton, J.M.; Fox, B. Bone marrow T cell changes by multiplex IHC after treatment with Flotetuzumab, a CD123 × CD3 bispecific DART® protein, in a primary refractory t-AML patient. Blood 2018, in press. [Google Scholar]
- Rettig, M.P.; Godwin, J.; Vey, N.; Fox, B.; Ballesteros-Merino, C.; Bifulco, C.B.; Li, D.; Primo, D.; Ballesteros, J.; Sun, J.; et al. Preliminary translational results from an ongoing phase 1 study of Flotetuzumab, a CD123 × CD3 DART®, in AML/MDS: Rationale for combining flotetuzumab and anti-PD-1/PD-L1 immunotherapies. Blood 2017, 130, 1365. [Google Scholar]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 2017, 169, 361. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.; Tasian, S.K.; Ruella, M.; Shestova, O.; Li, Y.; Porter, D.L.; Carroll, M.; Danet-Desnoyers, G.; Scholler, J.; Grupp, S.A.; et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood 2014, 123, 2343–2354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Auslander, N.; Zhang, G.; Lee, J.S.; Frederick, D.T.; Miao, B.; Moll, T.; Tian, T.; Wei, Z.; Madan, S.; Sullivan, R.J.; et al. Robust prediction of response to immune checkpoint blockade therapy in metastatic melanoma. Nat. Med. 2018. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Disease Stage | Therapeutic Agents | Study Design | Participants | Estimated Completion Date | Principal INVESTIGATOR | Clinicaltrials Gov. Identifier |
|---|---|---|---|---|---|---|
| Newly diagnosed AML age ≥ 60 years not eligible for intensive chemotherapy or HR MDS | Azacitidine monotherapy (days 1–7 every 28 days), or Azacitidine (days 1–7 every 28 days) + Nivolumab (every 2 weeks) or Azacitadine (days 1–7 every 28 days) ± Midostaurin (BID days 8–21 every 28 days), or Decitabine (days 1–5 every 28 days) and Cytarabine (days 6–11 every 28 days) | Randomized (stratified by FLT3 mutational status) Open-label Phase 2–3 | n = 1670 | August 2023 | Laura Michaelis, MD | NCT03092674 |
| Newly diagnosed AML age ≥ 60 years in first CR not eligible for HSCT | Pembrolizumab (200 mg every 3 weeks) | Non-randomized Open-label Phase 2 | n = 40 | October 2020 | Michael Boyiadzis, MD, MHSc | NCT02708641 |
| Previously untreated AML age ≥ 65 not eligible for HSCT or Previously untreated MDS | Durvalumab (1500 mg day 1 every 4 weeks) and Azacytidine (75 mg/m2 for 7 days every 4 weeks) vs. Azacytidine monotherapy (75 mg/m2 for 7 days every 4 weeks) | Randomized Open-label Phase 2 | n = 213 | April 2019 | Not listed/Celgene | NCT02775903 |
| Previously untreated AML not suitable for intensive chemotherapy | Avelumab (10 mg/kg, day 1, every 14 days) and Decitabine (20 mg/m2 IV days 1–5, every 28 days) | Non-randomized Open-label Phase 1 | n = 15 | December 2020 | Hong Zheng, MD | NCT03395873 |
| HR AML | Pembrolizumab on day +1 following lymphodepleting chemotherapy with FLU/MEL and autologous HSCT | Non-randomized Open-label Phase 2 | n = 20 | June 2021 | Scott Solomon, MD | NCT02771197 |
| Newly diagnosed AML age ≥ 65 years or R/R AML | Azacitidine (75 mg/m2 days 1–7 every 28 days) + pembrolizumab (200 mg every 3 weeks starting on day 8 of cycle 1) | Non-randomized Open-label Phase 2 | n = 40 | July 2020 | Ivana Gojo, MD | NCT02845297 |
| Newly diagnosed elderly AML (≥65 years) or R/R AML | Azacitidine + Nivolumab dose escalation starting at 75 mg/m2 (SQ) on days 1–7 of every 28 day cycle + 3.0 mg/kg on day 1 and day 14 every 28 days for the first 4 cycles or until CR (whichever occurs earlier) followed by a maintenance regimen (one dose of nivolumab on day 1 of each cycle of 5-azacytidine). Dose expansion with maximum tolerated dose (MTD)); or Azacitidine + Nivolumab + Ipilimumab dose escalation with Azacitidine + Nivolumab doses per above and Ipilumab starting at 1 mg/kg every 12 weeks. Dose expansion with MTD. | Non-randomized Open-label Phase II | n = 182 | April 2020 | Naval Daver, MD | NCT02397720 |
| AML (newly diagnosed for dose-expansion; newly diagnosed or R/R for dose escalation) and HR-MDS | Idarubicin (12 mg/m2 days 1–3 of 28 day cycle), cytarabine (1.5 g/m2 days 1–4 of 28 day cycle) with Solumedrol 50 mg; or Dexamethasone 10 mg for 3–4 days on days 1–4 and nivolumab (starting dose of 1 mg/kg on day 24 of 28 day cycle and dose escalated in successive cohorts to MTD) | Non-randomized Open label Phase 1/2 | n = 75 | July 2019 | Farhad Ravandi-Kashani, MD | NCT02464657 |
| AML (newly diagnosed elderly AML unfit for induction chemotherapy and R/R for dose-expansion; R/R for dose escalation) | Atezolizumab (840 mg on days 8 and 22 of every 28-day cycle) and guadecitabine (60 mg/m2 on days 1–5 of every 28-day cycle) | Non-randomized Open label Phase 1b | n = 40 | January 2019 | Not listed/Hoffmann-La Roche | NCT02892318 |
| AML (newly diagnosed AML not suitable for standard induction) or R/R AML or HR-MDS or HR-MDS who have failed hypo-methylating agent therapy | Decitabine + PDR001 (anti-PD-1) or Decitabine + MBG453 (anti-TIM3) or Decitabine + PDR001 + MBG453 or MBG453 monotherapy or MBG453 + PDR001 | Non-randomized Open label Phase 1b | n = 175 | April 2020 | Andrew M. Brunner, MD | NCT03066648 |
| AML in remission at HR for relapse | Nivolumab (3 mg/kg days 1 and 15 every 28 days, after cycle 6 day 1 every 28 days, after cycle 12 reduce to 1 time every 12 weeks) | Non-randomized Open-label Phase 2 | n = 30 | October 2020 | Tapan Kadia, MD | NCT02532231 |
| AML in remission after chemotherapy | Nivolumab (every 2 weeks for 46 courses) | Randomized Open-label, with cross-over upon relapse Phase 2 | n = 80 | June 2019 | Hongtao Liu, MD, PhD | NCT02275533 |
| Eldery AML (≥ 60 years) with CR or CRI after induction/consolidation and MRD positive status not planned for HSCT | Atezolizumab (1200 mg every cycle) and BL-8040 (1.25 mg/kg days 1–3 of cycle) | Randomized Open label Phase 1b/2 60 participants | n = 60 | March 2022 | Not listed/BioLineRx | NCT03154827 |
| Refractory AML | Pembrolizumab (200 mg every 3 weeks) | Non-randomized Open-label Phase 0 pilot study | n = 10 | August 2022 | Michael Boyiadzis, MD, MHSc | NCT03291353 |
| R/R AML | Decitabine (20 mg/m2 day 8 through 12 and 15 through 19 on alternative cycles) + pembrolizumab (200 mg; every cycle (21 days)) | Non-randomized Open-label Phase 1–2 | n = 15 | July 2019 | Christopher S Hourigan, MD | NCT02996474 |
| R/R AML | HiDAC salvage induction therapy followed by pembrolizumab monotherapy on day 14 (200 mg) and every 3 weeks | Non-randomized Open-label Phase 2 | n = 37 | September 2025 | Joshua F Zeidner, MD | NCT02768792 |
| Elderly AML age ≥ 55 | Cytarabine (500–1000 mg/m2 bid days −4, −3, −2) + G-CSF mobilized HLA-haploidentical donor peripheral blood stem cells (day 0) + Nivolumab (40 mg day +5 for 2–3 cycles) or Cytarabine (500–1000 mg/m2 bid days +1, +2, +3) + Nivolumab (40 mg day +1 for 2–3 cycles) | Randomized Open-label Haploidentical T cells, cytarabine and nivolumab vs. cytarabine and nivolumab Phase 2 | n = 52 | October 2020 | Boris Afanasyev, MD, Prof. & Anna Smirnova, PhD | NCT03381118 |
| AML, ALL, or MDS with relapse after allogeneic HSCT | Pembrolizumab (200 mg every 3 weeks) | Non-randomized Open-label Phase 1b | n = 20 | October 2021 | John M Magenau, MD | NCT03286114 |
| AML and other hematological malignancies with relapse after allogeneic HSCT | Pembrolizumab (200 mg every 3 weeks for up to 24 months) | Non-randomized Open-label Phase 1 pilot study | n = 26 | February 2020 | Justin Kline, MD | NCT02981914 |
| AML and MDS after allogeneic HSCT at HR for post-transplant recurrence | Nivolumab (1 or 3 mg/kg every 3 weeks for up to 34 weeks) or Ipilimumab (0.3 mg/kg, 1 mg/kg or 3 mg/kg every 3 weeks for up to 16 weeks) or Nivolumab + Ipilimumab (3 mg/kg every 3 weeks for up to 34 weeks and 0.3 mg/kg, 0.6 mg/kg or 1.0 mg/kg every 3 weeks for up to 16 weeks respectively) | Non-randomized Open-label Phase 1 | n = 21 | July 2023 | Andrew Pecora, MD & James McCloskey, MD & Jamie Koprivnika, MD | NCT02846376 |
| HR R/R AML following allogeneic HSCT | Nivolumab (days 1 and 15 every 28 days) up to 6 courses or Ipilimumab (day 1 every 21 days) up to 6 courses or Nivolumab (days 1 and 14 every 28 days) + Ipilumab (day 1 every 28 days) up to 6 courses | Non-randomized Open-label Phase 1 | n = 55 | January 2020 | Gheath Al-Atrash, DO, PhD | NCT03600155 |
| R/R AML and HR-MDS | Cyclophosphamide (50 mg orally) + nivolumab (3 mg/kg (or if prior alloHSCT, 1 mg/kg) every 14 days on Days 1 and 15 for up to four 28-day courses) or Cyclophosphamide (350 mg orally) + nivolumab (3 mg/kg (or if prior alloHSCT, 1 mg/kg) every 14 days on Days 1 and 15 for up to four 28-day courses) | Randomized Open-label Phase 2 | n = 32 | February 2023 | Daniel J Weisdorf, MD | NCT03417154 |
| R/R AML | PF-04518600 (anti-Ox40) monotherapy (dose escalation starting dose of 0.3 (units not given) on days 1 and 14 of a 28 day cycle) or PF-04518600 (dose escalation per above) and avelumab (10 mg/kg on days 1 and 14 of a 28 day cycle) or PF-04518600 (dose escalation per above) + Azacitidine (75 mg/m2 on days 1–5 or 1–7) or PF-04518600 (dose escalation per above) + Utomilumab (anti-CD137) (100 mg on days 1 and 14 of a 28 day cycle) or Avelumab (10 mg/kg on days 1 and 14 of a 28 day cycle) + Utomilumab (100 mg on days 1 and 14 of a 28 day cycle) or PF-04518600 (dose escalation per above) + Avelumab (10 mg/kg on days 1 and 14 of a 28 day cycle) + Azacitidine (75 mg/m2 on days 1–5 or 1–7) or Gemtuzumab Ozogamicin (3 mg/m2 on Days 1, 4, and 7 of each 28 day cycle) + Glasdegib (smoothened inhibitor) (100 mg oral daily) or Avelumab (10 mg/kg on days 1 and 14 of a 28 day cycle) + Glasdegib (100 mg oral daily) | Non-randomized Open-label Phase 1b/2 | n = 138 | December 2024 | Naval G. Daver, MD | NCT03390296 |
| R/R AML | Avelumab (starting dose for dose escalation 3.0 mg/kg on days 1 and 14 of 28 day cycle) and Azacytidine (75 mg/m2 days 1–7 or days 1–5, 8–9 of 28 day cycle) | Non-randomized Open-label Phase 1b/2 | n = 58 | February 2021 | Naval G. Daver, MD | NCT02953561 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davidson-Moncada, J.; Viboch, E.; Church, S.E.; Warren, S.E.; Rutella, S. Dissecting the Immune Landscape of Acute Myeloid Leukemia. Biomedicines 2018, 6, 110. https://doi.org/10.3390/biomedicines6040110
Davidson-Moncada J, Viboch E, Church SE, Warren SE, Rutella S. Dissecting the Immune Landscape of Acute Myeloid Leukemia. Biomedicines. 2018; 6(4):110. https://doi.org/10.3390/biomedicines6040110
Chicago/Turabian StyleDavidson-Moncada, Jan, Elena Viboch, Sarah E. Church, Sarah E. Warren, and Sergio Rutella. 2018. "Dissecting the Immune Landscape of Acute Myeloid Leukemia" Biomedicines 6, no. 4: 110. https://doi.org/10.3390/biomedicines6040110
APA StyleDavidson-Moncada, J., Viboch, E., Church, S. E., Warren, S. E., & Rutella, S. (2018). Dissecting the Immune Landscape of Acute Myeloid Leukemia. Biomedicines, 6(4), 110. https://doi.org/10.3390/biomedicines6040110