Recombinant Viruses for Cancer Therapy

 , , ,
, , , 


Abstract
:
1. Introduction
2. Recombinant Virus-Based Therapeutic Vaccines
3. Virus-Based Engineering of Cell-Based Vaccines
4. Oncolytic Viruses
5. Virus-Mediated Immunomodulation and Oncosuppression
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Padma, V.V. An overview of targeted cancer therapy. BioMedicine 2015, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Menezes, M.E.; Bhatia, S.; Wang, X.Y.; Emdad, L.; Sarkar, D.; Fisher, P.B. Gene therapies for cancer: Strategies, challenges and successes. J. Cell. Physiol. 2015, 230, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Banchereau, J. Dendritic-cell-based therapeutic cancer vaccines. Immunity 2013, 39, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yin, B.; Wang, H.Y.; Wang, R.F. Current advances in T-cell-based cancer immunotherapy. Immunotherapy 2014, 6, 1265–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivatsan, S.; Patel, J.M.; Bozeman, E.N.; Imasuen, I.E.; He, S.; Daniels, D.; Selvaraj, P. Allogeneic tumor cell vaccines: The promise and limitations in clinical trials. Hum. Vaccines Immunother. 2014, 10, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Gilazieva, Z.E.; Tazetdinova, L.G.; Arkhipova, S.S.; Solovyeva, V.V.; Rizvanov, A.A. Effect of cisplatin on ultrastructure and viability of adipose-derived mesenchymal stem cells. BioNanoScience 2016, 6, 534–539. [Google Scholar] [CrossRef]
- Chulpanova, D.S.; Kitaeva, K.V.; Tazetdinova, L.G.; James, V.; Rizvanov, A.A.; Solovyeva, V.V. Application of mesenchymal stem cells for therapeutic agent delivery in anti-tumor treatment. Front. Pharmacol. 2018, 9, 259. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.J.; Tang, Y.M. Cytokine release syndrome in cancer immunotherapy with chimeric antigen receptor engineered T cells. Cancer Lett. 2014, 343, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Chulpanova, D.S.; Kitaeva, K.V.; James, V.; Rizvanov, A.A.; Solovyeva, V.V. Therapeutic prospects of extracellular vesicles in cancer treatment. Front. Immunol. 2018, 9, 1534. [Google Scholar] [CrossRef] [PubMed]
- Gilligan, K.E.; Dwyer, R.M. Engineering exosomes for cancer therapy. Int. J. Mol. Sci. 2017, 18, 1122. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, K. Latest development in viral vectors for gene therapy. Trends Biotechnol. 2003, 21, 117–122. [Google Scholar] [CrossRef]
- Rollier, C.S.; Reyes-Sandoval, A.; Cottingham, M.G.; Ewer, K.; Hill, A.V. Viral vectors as vaccine platforms: Deployment in sight. Curr. Opin. Immunol. 2011, 23, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, H.; Ino, Y.; Todo, T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016, 107, 1373–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lizee, G.; Gonzales, M.I.; Topalian, S.L. Lentivirus vector-mediated expression of tumor-associated epitopes by human antigen presenting cells. Hum. Gene Ther. 2004, 15, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Tsang, K.Y.; Schlom, J. Induction of higher-avidity human ctls by vector-mediated enhanced costimulation of antigen-presenting cells. Clin. Cancer Res. 2005, 11, 5603–5615. [Google Scholar] [CrossRef] [PubMed]
- Larocca, C.; Schlom, J. Viral vector-based therapeutic cancer vaccines. Cancer J. 2011, 17, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Coulie, P.G.; Lehmann, F.; Lethe, B.; Herman, J.; Lurquin, C.; Andrawiss, M.; Boon, T. A mutated intron sequence codes for an antigenic peptide recognized by cytolytic T lymphocytes on a human melanoma. Proc. Natl. Acad. Sci. USA 1995, 92, 7976–7980. [Google Scholar] [CrossRef] [PubMed]
- El-Sharkawy, A.; Al Zaidan, L.; Malki, A. Epstein-barr virus-associated malignancies: Roles of viral oncoproteins in carcinogenesis. Front. Oncol. 2018, 8, 265. [Google Scholar] [CrossRef] [PubMed]
- Neek, M.; Tucker, J.A.; Kim, T.I.; Molino, N.M.; Nelson, E.L.; Wang, S.W. Co-delivery of human cancer-testis antigens with adjuvant in protein nanoparticles induces higher cell-mediated immune responses. Biomaterials 2018, 156, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Cao, S.; Li, J.; Meng, Q.; Wang, C.; Yao, L.; Lang, Y.; Cao, J.; Shen, J.; Pan, B.; et al. Cancer/testis antigens (CTAs) expression in resected lung cancer. OncoTargets Ther. 2018, 11, 4491–4499. [Google Scholar] [CrossRef] [PubMed]
- Scanlan, M.J.; Gure, A.O.; Jungbluth, A.A.; Old, L.J.; Chen, Y.T. Cancer/testis antigens: An expanding family of targets for cancer immunotherapy. Immunol. Rev. 2002, 188, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, M.; Schlom, J.; Hodge, J.W. The combined activation of positive costimulatory signals with modulation of a negative costimulatory signal for the enhancement of vaccine-mediated T-cell responses. Cancer Immunol. Immunother. 2007, 56, 1471–1484. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Zhu, Z.; Chang, J.; Wang, J.; Shen, X. Lentivirus-mediated silencing of myosin VI inhibits proliferation and cell cycle progression in human lung cancer cells. Chem. Biol. Drug Des. 2015, 86, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Li, L.X.; Zhang, Y.L.; Zhou, L.; Ke, M.L.; Chen, J.M.; Fu, X.; Ye, C.L.; Wu, J.X.; Liu, R.Y.; Huang, W. Antitumor efficacy of a recombinant adenovirus encoding endostatin combined with an E1B55KD-deficient adenovirus in gastric cancer cells. J. Transl. Med. 2013, 11, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermeij, J.; Zeinoun, Z.; Neyns, B.; Teugels, E.; Bourgain, C.; De Greve, J. Transduction of ovarian cancer cells: A recombinant adeno-associated viral vector compared to an adenoviral vector. Br. J. Cancer 2001, 85, 1592–1599. [Google Scholar] [CrossRef] [PubMed]
- Parato, K.A.; Breitbach, C.J.; Le Boeuf, F.; Wang, J.; Storbeck, C.; Ilkow, C.; Diallo, J.S.; Falls, T.; Burns, J.; Garcia, V.; et al. The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol. Ther. 2012, 20, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Goshima, F.; Esaki, S.; Luo, C.; Kamakura, M.; Kimura, H.; Nishiyama, Y. Oncolytic viral therapy with a combination of HF10, a herpes simplex virus type 1 variant and granulocyte-macrophage colony-stimulating factor for murine ovarian cancer. Int. J. Cancer 2014, 134, 2865–2877. [Google Scholar] [CrossRef] [PubMed]
- Seth, P. Vector-mediated cancer gene therapy: An overview. Cancer Biol. Ther. 2005, 4, 512–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Rosa, C.; Longmate, J.; Martinez, J.; Zhou, Q.; Kaltcheva, T.I.; Tsai, W.; Drake, J.; Carroll, M.; Wussow, F.; Chiuppesi, F.; et al. MVA vaccine encoding CMV antigens safely induces durable expansion of CMV-specific T cells in healthy adults. Blood 2017, 129, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, D.G.; McNeil, B.; Visan, L.; Rodrigues, L.; Dunn, P.; Shewen, P.E.; Macallum, G.E.; Turner, P.V.; Vogel, T.U. Murine responses to recombinant MVA versus ALVAC vaccines against tumor-associated antigens, gp100 and 5T4. J. Immunother. 2013, 36, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Kwilas, A.R.; Ardiani, A.; Dirmeier, U.; Wottawah, C.; Schlom, J.; Hodge, J.W. A poxviral-based cancer vaccine the transcription factor twist inhibits primary tumor growth and metastases in a model of metastatic breast cancer and improves survival in a spontaneous prostate cancer model. Oncotarget 2015, 6, 28194–28210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acres, B.; Bonnefoy, J.Y. Clinical development of MVA-based therapeutic cancer vaccines. Expert Rev. Vaccine. 2008, 7, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Amato, R.J.; Shingler, W.; Goonewardena, M.; de Belin, J.; Naylor, S.; Jac, J.; Willis, J.; Saxena, S.; Hernandez-McClain, J.; Harrop, R. Vaccination of renal cell cancer patients with modified vaccinia Ankara delivering the tumor antigen 5T4 (TroVax) alone or administered in combination with interferon-alpha (IFN-alpha): A phase 2 trial. J. Immunother. 2009, 32, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Rochlitz, C.; Figlin, R.; Squiban, P.; Salzberg, M.; Pless, M.; Herrmann, R.; Tartour, E.; Zhao, Y.; Bizouarne, N.; Baudin, M.; et al. Phase I immunotherapy with a modified vaccinia virus (MVA) expressing human MUC1 as antigen-specific immunotherapy in patients with MUC1-positive advanced cancer. J. Gene Med. 2003, 5, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Madan, R.A.; Mohebtash, M.; Arlen, P.M.; Vergati, M.; Rauckhorst, M.; Steinberg, S.M.; Tsang, K.Y.; Poole, D.J.; Parnes, H.L.; Wright, J.J.; et al. Ipilimumab and a poxviral vaccine targeting prostate-specific antigen in metastatic castration-resistant prostate cancer: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 501–508. [Google Scholar] [CrossRef]
- Scurr, M.; Pembroke, T.; Bloom, A.; Roberts, D.; Thomson, A.; Smart, K.; Bridgeman, H.; Adams, R.; Brewster, A.; Jones, R.; et al. Effect of modified vaccinia Ankara-5T4 and low-dose cyclophosphamide on antitumor immunity in metastatic colorectal cancer: A randomized clinical trial. JAMA Oncol. 2017, 3, e172579. [Google Scholar] [CrossRef] [PubMed]
- Hui, E.P.; Taylor, G.S.; Jia, H.; Ma, B.B.; Chan, S.L.; Ho, R.; Wong, W.L.; Wilson, S.; Johnson, B.F.; Edwards, C.; et al. Phase I trial of recombinant modified vaccinia ankara encoding epstein-barr viral tumor antigens in nasopharyngeal carcinoma patients. Cancer Res. 2013, 73, 1676–1688. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Arriola, E.; Ottensmeier, C. TG4010: A vaccine with a therapeutic role in cancer. Immunotherapy 2016, 8, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Oudard, S.; Rixe, O.; Beuselinck, B.; Linassier, C.; Banu, E.; Machiels, J.P.; Baudard, M.; Ringeisen, F.; Velu, T.; Lefrere-Belda, M.A.; et al. A phase II study of the cancer vaccine TG4010 alone and in combination with cytokines in patients with metastatic renal clear-cell carcinoma: Clinical and immunological findings. Cancer Immunol. Immunother. 2011, 60, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Negrier, S.; Escudier, B.; Lasset, C.; Douillard, J.Y.; Savary, J.; Chevreau, C.; Ravaud, A.; Mercatello, A.; Peny, J.; Mousseau, M.; et al. Recombinant human interleukin-2, recombinant human interferon alfa-2a, or both in metastatic renal-cell carcinoma. N. Engl. J. Med. 1998, 338, 1272–1278. [Google Scholar] [CrossRef] [PubMed]
- Duggan, M.C.; Jochems, C.; Donahue, R.N.; Richards, J.; Karpa, V.; Foust, E.; Paul, B.; Brooks, T.; Tridandapani, S.; Olencki, T.; et al. A phase I study of recombinant (r) vaccinia-CEA(6D)-TRICOM and rFowlpox-CEA(6D)-TRICOM vaccines with GM-CSF and IFN-α-2b in patients with CEA-expressing carcinomas. Cancer Immunol. Immunother. 2016, 65, 1353–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remy-Ziller, C.; Thioudellet, C.; Hortelano, J.; Gantzer, M.; Nourtier, V.; Claudepierre, M.C.; Sansas, B.; Preville, X.; Bendjama, K.; Quemeneur, E.; et al. Sequential administration of MVA-based vaccines and PD-1/PD-L1-blocking antibodies confers measurable benefits on tumor growth and survival: Preclinical studies with MVA-βGal and MVA-MUC1 (TG4010) in a murine tumor model. Hum. Vaccines Immunother. 2018, 14, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Quoix, E.; Ramlau, R.; Westeel, V.; Papai, Z.; Madroszyk, A.; Riviere, A.; Koralewski, P.; Breton, J.L.; Stoelben, E.; Braun, D.; et al. Therapeutic vaccination with TG4010 and first-line chemotherapy in advanced non-small-cell lung cancer: A controlled phase 2B trial. Lancet Oncol. 2011, 12, 1125–1133. [Google Scholar] [CrossRef]
- Hillman, G.G.; Reich, L.A.; Rothstein, S.E.; Abernathy, L.M.; Fountain, M.D.; Hankerd, K.; Yunker, C.K.; Rakowski, J.T.; Quemeneur, E.; Slos, P. Radiotherapy and MVA-MUC1-IL-2 vaccine act synergistically for inducing specific immunity to MUC-1 tumor antigen. J. Immunother. Cancer 2017, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Hardwick, N.R.; Carroll, M.; Kaltcheva, T.; Qian, D.; Lim, D.; Leong, L.; Chu, P.; Kim, J.; Chao, J.; Fakih, M.; et al. p53MVA therapy in patients with refractory gastrointestinal malignancies elevates p53-specific CD8+ T-cell responses. Clin. Cancer Res. 2014, 20, 4459–4470. [Google Scholar] [CrossRef] [PubMed]
- Cawood, R.; Hills, T.; Wong, S.L.; Alamoudi, A.A.; Beadle, S.; Fisher, K.D.; Seymour, L.W. Recombinant viral vaccines for cancer. Trends Mol. Med. 2012, 18, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Cappuccini, F.; Stribbling, S.; Pollock, E.; Hill, A.V.; Redchenko, I. Immunogenicity and efficacy of the novel cancer vaccine based on simian adenovirus and MVA vectors alone and in combination with PD-1 mAB in a mouse model of prostate cancer. Cancer Immunol. Immunother. 2016, 65, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Schietinger, A.; Greenberg, P.D. Tolerance and exhaustion: Defining mechanisms of T cell dysfunction. Trends Immunol. 2014, 35, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Anassi, E.; Ndefo, U.A. Sipuleucel-T (provenge) injection: The first immunotherapy agent (vaccine) for hormone-refractory prostate cancer. Pharm. Ther. 2011, 36, 197–202. [Google Scholar]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.L.; Robinson, M.; Han, Z.Q.; Branston, R.H.; English, C.; Reay, P.; McGrath, Y.; Thomas, S.K.; Thornton, M.; Bullock, P.; et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003, 10, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Twumasi-Boateng, K.; Pettigrew, J.L.; Kwok, Y.Y.E.; Bell, J.C.; Nelson, B.H. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat. Rev. Cancer 2018, 18, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Mathis, J.M.; Stoff-Khalili, M.A.; Curiel, D.T. Oncolytic adenoviruses—Selective retargeting to tumor cells. Oncogene 2005, 24, 7775–7791. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Tao, L.; Wang, P.Y.; Cripe, T.P.; Zhang, X. Comparison of infectivity and spread between HSV-1 and HSV-2 based oncolytic viruses on tumor cells with different receptor expression profiles. Oncotarget 2018, 9, 21348–21358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waters, A.M.; Johnston, J.M.; Reddy, A.T.; Fiveash, J.; Madan-Swain, A.; Kachurak, K.; Bag, A.K.; Gillespie, G.Y.; Markert, J.M.; Friedman, G.K. Rationale and design of a phase 1 clinical trial to evaluate HSV G207 alone or with a single radiation dose in children with progressive or recurrent malignant supratentorial brain tumors. Hum. Gene Ther. Clin. Dev. 2017, 28, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Todo, T.; Martuza, R.L.; Rabkin, S.D.; Johnson, P.A. Oncolytic herpes simplex virus vector with enhanced mhc class i presentation and tumor cell killing. Proc. Natl. Acad. Sci. USA 2001, 98, 6396–6401. [Google Scholar] [CrossRef] [PubMed]
- Breitbach, C.J.; Moon, A.; Burke, J.; Hwang, T.H.; Kirn, D.H. A phase 2, open-label, randomized study of Pexa-Vec (JX-594) administered by intratumoral injection in patients with unresectable primary hepatocellular carcinoma. Gene Ther. Solid Cancers 2015, 1317, 343–357. [Google Scholar]
- Ranki, T.; Pesonen, S.; Hemminki, A.; Partanen, K.; Kairemo, K.; Alanko, T.; Lundin, J.; Linder, N.; Turkki, R.; Ristimaki, A.; et al. Phase I study with ONCOS-102 for the treatment of solid tumors—An evaluation of clinical response and exploratory analyses of immune markers. J. Immunother. Cancer 2016, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Nokisalmi, P.; Pesonen, S.; Escutenaire, S.; Sarkioja, M.; Raki, M.; Cerullo, V.; Laasonen, L.; Alemany, R.; Rojas, J.; Cascallo, M.; et al. Oncolytic adenovirus ICOVIR-7 in patients with advanced and refractory solid tumors. Clin. Cancer Res. 2010, 16, 3035–3043. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.N.; Hu, P.; Zeng, M.S.; Liu, R.B. Anti-tumor effect of oncolytic herpes simplex virus G47delta on human nasopharyngeal carcinoma. Chin. J. Cancer 2011, 30, 831–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Xu, L.; Zeng, W.; Hu, P.; Zeng, M.; Rabkin, S.D.; Liu, R. Treatment of human hepatocellular carcinoma by the oncolytic herpes simplex virus G47delta. Cancer Cell Int. 2014, 14, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatake, R.; Kaibori, M.; Nakamura, Y.; Tanaka, Y.; Matushima, H.; Okumura, T.; Murakami, T.; Ino, Y.; Todo, T.; Kon, M. Third-generation oncolytic herpes simplex virus inhibits the growth of liver tumors in mice. Cancer Sci. 2018, 109, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Hwang, T.H.; Moon, A.; Burke, J.; Ribas, A.; Stephenson, J.; Breitbach, C.J.; Daneshmand, M.; De Silva, N.; Parato, K.; Diallo, J.S.; et al. A mechanistic proof-of-concept clinical trial with JX-594, a targeted multi-mechanistic oncolytic poxvirus, in patients with metastatic melanoma. Mol. Ther. 2011, 19, 1913–1922. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, C.; Huebener, P.; Schwabe, R.F. Damage-associated molecular patterns in cancer: A double-edged sword. Oncogene 2016, 35, 5931–5941. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, R.J.; Errington, F.; Diaz, R.M.; Pandha, H.S.; Harrington, K.J.; Melcher, A.A.; Vile, R.G. The case of oncolytic viruses versus the immune system: Waiting on the judgment of solomon. Hum. Gene Ther. 2009, 20, 1119–1132. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Martin, S.; Golab, J.; Agostinis, P. Danger signalling during cancer cell death: Origins, plasticity and regulation. Cell Death Differ. 2014, 21, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Hobohm, U.; Stanford, J.L.; Grange, J.M. Pathogen-associated molecular pattern in cancer immunotherapy. Crit. Rev. Immunol. 2008, 28, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kim, D.W.; DeRaffele, G.; Mitcham, J.; Coffin, R.S.; Kim-Schulze, S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann. Surg. Oncol. 2010, 17, 718–730. [Google Scholar] [CrossRef] [PubMed]
- Breitbach, C.J.; Arulanandam, R.; De Silva, N.; Thorne, S.H.; Patt, R.; Daneshmand, M.; Moon, A.; Ilkow, C.; Burke, J.; Hwang, T.H.; et al. Oncolytic vaccinia virus disrupts tumor-associated vasculature in humans. Cancer Res. 2013, 73, 1265–1275. [Google Scholar] [CrossRef] [PubMed]
- Dispenzieri, A.; Tong, C.; LaPlant, B.; Lacy, M.Q.; Laumann, K.; Dingli, D.; Zhou, Y.; Federspiel, M.J.; Gertz, M.A.; Hayman, S.; et al. Phase I trial of systemic administration of edmonston strain of measles virus genetically engineered to express the sodium iodide symporter in patients with recurrent or refractory multiple myeloma. Leukemia 2017, 31, 2791–2798. [Google Scholar] [CrossRef] [PubMed]
- Ong, H.T.; Federspiel, M.J.; Guo, C.M.; Ooi, L.L.; Russell, S.J.; Peng, K.W.; Hui, K.M. Systemically delivered measles virus-infected mesenchymal stem cells can evade host immunity to inhibit liver cancer growth. J. Hepatol. 2013, 59, 999–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, J.; Reid, T.; Ruo, L.; Breitbach, C.J.; Rose, S.; Bloomston, M.; Cho, M.; Lim, H.Y.; Chung, H.C.; Kim, C.W.; et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med. 2013, 19, 329–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chesney, J.; Puzanov, I.; Collichio, F.; Singh, P.; Milhem, M.M.; Glaspy, J.; Hamid, O.; Ross, M.; Friedlander, P.; Garbe, C.; et al. Randomized, open-label phase II study evaluating the efficacy and safety of talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone in patients with advanced, unresectable melanoma. J. Clin. Oncol. 2018, 36, 1658–1667. [Google Scholar] [CrossRef] [PubMed]
- Carew, J.S.; Espitia, C.M.; Zhao, W.; Kelly, K.R.; Coffey, M.; Freeman, J.W.; Nawrocki, S.T. Reolysin is a novel reovirus-based agent that induces endoplasmic reticular stress-mediated apoptosis in pancreatic cancer. Cell Death Dis. 2013, 4, e728. [Google Scholar] [CrossRef] [PubMed]
- Cohn, D.E.; Sill, M.W.; Walker, J.L.; O’Malley, D.; Nagel, C.I.; Rutledge, T.L.; Bradley, W.; Richardson, D.L.; Moxley, K.M.; Aghajanian, C. Randomized phase IIB evaluation of weekly paclitaxel versus weekly paclitaxel with oncolytic reovirus (Reolysin®) in recurrent ovarian, tubal, or peritoneal cancer: An nrg oncology/gynecologic oncology group study. Gynecol. Oncol. 2017, 146, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Noonan, A.M.; Farren, M.R.; Geyer, S.M.; Huang, Y.; Tahiri, S.; Ahn, D.; Mikhail, S.; Ciombor, K.K.; Pant, S.; Aparo, S.; et al. Randomized phase 2 trial of the oncolytic virus pelareorep (reolysin) in upfront treatment of metastatic pancreatic adenocarcinoma. Mol. Ther. 2016, 24, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Villalona-Calero, M.A.; Lam, E.; Otterson, G.A.; Zhao, W.; Timmons, M.; Subramaniam, D.; Hade, E.M.; Gill, G.M.; Coffey, M.; Selvaggi, G.; et al. Oncolytic reovirus in combination with chemotherapy in metastatic or recurrent non-small cell lung cancer patients with kras-activated tumors. Cancer 2016, 122, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Rossowska, J.; Anger, N.; Szczygiel, A.; Mierzejewska, J.; Pajtasz-Piasecka, E. Intratumoral lentivector-mediated TGF-β1 gene downregulation as a potent strategy for enhancing the antitumor effect of therapy composed of cyclophosphamide and dendritic cells. Front. Immunol. 2017, 8, 713. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Zhang, Y.; Peng, K.; Wang, Q.; Hong, X.; Li, H.; Fan, G.; Zhang, Z.; Gong, T.; Sun, X. Combined delivery of a TGF-β inhibitor and an adenoviral vector expressing interleukin-12 potentiates cancer immunotherapy. Acta Biomater. 2017, 61, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ji, X.; Liu, X.; Yao, R.; Chi, J.; Liu, S.; Wang, Y.; Cao, W.; Zhou, Q. Lentivirus-mediated inhibition of USP39 suppresses the growth of breast cancer cells in vitro. Oncol. Rep. 2013, 30, 2871–2877. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Cao, J.; Liu, L.; Zhu, Z.A.; Wu, K.J.; Fei, S.J. Lentivirus-mediated gene transfer of human sTRAIL induced apoptosis in SGC-7901 cells. Sichuan Da Xue Xue Bao Yi Xue Ban 2013, 44, 348–351. (In Chinese) [Google Scholar] [PubMed]
- Ru, Q.; Li, W.; Wang, X.; Zhang, S.; Chen, L.; Zhang, Y.; Ge, Y.; Zu, Y.; Liu, Y.; Zheng, D. Preclinical study of rAAV2-sTRAIL: Pharmaceutical efficacy, biodistribution and safety in animals. Cancer Gene Ther. 2017, 24, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Liu, Y.; Liu, S.; Xu, R.; Zheng, D. Oral adeno-associated virus-sTRAIL gene therapy suppresses human hepatocellular carcinoma growth in mice. Hepatology 2005, 42, 1355–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nan, Y.; Guo, L.; Song, Y.; Wang, L.; Yu, K.; Huang, Q.; Zhong, Y. Combinatorial therapy with adenoviral-mediated PTEN and a PI3K inhibitor suppresses malignant glioma cell growth in vitro and in vivo by regulating the PI3K/AKT signaling pathway. J. Cancer Res. Clin. Oncol. 2017, 143, 1477–1487. [Google Scholar] [CrossRef] [PubMed]
- Mori, A.; Watanabe, M.; Sadahira, T.; Kobayashi, Y.; Ariyoshi, Y.; Ueki, H.; Wada, K.; Ochiai, K.; Li, S.A.; Nasu, Y. The downregulation of the expression of CD147 by tumor suppressor REIC/Dkk-3, and its implication in human prostate cancer cell growth inhibition. Acta Med. Okayama 2017, 71, 135–142. [Google Scholar] [PubMed]
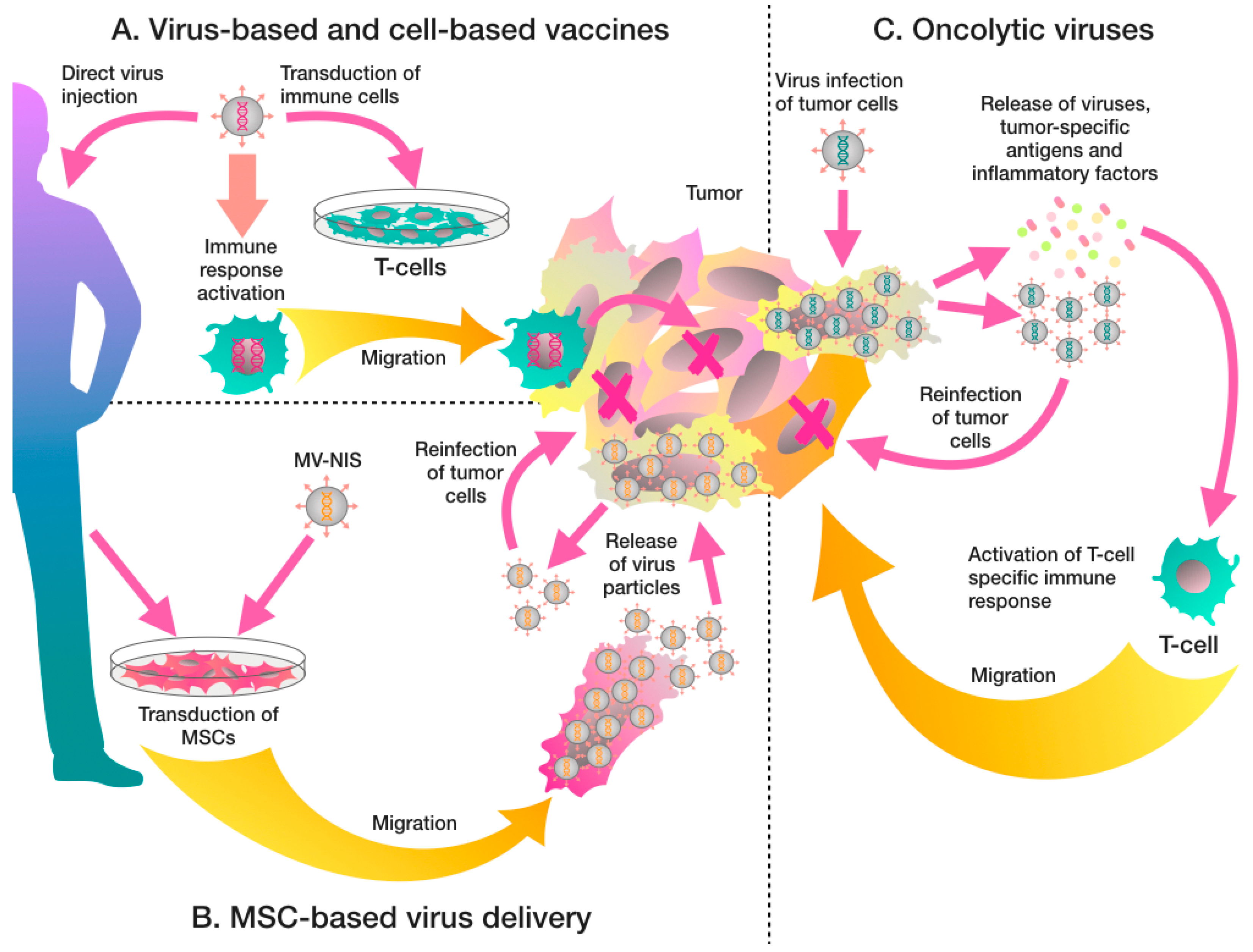

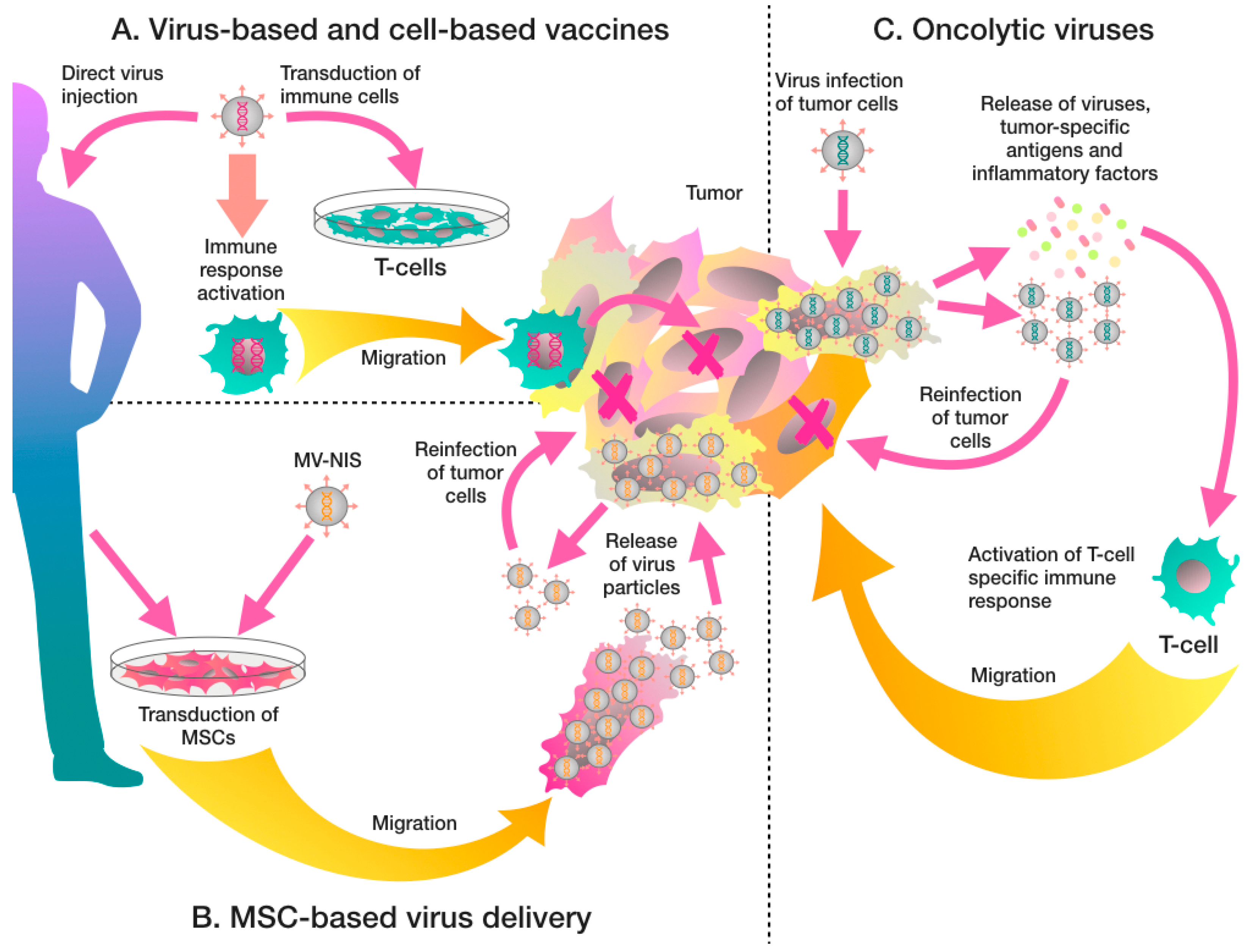
{kind=link}
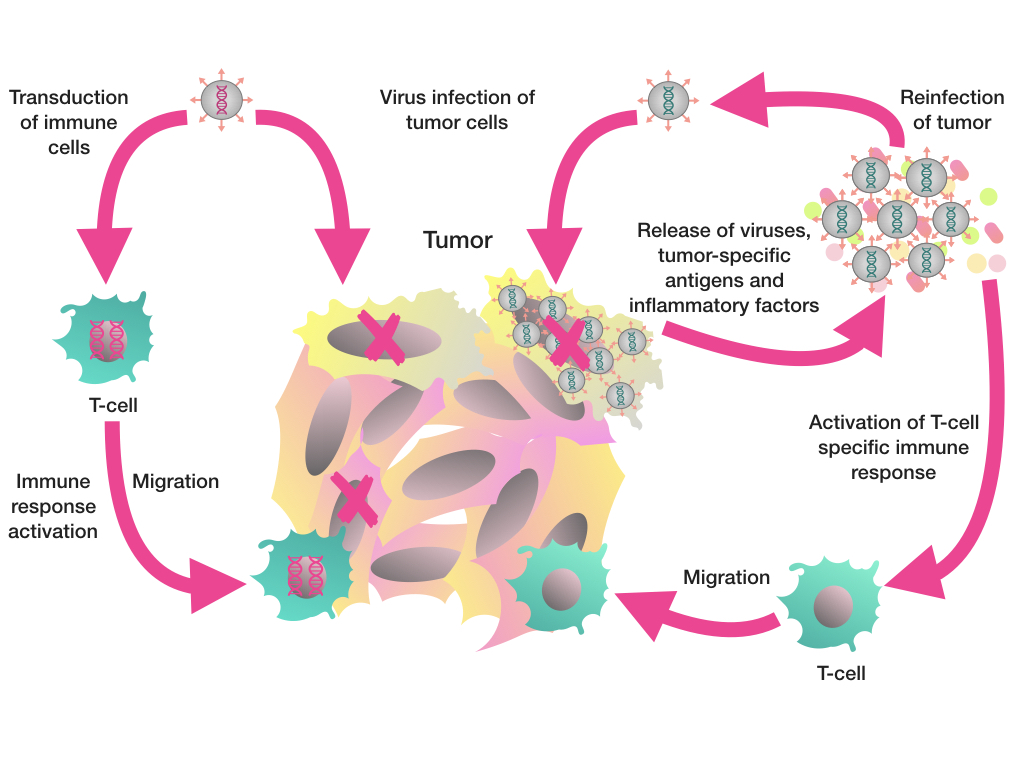
{kind=link}
| Clinical Trial Phase | ||||||
|---|---|---|---|---|---|---|
| Therapeutic agent | I, I/II | II, II/III | III | IV | N/A * | Total |
| Oncolytic virus | 73 | 31 | 7 | 0 | 4 | 115 |
| Virus-based vaccine | 39 | 38 | 0 | 0 | 0 | 77 |
| Virus-engineered CAR T-cell | 311 | 22 | 3 | 1 | 23 | 360 |
| Other virus-engineered cell-based vaccines (DCs, MSCs) | 11 | 4 | 0 | 0 | 0 | 15 |
| Drug | IMLYGIC (Talimogene Laherparepvec, T-Vec), Oncolytic Virus | YESCARTA (Axicabtagene Ciloleucel), Genetically Modified Autologous T-cell | KYMRIAH (Tisagenlecleucel), Genetically Modified Autologous T-cell |
|---|---|---|---|
| Approval date | 2015 | 2017 | 2018 |
| Viral vector | HSV-1 | Retrovirus | Lentivirus |
| Genetic modification | Deletions in γ34.5 and α47 genes and insertion of GM-CSF gene | Insertion of anti-CD19 CAR | Insertion of anti-CD19 CAR |
| Application | In patients with melanoma recurrent after initial surgery | Diffuse large B-cell lymphoma (DLBCL), TFL and high-grade B-cell lymphoma | B-cell precursor acute lymphoblastic leukemia (ALL), DLBCL, high grade B-cell lymphoma |
| Mechanism of action | Causes lysis of tumor, followed by release of tumor-derived antigens, which together with virally derived GM-CSF may promote an antitumor immune response | T-cell activation, proliferation, acquisition of effector functions and secretion of inflammatory cytokines and chemokines. This sequence of events leads to killing of CD19-expressing cells | Identify and eliminate CD19-expressing malignant and normal cells |
| Adverse reactions | Fatigue, chills, pyrexia, nausea, influenza-like illness, and injection site pain | Cytokine release syndrome, neurological toxicities, infections and febrile neutropenia, prolonged cytopenia, hypogammaglobulinemia | Cytokine release syndrome, neurological toxicities, infections and febrile neutropenia, prolonged cytopenia, hypogammaglobulinemia |
| Clinical studies | Randomized phase III trial (NCT00769704). Patients with stage IIIB–IV melanoma were injected with T-Vec or GM-CSF. OS in GM-CSF arm was 18.9 months, and T-Vec arm was 23.3 months; objective response in both arms was 5.7% and 26.4% of patients | In Phase II clinical trial (NCT02445248) efficacy was established based on complete remission (CR). Half of the patients achieved CR, while 21% achieved a partial response | ALL: In Phase II clinical trial (NCT02228096), efficacy of KYMRIAH was established based on complete remission (CR) within 3 months after infusion. Overall, 83% of patients achieved CR. DLBCL: In Phase II clinical trial (NCT02445248), efficacy was established based on complete response (CR) and partial response (PR). Overall 50% of patients achieved CR or PR |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chulpanova, D.S.; Solovyeva, V.V.; Kitaeva, K.V.; Dunham, S.P.; Khaiboullina, S.F.; Rizvanov, A.A. Recombinant Viruses for Cancer Therapy. Biomedicines 2018, 6, 94. https://doi.org/10.3390/biomedicines6040094
Chulpanova DS, Solovyeva VV, Kitaeva KV, Dunham SP, Khaiboullina SF, Rizvanov AA. Recombinant Viruses for Cancer Therapy. Biomedicines. 2018; 6(4):94. https://doi.org/10.3390/biomedicines6040094
Chicago/Turabian StyleChulpanova, Daria S., Valeriya V. Solovyeva, Kristina V. Kitaeva, Stephen P. Dunham, Svetlana F. Khaiboullina, and Albert A. Rizvanov. 2018. "Recombinant Viruses for Cancer Therapy" Biomedicines 6, no. 4: 94. https://doi.org/10.3390/biomedicines6040094
APA StyleChulpanova, D. S., Solovyeva, V. V., Kitaeva, K. V., Dunham, S. P., Khaiboullina, S. F., & Rizvanov, A. A. (2018). Recombinant Viruses for Cancer Therapy. Biomedicines, 6(4), 94. https://doi.org/10.3390/biomedicines6040094