Neuromyelitis Optica Spectrum Disorder and Anti-MOG Syndromes

Abstract
1. Introduction
2. Neuromyelitis Optica Spectrum Disorders
2.1. Pathophysiology
2.2. Epidemiology
2.3. Clinical Manifestations
2.4. Laboratorial Characteristics
2.5. Diagnostic Criteria
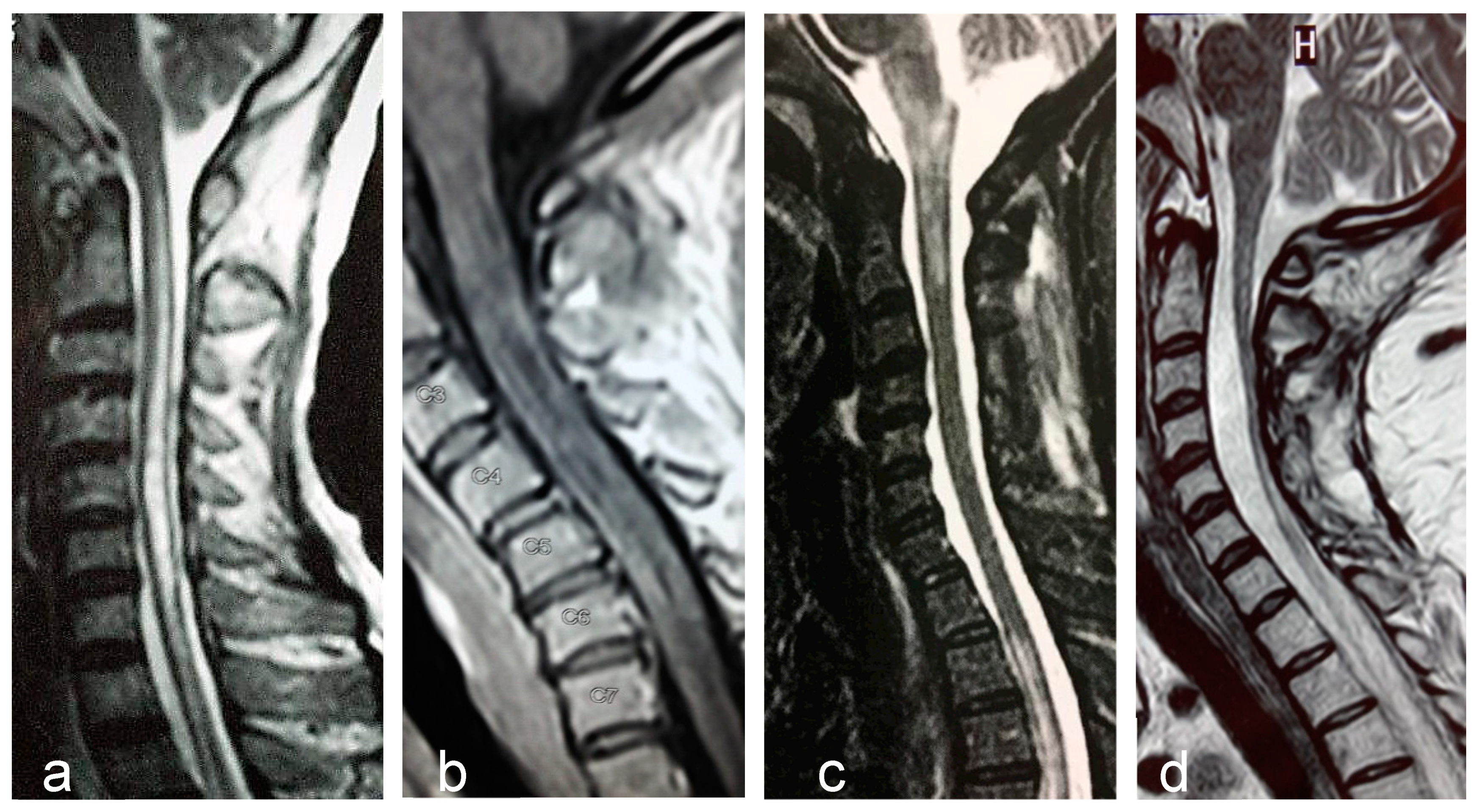
2.6. Magnetic Resonance Imaging
2.7. Treatment
2.7.1. Therapy of Acute Relapses
2.7.2. Therapy for Relapses Prevention
3. Anti-Myelin Oligodendrocyte Glycoprotein Syndromes
3.1. Pathophysiology
3.2. Epidemiology
3.3. Clinical Manifestations
3.4. Anti-MOG Testing
3.5. Cerebrospinal Fluid Analysis
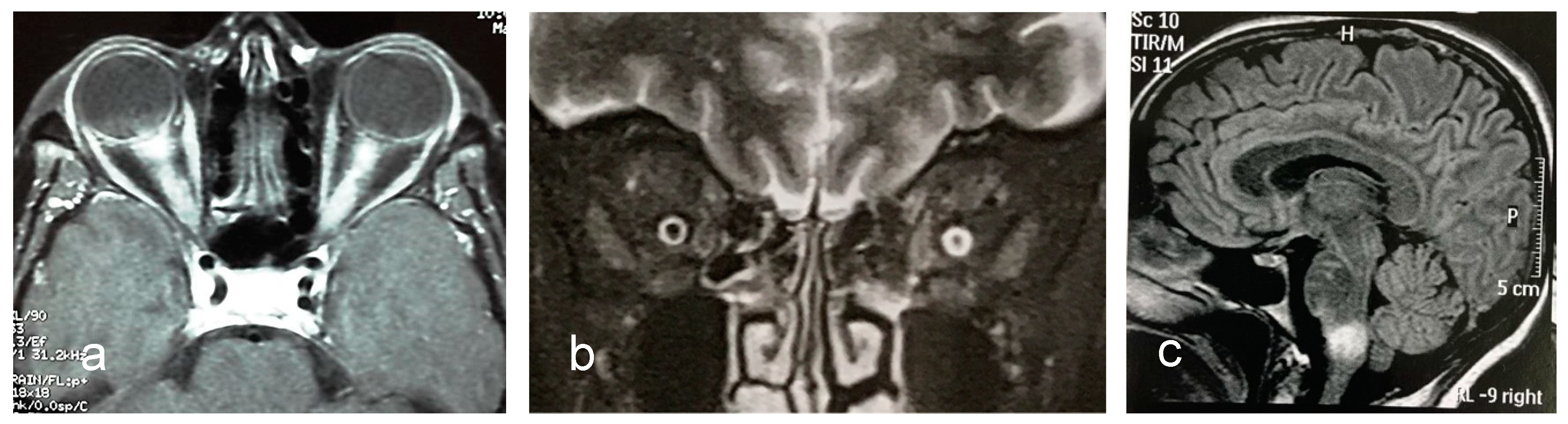
3.6. MRI Features
3.7. Diagnosis
- Monophasic or relapsing acute ON, myelitis, brainstem encephalitis, or any combination of these symptoms
- MRI or electrophysiological (visual evoked potentials in patients with isolated ON) findings compatible with CNS demyelination
- Seropositivity for MOG-IgG as detected by means of a cell-based assay employing full length human MOG as target antigen.
- Chronic progressive course (progressive MS, sarcoidosis and tumors) or acute onset (ischemia);
- Clinical and paraclinical findings suggesting other conditions such as:
- Tuberculosis, borreliosis, syphilis, Behçet’s disease, subacute combined degeneration of the spinal cord, Leber’s hereditary optic neuropathy, lymphoma, and paraneoplastic disorders;
- Peripheral demyelination
- Brain MRI abnormalities such as:
- Lesion adjacent to lateral ventricle associated with inferior temporal lobe lesion, or Dawson’s finger-type lesion;
- Increasing number of lesions between relapses.
- Serum MOG-IgG at low titers.
3.8. Treatment
3.9. Conclusions
Funding
Conflicts of Interest
References
- Kleiter, I.; Hellwig, K.; Berthele, A.; Kumpfel, T.; Linker, R.A.; Hartung, H.P.; Paul, F.; Aktas, O. Failure of natalizumab to prevent relapses in neuromyelitis optica. Arch. Neurol. 2012, 69, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Min, J.H.; Kim, B.J.; Lee, K.H. Development of extensive brain lesions following fingolimod (fty720) treatment in a patient with neuromyelitis optica spectrum disorder. Mult. Scler. 2012, 18, 113–115. [Google Scholar] [CrossRef] [PubMed]
- Trebst, C.; Jarius, S.; Berthele, A.; Paul, F.; Schippling, S.; Wildemann, B.; Borisow, N.; Kleiter, I.; Aktas, O.; Kumpfel, T. Update on the diagnosis and treatment of neuromyelitis optica: Recommendations of the neuromyelitis optica study group (nemos). J. Neurol. 2014, 261, 1–16. [Google Scholar] [CrossRef] [PubMed]
- van Pelt, E.D.; Wong, Y.Y.; Ketelslegers, I.A.; Hamann, D.; Hintzen, R.Q. Neuromyelitis optica spectrum disorders: Comparison of clinical and magnetic resonance imaging characteristics of aqp4-igg versus mog-igg seropositive cases in the netherlands. Eur. J. Neurol. 2016, 23, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Narayan, R.; Simpson, A.; Fritsche, K.; Salama, S.; Pardo, S.; Mealy, M.; Paul, F.; Levy, M. Mog antibody disease: A review of mog antibody seropositive neuromyelitis optica spectrum disorder. Mult. Scler. Relat. Disord. 2018, 25, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Metz, I.; Konig, F.B.; Ruprecht, K.; Reindl, M.; Paul, F.; Bruck, W.; Wildemann, B. Screening for mog-igg and 27 other anti-glial and anti-neuronal autoantibodies in ‘pattern ii multiple sclerosis’ and brain biopsy findings in a mog-igg-positive case. Mult. Scler. 2016, 22, 1541–1549. [Google Scholar] [CrossRef] [PubMed]
- Spadaro, M.; Gerdes, L.A.; Mayer, M.C.; Ertl-Wagner, B.; Laurent, S.; Krumbholz, M.; Breithaupt, C.; Hogen, T.; Straube, A.; Giese, A.; et al. Histopathology and clinical course of mog-antibody-associated encephalomyelitis. Ann. Clin. Transl. Neurol. 2015, 2, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Misu, T.; Hoftberger, R.; Fujihara, K.; Wimmer, I.; Takai, Y.; Nishiyama, S.; Nakashima, I.; Konno, H.; Bradl, M.; Garzuly, F.; et al. Presence of six different lesion types suggests diverse mechanisms of tissue injury in neuromyelitis optica. Acta Neuropathol. 2013, 125, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Reindl, M.; Rostasy, K. Mog antibody-associated diseases. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e60. [Google Scholar] [CrossRef]
- Di Pauli, F.; Hoftberger, R.; Reindl, M.; Beer, R.; Rhomberg, P.; Schanda, K.; Sato, D.; Fujihara, K.; Lassmann, H.; Schmutzhard, E.; et al. Fulminant demyelinating encephalomyelitis: Insights from antibody studies and neuropathology. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e175. [Google Scholar] [CrossRef]
- Ramanathan, S.; Dale, R.C.; Brilot, F. Anti-mog antibody: The history, clinical phenotype, and pathogenicity of a serum biomarker for demyelination. Autoimmun. Rev. 2016, 15, 307–324. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Probst, C.; Borowski, K.; Franciotta, D.; Wildemann, B.; Stoecker, W.; Wandinger, K.P. Standardized method for the detection of antibodies to aquaporin-4 based on a highly sensitive immunofluorescence assay employing recombinant target antigen. J. Neurol. Sci. 2010, 291, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Waters, P.J.; McKeon, A.; Leite, M.I.; Rajasekharan, S.; Lennon, V.A.; Villalobos, A.; Palace, J.; Mandrekar, J.N.; Vincent, A.; Bar-Or, A.; et al. Serologic diagnosis of nmo: A multicenter comparison of aquaporin-4-igg assays. Neurology 2012, 78, 665–671; discussion 669. [Google Scholar] [CrossRef] [PubMed]
- Waters, P.J.; Pittock, S.J.; Bennett, J.L.; Jarius, S.; Weinshenker, B.G.; Wingerchuk, D.M. Evaluation of aquaporin-4 antibody assays. Clin. Exp. Neuroimmunol. 2014, 5, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Ruprecht, K.; Kleiter, I.; Borisow, N.; Asgari, N.; Pitarokoili, K.; Pache, F.; Stich, O.; Beume, L.A.; Hummert, M.W.; et al. Mog-igg in nmo and related disorders: A multicenter study of 50 patients. Part 1: Frequency, syndrome specificity, influence of disease activity, long-term course, association with aqp4-igg, and origin. J. Neuroinflamm. 2016, 13, 279. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, N.; Tajima, G.; Hyodo, S.; Takahashi, Y.; Kobayashi, M. Detection of autoantibodies against nmda-type glutamate receptor in a patient with recurrent optic neuritis and transient cerebral lesions. Neuropediatrics 2007, 38, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Kruer, M.C.; Koch, T.K.; Bourdette, D.N.; Chabas, D.; Waubant, E.; Mueller, S.; Moscarello, M.A.; Dalmau, J.; Woltjer, R.L.; Adamus, G. Nmda receptor encephalitis mimicking seronegative neuromyelitis optica. Neurology 2010, 74, 1473–1475. [Google Scholar] [CrossRef] [PubMed]
- Mader, S.; Gredler, V.; Schanda, K.; Rostasy, K.; Dujmovic, I.; Pfaller, K.; Lutterotti, A.; Jarius, S.; Di Pauli, F.; Kuenz, B.; et al. Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J. Neuroinflamm. 2011, 8, 184. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Wandinger, K.P.; Borowski, K.; Stoecker, W.; Wildemann, B. Antibodies to cv2/crmp5 in neuromyelitis optica-like disease: Case report and review of the literature. Clin. Neurol. Neurosurg. 2012, 114, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Devic, E. Myelite subaigue compliquee de nevrite optique. Bull. Med. 1894, 8, 1033. [Google Scholar]
- Gault, F. De la neuromyélite optique aiguë. Ph.D. Thesis, Alexandre Rey, imprimeur de la faculté de médecine, Faculté de Medicine et de Pharmacie de Lyon, Lyon, France, 1894. [Google Scholar]
- Acchiote, P. Sur un cas de neuromyélite subaiguë ou maladie de devic. Rev. Neurol. 1907, 20, 775–777. [Google Scholar]
- Jarius, S.; Wildemann, B. The case of the marquis de causan (1804): An early account of visual loss associated with spinal cord inflammation. J. Neurol. 2012, 259, 1354–1357. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Wildemann, B. ‘Noteomielite’ accompanied by acute amaurosis (1844). An early case of neuromyelitis optica. J. Neurol. Sci. 2012, 313, 182–184. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Wildemann, B. An early british case of neuromyelitis optica (1850). BMJ Clin. Res. Ed. 2012, 345, e6430. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Wildemann, B. An early case of neuromyelitis optica: On a forgotten report by jacob lockhart clarke, frs. Mult. Scler. 2011, 17, 1384–1386. [Google Scholar] [CrossRef] [PubMed]
- Allbutt, T.C. On the ophthalmoscopic signs of spinal disease. Lancet 1870, 95, 76–78. [Google Scholar] [CrossRef]
- Erb, W. Ueber das zusammenvorkommen von neuritis optica und myelitis subacuta. Eur. Arch. Psychiatry Clin. Neurosci. 1880, 10, 146–157. [Google Scholar] [CrossRef]
- Seguin, E.C. Art. I.—On the coincidence of optic neuritis and subacute transverse myelitis. J. Nerv. Ment. Dis. 1880, 7, 177–188. [Google Scholar] [CrossRef][Green Version]
- Marques, A. Da neuromielite ótica: Contribuição clínica e etiológica. Hospital 1943, 24, 49–63. [Google Scholar]
- Lennon, V.A.; Wingerchuk, D.M.; Kryzer, T.J.; Pittock, S.J.; Lucchinetti, C.F.; Fujihara, K.; Nakashima, I.; Weinshenker, B.G. A serum autoantibody marker of neuromyelitis optica: Distinction from multiple sclerosis. Lancet 2004, 364, 2106–2112. [Google Scholar] [CrossRef]
- Lennon, V.A.; Kryzer, T.J.; Pittock, S.J.; Verkman, A.S.; Hinson, S.R. Igg marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J. Exp. Med. 2005, 202, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Wingerchuk, D.M.; Lennon, V.A.; Lucchinetti, C.F.; Pittock, S.J.; Weinshenker, B.G. The spectrum of neuromyelitis optica. Lancet Neurol. 2007, 6, 805–815. [Google Scholar] [CrossRef]
- Lana-Peixoto, M.A.; Callegaro, D. The expanded spectrum of neuromyelitis optica: Evidences for a new definition. Arq. Neuro-Psiquiatr. 2012, 70, 807–813. [Google Scholar] [CrossRef]
- Wingerchuk, D.M.; Banwell, B.; Bennett, J.L.; Cabre, P.; Carroll, W.; Chitnis, T.; de Seze, J.; Fujihara, K.; Greenberg, B.; Jacob, A.; et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015, 85, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.J.; Rossi, A.; Verkman, A.S. Model of aquaporin-4 supramolecular assembly in orthogonal arrays based on heterotetrameric association of m1-m23 isoforms. Biophys. J. 2011, 100, 2936–2945. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin 4 and neuromyelitis optica. Lancet Neurol. 2012, 11, 535–544. [Google Scholar] [CrossRef]
- Verkman, A.S.; Anderson, M.O.; Papadopoulos, M.C. Aquaporins: Important but elusive drug targets. Nat. Rev. Drug Discov. 2014, 13, 259–277. [Google Scholar] [CrossRef]
- Saadoun, S.; Waters, P.; Bell, B.A.; Vincent, A.; Verkman, A.S.; Papadopoulos, M.C. Intra-cerebral injection of neuromyelitis optica immunoglobulin g and human complement produces neuromyelitis optica lesions in mice. Brain J. Neurol. 2010, 133, 349–361. [Google Scholar] [CrossRef]
- Lucchinetti, C.F.; Mandler, R.N.; McGavern, D.; Bruck, W.; Gleich, G.; Ransohoff, R.M.; Trebst, C.; Weinshenker, B.; Wingerchuk, D.; Parisi, J.E.; et al. A role for humoral mechanisms in the pathogenesis of devic’s neuromyelitis optica. Brain J. Neurol. 2002, 125, 1450–1461. [Google Scholar] [CrossRef]
- Jarius, S.; Franciotta, D.; Paul, F.; Ruprecht, K.; Bergamaschi, R.; Rommer, P.S.; Reuss, R.; Probst, C.; Kristoferitsch, W.; Wandinger, K.P.; et al. Cerebrospinal fluid antibodies to aquaporin-4 in neuromyelitis optica and related disorders: Frequency, origin, and diagnostic relevance. J. Neuroinflamm. 2010, 7, 52. [Google Scholar] [CrossRef]
- Chihara, N.; Aranami, T.; Sato, W.; Miyazaki, Y.; Miyake, S.; Okamoto, T.; Ogawa, M.; Toda, T.; Yamamura, T. Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc. Natl. Acad. Sci. USA 2011, 108, 3701–3706. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, F.; Schaller, K.L.; Owens, G.P.; Cotleur, A.C.; Kellner, D.; Takeshita, Y.; Obermeier, B.; Kryzer, T.J.; Sano, Y.; Kanda, T.; et al. Glucose-regulated protein 78 autoantibody associates with blood-brain barrier disruption in neuromyelitis optica. Sci. Transl. Med. 2017, 9, eaai9111. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, F.; Nishihara, H.; Kanda, T. Blood-brain barrier dysfunction in immuno-mediated neurological diseases. Immunol. Med. 2018, 41, 120–128. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jarius, S.; Wildemann, B.; Paul, F. Neuromyelitis optica: Clinical features, immunopathogenesis and treatment. Clin. Exp. Immunol. 2014, 176, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Weinshenker, B.G.; Wingerchuk, D.M. Neuromyelitis spectrum disorders. Mayo Clin. Proc. 2017, 92, 663–679. [Google Scholar] [CrossRef] [PubMed]
- Hinson, S.R.; Pittock, S.J.; Lucchinetti, C.F.; Roemer, S.F.; Fryer, J.P.; Kryzer, T.J.; Lennon, V.A. Pathogenic potential of igg binding to water channel extracellular domain in neuromyelitis optica. Neurology 2007, 69, 2221–2231. [Google Scholar] [CrossRef]
- Bennett, J.L.; Lam, C.; Kalluri, S.R.; Saikali, P.; Bautista, K.; Dupree, C.; Glogowska, M.; Case, D.; Antel, J.P.; Owens, G.P.; et al. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann. Neurol. 2009, 66, 617–629. [Google Scholar] [CrossRef]
- Bradl, M.; Misu, T.; Takahashi, T.; Watanabe, M.; Mader, S.; Reindl, M.; Adzemovic, M.; Bauer, J.; Berger, T.; Fujihara, K.; et al. Neuromyelitis optica: Pathogenicity of patient immunoglobulin in vivo. Ann. Neurol. 2009, 66, 630–643. [Google Scholar] [CrossRef]
- Kinoshita, M.; Nakatsuji, Y.; Kimura, T.; Moriya, M.; Takata, K.; Okuno, T.; Kumanogoh, A.; Kajiyama, K.; Yoshikawa, H.; Sakoda, S. Neuromyelitis optica: Passive transfer to rats by human immunoglobulin. Biochem. Biophys. Res. Commun. 2009, 386, 623–627. [Google Scholar] [CrossRef]
- Kinoshita, M.; Nakatsuji, Y.; Kimura, T.; Moriya, M.; Takata, K.; Okuno, T.; Kumanogoh, A.; Kajiyama, K.; Yoshikawa, H.; Sakoda, S. Anti-aquaporin-4 antibody induces astrocytic cytotoxicity in the absence of cns antigen-specific t cells. Biochem. Biophys. Res. Commun. 2010, 394, 205–210. [Google Scholar] [CrossRef]
- Etemadifar, M.; Nasr, Z.; Khalili, B.; Taherioun, M.; Vosoughi, R. Epidemiology of neuromyelitis optica in the world: A systematic review and meta-analysis. Mult. Scler. Int. 2015, 2015, 174720. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, E.P.; Cabre, P.; Weinshenker, B.G.; Sauver, J.S.; Jacobson, D.J.; Majed, M.; Lennon, V.A.; Lucchinetti, C.F.; McKeon, A.; Matiello, M.; et al. Epidemiology of aquaporin-4 autoimmunity and neuromyelitis optica spectrum. Ann. Neurol. 2016, 79, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Rivera, J.F.; Kurtzke, J.F.; Booth, V.A.; Corona, T. Characteristics of devic’s disease (neuromyelitis optica) in mexico. J. Neurol. 2008, 255, 710–715. [Google Scholar] [CrossRef] [PubMed]
- Asgari, N.; Lillevang, S.T.; Skejoe, H.P.; Falah, M.; Stenager, E.; Kyvik, K.O. A population-based study of neuromyelitis optica in caucasians. Neurology 2011, 76, 1589–1595. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Gomez, J.A.; Kurtzke, J.F.; Gonzalez-Quevedo, A.; Lara-Rodriguez, R. An epidemiological study of neuromyelitis optica in cuba. J. Neurol. 2009, 256, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Aboul-Enein, F.; Seifert-Held, T.; Mader, S.; Kuenz, B.; Lutterotti, A.; Rauschka, H.; Rommer, P.; Leutmezer, F.; Vass, K.; Flamm-Horak, A.; et al. Neuromyelitis optica in austria in 2011: To bridge the gap between neuroepidemiological research and practice in a study population of 8.4 million people. PLoS ONE 2013, 8, e79649. [Google Scholar] [CrossRef] [PubMed]
- Cossburn, M.; Tackley, G.; Baker, K.; Ingram, G.; Burtonwood, M.; Malik, G.; Pickersgill, T.; te Water Naude, J.; Robertson, N. The prevalence of neuromyelitis optica in south east wales. Eur. J. Neurol. 2012, 19, 655–659. [Google Scholar] [CrossRef] [PubMed]
- Houzen, H.; Niino, M.; Hirotani, M.; Fukazawa, T.; Kikuchi, S.; Tanaka, K.; Sasaki, H. Increased prevalence, incidence, and female predominance of multiple sclerosis in northern japan. J. Neurol. Sci. 2012, 323, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.; Panicker, J.; Lythgoe, D.; Elsone, L.; Mutch, K.; Wilson, M.; Das, K.; Boggild, M. The epidemiology of neuromyelitis optica amongst adults in the merseyside county of united kingdom. J. Neurol. 2013, 260, 2134–2137. [Google Scholar] [CrossRef] [PubMed]
- Etemadifar, M.; Dashti, M.; Vosoughi, R.; Abtahi, S.H.; Ramagopalan, S.V.; Nasr, Z. An epidemiological study of neuromyelitis optica in isfahan. Mult. Scler. 2014, 20, 1920–1922. [Google Scholar] [CrossRef] [PubMed]
- Pandit, L.; Kundapur, R. Prevalence and patterns of demyelinating central nervous system disorders in urban mangalore, south india. Mult. Scler. 2014, 20, 1651–1653. [Google Scholar] [CrossRef] [PubMed]
- Kashipazha, D.; Mohammadianinejad, S.E.; Majdinasab, N.; Azizi, M.; Jafari, M. A descriptive study of prevalence, clinical features and other findings of neuromyelitis optica and neuromyelitis optica spectrum disorder in khuzestan province, iran. Iran. J. Neurol. 2015, 14, 204–210. [Google Scholar] [PubMed]
- Danielle van Pelt, E.; Wong, Y.Y.M.; Ketelslegers, I.A.; Siepman, D.A.; Hamann, D.; Hintzen, R.Q. Incidence of aqp4-igg seropositive neuromyelitis optica spectrum disorders in the netherlands: About one in a million. Mult. Scler. J. Exp. Transl. Clin. 2016, 2, 2055217315625652. [Google Scholar] [CrossRef] [PubMed]
- Houzen, H.; Kondo, K.; Niino, M.; Horiuchi, K.; Takahashi, T.; Nakashima, I.; Tanaka, K. Prevalence and clinical features of neuromyelitis optica spectrum disorders in northern japan. Neurology 2017, 89, 1995–2001. [Google Scholar] [CrossRef] [PubMed]
- Hor, J.Y.; Lim, T.T.; Chia, Y.K.; Ching, Y.M.; Cheah, C.F.; Tan, K.; Chow, H.B.; Arip, M.; Eow, G.B.; Easaw, P.E.S.; et al. Prevalence of neuromyelitis optica spectrum disorder in the multi-ethnic penang island, malaysia, and a review of worldwide prevalence. Mult. Scler. Relat. Disord. 2018, 19, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, W.; Prain, K.M.; Waters, P.; Woodhall, M.; O’Gorman, C.M.; Clarke, L.; Silvestrini, R.A.; Bundell, C.S.; Abernethy, D.; Bhuta, S.; et al. Incidence and prevalence of nmosd in australia and new zealand. J. Neurol. Neurosurg. Psychiatry 2017, 88, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, M.; Aldea, M.; Escudero, D.; Llufriu, S.; Arrambide, G.; Otero-Romero, S.; Sastre-Garriga, J.; Romero-Pinel, L.; Martinez-Yelamos, S.; Sola-Valls, N.; et al. Epidemiology of nmosd in catalonia: Influence of the new 2015 criteria in incidence and prevalence estimates. Mult. Scler. 2017, 24, 1843–1851. [Google Scholar] [CrossRef] [PubMed]
- Holroyd, K.B.; Aziz, F.; Szolics, M.; Alsaadi, T.; Levy, M.; Schiess, N. Prevalence and characteristics of transverse myelitis and neuromyelitis optica spectrum disorders in the united arab emirates: A multicenter, retrospective study. Clin. Exp. Neuroimmunol. 2018, 9, 155–161. [Google Scholar] [CrossRef]
- Mori, M.; Kuwabara, S.; Paul, F. Worldwide prevalence of neuromyelitis optica spectrum disorders. J. Neurol. Neurosurg. Psychiatry 2018, 89, 555–556. [Google Scholar] [CrossRef]
- Kim, S.H.; Mealy, M.A.; Levy, M.; Schmidt, F.; Ruprecht, K.; Paul, F.; Ringelstein, M.; Aktas, O.; Hartung, H.P.; Asgari, N.; et al. Racial differences in neuromyelitis optica spectrum disorder. Neurology 2018, 91, e2089–e2099. [Google Scholar] [CrossRef]
- Kitley, J.; Leite, M.I.; Nakashima, I.; Waters, P.; McNeillis, B.; Brown, R.; Takai, Y.; Takahashi, T.; Misu, T.; Elsone, L.; et al. Prognostic factors and disease course in aquaporin-4 antibody-positive patients with neuromyelitis optica spectrum disorder from the united kingdom and japan. Brain J. Neurol. 2012, 135, 1834–1849. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, M.; Armangue, T.; Sola-Valls, N.; Arrambide, G.; Meca-Lallana, J.E.; Oreja-Guevara, C.; Mendibe, M.; Alvarez de Arcaya, A.; Aladro, Y.; Casanova, B.; et al. Neuromyelitis optica spectrum disorders: Comparison according to the phenotype and serostatus. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e225. [Google Scholar] [CrossRef] [PubMed]
- McKeon, A.; Lennon, V.A.; Lotze, T.; Tenenbaum, S.; Ness, J.M.; Rensel, M.; Kuntz, N.L.; Fryer, J.P.; Homburger, H.; Hunter, J.; et al. Cns aquaporin-4 autoimmunity in children. Neurology 2008, 71, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Matiello, M.; Kim, H.J.; Kim, W.; Brum, D.G.; Barreira, A.A.; Kingsbury, D.J.; Plant, G.T.; Adoni, T.; Weinshenker, B.G. Familial neuromyelitis optica. Neurology 2010, 75, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, T.; Matsuoka, T.; Isobe, N.; Kawano, Y.; Minohara, M.; Shi, N.; Nishimura, Y.; Ochi, H.; Kira, J. Association of the hla-dpb1*0501 allele with anti-aquaporin-4 antibody positivity in japanese patients with idiopathic central nervous system demyelinating disorders. Tissue Antigens 2009, 73, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Dai, Y.; Qiu, W.; Zhong, X.; Wu, A.; Wang, Y.; Lu, Z.; Bao, J.; Hu, X. Hla-dpb1 0501 is associated with susceptibility to anti-aquaporin-4 antibodies positive neuromyelitis optica in southern han chinese. J. Neuroimmunol. 2011, 233, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Blanco, Y.; Ercilla-Gonzalez, G.; Llufriu, S.; Casanova-Estruch, B.; Magraner, M.J.; Ramio-Torrenta, L.; Mendibe-Bilbao, M.M.; Ucles-Sanchez, A.J.; Casado-Chocan, J.L.; Lopez de Munain, A.; et al. hla-drb1 typing in caucasians patients with neuromyelitis optica. Rev. De Neurol. 2011, 53, 146–152. [Google Scholar]
- Pandit, L.; Malli, C.; D’Cunha, A.; Mustafa, S. Human leukocyte antigen association with neuromyelitis optica in a south indian population. Mult. Scler. 2015, 21, 1217–1218. [Google Scholar] [CrossRef]
- Deschamps, R.; Paturel, L.; Jeannin, S.; Chausson, N.; Olindo, S.; Bera, O.; Bellance, R.; Smadja, D.; Cesaire, D.; Cabre, P. Different hla class ii (drb1 and dqb1) alleles determine either susceptibility or resistance to nmo and multiple sclerosis among the french afro-caribbean population. Mult. Scler. 2011, 17, 24–31. [Google Scholar] [CrossRef]
- Zephir, H.; Fajardy, I.; Outteryck, O.; Blanc, F.; Roger, N.; Fleury, M.; Rudolf, G.; Marignier, R.; Vukusic, S.; Confavreux, C.; et al. Is neuromyelitis optica associated with human leukocyte antigen? Mult. Scler. 2009, 15, 571–579. [Google Scholar] [CrossRef]
- Popescu, B.F.; Lennon, V.A.; Parisi, J.E.; Howe, C.L.; Weigand, S.D.; Cabrera-Gomez, J.A.; Newell, K.; Mandler, R.N.; Pittock, S.J.; Weinshenker, B.G.; et al. Neuromyelitis optica unique area postrema lesions: Nausea, vomiting, and pathogenic implications. Neurology 2011, 76, 1229–1237. [Google Scholar] [CrossRef] [PubMed]
- Dubey, D.; Pittock, S.J.; Krecke, K.N.; Flanagan, E.P. Association of extension of cervical cord lesion and area postrema syndrome with neuromyelitis optica spectrum disorder. JAMA Neurol. 2017, 74, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Kremer, L.; Mealy, M.; Jacob, A.; Nakashima, I.; Cabre, P.; Bigi, S.; Paul, F.; Jarius, S.; Aktas, O.; Elsone, L.; et al. Brainstem manifestations in neuromyelitis optica: A multicenter study of 258 patients. Mult. Scler. 2014, 20, 843–847. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Wildemann, B. Aquaporin-4 antibodies (nmo-igg) as a serological marker of neuromyelitis optica: A critical review of the literature. Brain Pathol. 2013, 23, 661–683. [Google Scholar] [CrossRef] [PubMed]
- Marignier, R.; Bernard-Valnet, R.; Giraudon, P.; Collongues, N.; Papeix, C.; Zephir, H.; Cavillon, G.; Rogemond, V.; Casey, R.; Frangoulis, B.; et al. Aquaporin-4 antibody-negative neuromyelitis optica: Distinct assay sensitivity-dependent entity. Neurology 2013, 80, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Pittock, S.J.; Lennon, V.A.; Bakshi, N.; Shen, L.; McKeon, A.; Quach, H.; Briggs, F.B.; Bernstein, A.L.; Schaefer, C.A.; Barcellos, L.F. Seroprevalence of aquaporin-4-igg in a northern california population representative cohort of multiple sclerosis. JAMA Neurol. 2014, 71, 1433–1436. [Google Scholar] [CrossRef] [PubMed]
- Waters, P.; Reindl, M.; Saiz, A.; Schanda, K.; Tuller, F.; Kral, V.; Nytrova, P.; Sobek, O.; Nielsen, H.H.; Barington, T.; et al. Multicentre comparison of a diagnostic assay: Aquaporin-4 antibodies in neuromyelitis optica. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.; Alvarez-Cermeno, J.; Bernardi, G.; Cogato, I.; Fredman, P.; Frederiksen, J.; Fredrikson, S.; Gallo, P.; Grimaldi, L.M.; Gronning, M.; et al. Cerebrospinal fluid in the diagnosis of multiple sclerosis: A consensus report. J. Neurol. Neurosurg. Psychiatry 1994, 57, 897–902. [Google Scholar] [CrossRef]
- Jarius, S.; Ruprecht, K.; Wildemann, B.; Kuempfel, T.; Ringelstein, M.; Geis, C.; Kleiter, I.; Kleinschnitz, C.; Berthele, A.; Brettschneider, J.; et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: A multicentre study of 175 patients. J. Neuroinflamm. 2012, 9, 14. [Google Scholar] [CrossRef]
- Kim, S.M.; Kim, S.J.; Lee, H.J.; Kuroda, H.; Palace, J.; Fujihara, K. Differential diagnosis of neuromyelitis optica spectrum disorders. Ther. Adv. Neurol. Disord. 2017, 10, 265–289. [Google Scholar] [CrossRef]
- Flanagan, E.P.; Weinshenker, B.G.; Krecke, K.N.; Lennon, V.A.; Lucchinetti, C.F.; McKeon, A.; Wingerchuk, D.M.; Shuster, E.A.; Jiao, Y.; Horta, E.S.; et al. Short myelitis lesions in aquaporin-4-igg-positive neuromyelitis optica spectrum disorders. JAMA Neurol. 2015, 72, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Pittock, S.J.; Weinshenker, B.G.; Lucchinetti, C.F.; Wingerchuk, D.M.; Corboy, J.R.; Lennon, V.A. Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch. Neurol. 2006, 63, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Matthews, L.; Marasco, R.; Jenkinson, M.; Kuker, W.; Luppe, S.; Leite, M.I.; Giorgio, A.; De Stefano, N.; Robertson, N.; Johansen-Berg, H.; et al. Distinction of seropositive nmo spectrum disorder and ms brain lesion distribution. Neurology 2013, 80, 1330–1337. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Paul, F.; Lana-Peixoto, M.A.; Tenembaum, S.; Asgari, N.; Palace, J.; Klawiter, E.C.; Sato, D.K.; de Seze, J.; Wuerfel, J.; et al. Mri characteristics of neuromyelitis optica spectrum disorder: An international update. Neurology 2015, 84, 1165–1173. [Google Scholar] [CrossRef] [PubMed]
- Jurynczyk, M.; Tackley, G.; Kong, Y.; Geraldes, R.; Matthews, L.; Woodhall, M.; Waters, P.; Kuker, W.; Craner, M.; Weir, A.; et al. Brain lesion distribution criteria distinguish ms from aqp4-antibody nmosd and mog-antibody disease. J. Neurol. Neurosurg. Psychiatry 2017, 88, 132–136. [Google Scholar] [CrossRef]
- Kira, J.I. Unexpected exacerbations following initiation of disease-modifying drugs in neuromyelitis optica spectrum disorder: Which factor is responsible, anti-aquaporin 4 antibodies, b cells, th1 cells, th2 cells, th17 cells, or others? Mult. Scler. 2017, 23, 1300–1302. [Google Scholar] [CrossRef] [PubMed]
- Kleiter, I.; Gahlen, A.; Borisow, N.; Fischer, K.; Wernecke, K.D.; Wegner, B.; Hellwig, K.; Pache, F.; Ruprecht, K.; Havla, J.; et al. Neuromyelitis optica: Evaluation of 871 attacks and 1,153 treatment courses. Ann. Neurol. 2016, 79, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Weinshenker, B.G. What is the optimal sequence of rescue treatments for attacks of neuromyelitis optica spectrum disorder? Ann. Neurol. 2016, 79, 204–205. [Google Scholar] [CrossRef]
- Wingerchuk, D.M.; Weinshenker, B.G. Neuromyelitis optica. Curr. Treat. Options Neurol. 2008, 10, 55–66. [Google Scholar] [CrossRef]
- Kim, S.H.; Hyun, J.W.; Kim, H.J. Individualized b cell-targeting therapy for neuromyelitis optica spectrum disorder. Neurochem. Int. 2018, in press. [Google Scholar] [CrossRef]
- Paul, F.; Murphy, O.; Pardo, S.; Levy, M. Investigational drugs in development to prevent neuromyelitis optica relapses. Expert Opin. Investig. Drugs 2018, 27, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.J.; Hochsmann, B.; Szer, J.; Kulasekararaj, A.; de Guibert, S.; Roth, A.; Weitz, I.C.; Armstrong, E.; Risitano, A.M.; Patriquin, C.J.; et al. Eculizumab in pregnant patients with paroxysmal nocturnal hemoglobinuria. N. Engl. J. Med. 2015, 373, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- McNamara, L.A.; Topaz, N.; Wang, X.; Hariri, S.; Fox, L.; MacNeil, J.R. High risk for invasive meningococcal disease among patients receiving eculizumab (soliris) despite receipt of meningococcal vaccine. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2017, 17, 2481–2484. [Google Scholar] [CrossRef]
- Traboulsee, A.; Greenberg, B.; Bennett, J.L.; Szczechowiski, L.; Fox, E.; Shkrobot, S.; Yamamura, T.; Terada, Y.; Kawata, Y.; Melia, A.; et al. A double-blind placebo-controlled study of satralizumab (sa 237), a recycling anti-il-6 receptor monoclonal antibody, as monotherapy for patients witn neuromyelitis optica spectrum disorder (nmosd). In Proceedings of the ECTRIMS, Berlin, Germany, 10–12 Octorber 2018. Poster 1278. [Google Scholar]
- Steinman, L.; Bar-Or, A.; Behne, J.M.; Benitez-Ribas, D.; Chin, P.S.; Clare-Salzler, M.; Healey, D.; Kim, J.I.; Kranz, D.M.; Lutterotti, A.; et al. Restoring immune tolerance in neuromyelitis optica: Part i. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e276. [Google Scholar] [CrossRef] [PubMed]
- Brunner, C.; Lassmann, H.; Waehneldt, T.V.; Matthieu, J.M.; Linington, C. Differential ultrastructural localization of myelin basic protein, myelin/oligodendroglial glycoprotein, and 2’,3’-cyclic nucleotide 3’-phosphodiesterase in the cns of adult rats. J. Neurochem. 1989, 52, 296–304. [Google Scholar] [CrossRef]
- Pham-Dinh, D.; Mattei, M.G.; Nussbaum, J.L.; Roussel, G.; Pontarotti, P.; Roeckel, N.; Mather, I.H.; Artzt, K.; Lindahl, K.F.; Dautigny, A. Myelin/oligodendrocyte glycoprotein is a member of a subset of the immunoglobulin superfamily encoded within the major histocompatibility complex. Proc. Natl. Acad. Sci. USA 1993, 90, 7990–7994. [Google Scholar] [CrossRef] [PubMed]
- Gardinier, M.V.; Amiguet, P.; Linington, C.; Matthieu, J.M. Myelin/oligodendrocyte glycoprotein is a unique member of the immunoglobulin superfamily. J. Neurosci. Res. 1992, 33, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Berger, T.; Rubner, P.; Schautzer, F.; Egg, R.; Ulmer, H.; Mayringer, I.; Dilitz, E.; Deisenhammer, F.; Reindl, M. Antimyelin antibodies as a predictor of clinically definite multiple sclerosis after a first demyelinating event. N. Engl. J. Med. 2003, 349, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Reindl, M.; Linington, C.; Brehm, U.; Egg, R.; Dilitz, E.; Deisenhammer, F.; Poewe, W.; Berger, T. Antibodies against the myelin oligodendrocyte glycoprotein and the myelin basic protein in multiple sclerosis and other neurological diseases: A comparative study. Brain J. Neurol. 1999, 122 Pt 11, 2047–2056. [Google Scholar] [CrossRef]
- Karni, A.; Bakimer-Kleiner, R.; Abramsky, O.; Ben-Nun, A. Elevated levels of antibody to myelin oligodendrocyte glycoprotein is not specific for patients with multiple sclerosis. Arch. Neurol. 1999, 56, 311–315. [Google Scholar] [CrossRef]
- Markovic, M.; Trajkovic, V.; Drulovic, J.; Mesaros, S.; Stojsavljevic, N.; Dujmovic, I.; Mostarica Stojkovic, M. Antibodies against myelin oligodendrocyte glycoprotein in the cerebrospinal fluid of multiple sclerosis patients. J. Neurol. Sci. 2003, 211, 67–73. [Google Scholar] [CrossRef]
- Gaertner, S.; de Graaf, K.L.; Greve, B.; Weissert, R. Antibodies against glycosylated native mog are elevated in patients with multiple sclerosis. Neurology 2004, 63, 2381–2383. [Google Scholar] [CrossRef] [PubMed]
- Lindert, R.B.; Haase, C.G.; Brehm, U.; Linington, C.; Wekerle, H.; Hohlfeld, R. Multiple sclerosis: B- and t-cell responses to the extracellular domain of the myelin oligodendrocyte glycoprotein. Brain J. Neurol. 1999, 122 Pt 11, 2089–2100. [Google Scholar] [CrossRef]
- Egg, R.; Reindl, M.; Deisenhammer, F.; Linington, C.; Berger, T. Anti-mog and anti-mbp antibody subclasses in multiple sclerosis. Mult. Scler. 2001, 7, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Kuhle, J.; Lindberg, R.L.; Regeniter, A.; Mehling, M.; Hoffmann, F.; Reindl, M.; Berger, T.; Radue, E.W.; Leppert, D.; Kappos, L. Antimyelin antibodies in clinically isolated syndromes correlate with inflammation in mri and csf. J. Neurol. 2007, 254, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Brilot, F.; Dale, R.C.; Selter, R.C.; Grummel, V.; Kalluri, S.R.; Aslam, M.; Busch, V.; Zhou, D.; Cepok, S.; Hemmer, B. Antibodies to native myelin oligodendrocyte glycoprotein in children with inflammatory demyelinating central nervous system disease. Ann. Neurol. 2009, 66, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Kitley, J.; Waters, P.; Woodhall, M.; Leite, M.I.; Murchison, A.; George, J.; Kuker, W.; Chandratre, S.; Vincent, A.; Palace, J. Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: A comparative study. JAMA Neurol. 2014, 71, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, S.; Reddel, S.W.; Henderson, A.; Parratt, J.D.; Barnett, M.; Gatt, P.N.; Merheb, V.; Kumaran, R.Y.; Pathmanandavel, K.; Sinmaz, N.; et al. Antibodies to myelin oligodendrocyte glycoprotein in bilateral and recurrent optic neuritis. Neurol. Neuroimmunol. Neuroinflamm. 2014, 1, e40. [Google Scholar] [CrossRef]
- Hoftberger, R.; Sepulveda, M.; Armangue, T.; Blanco, Y.; Rostasy, K.; Calvo, A.C.; Olascoaga, J.; Ramio-Torrenta, L.; Reindl, M.; Benito-Leon, J.; et al. Antibodies to mog and aqp4 in adults with neuromyelitis optica and suspected limited forms of the disease. Mult. Scler. 2015, 21, 866–874. [Google Scholar] [CrossRef]
- Jarius, S.; Kleiter, I.; Ruprecht, K.; Asgari, N.; Pitarokoili, K.; Borisow, N.; Hummert, M.W.; Trebst, C.; Pache, F.; Winkelmann, A.; et al. Mog-igg in nmo and related disorders: A multicenter study of 50 patients. Part 3: Brainstem involvement—Frequency, presentation and outcome. J. Neuroinflamm. 2016, 13, 281. [Google Scholar] [CrossRef]
- Spadaro, M.; Gerdes, L.A.; Krumbholz, M.; Ertl-Wagner, B.; Thaler, F.S.; Schuh, E.; Metz, I.; Blaschek, A.; Dick, A.; Bruck, W.; et al. Autoantibodies to mog in a distinct subgroup of adult multiple sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e257. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, S.; Mohammad, S.; Tantsis, E.; Nguyen, T.K.; Merheb, V.; Fung, V.S.C.; White, O.B.; Broadley, S.; Lechner-Scott, J.; Vucic, S.; et al. Clinical course, therapeutic responses and outcomes in relapsing mog antibody-associated demyelination. J. Neurol. Neurosurg. Psychiatry 2018, 89, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Reindl, M.; Di Pauli, F.; Rostasy, K.; Berger, T. The spectrum of mog autoantibody-associated demyelinating diseases. Nat. Rev. Neurol. 2013, 9, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Kerlero de Rosbo, N.; Honegger, P.; Lassmann, H.; Matthieu, J.M. Demyelination induced in aggregating brain cell cultures by a monoclonal antibody against myelin/oligodendrocyte glycoprotein. J. Neurochem. 1990, 55, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Piddlesden, S.J.; Lassmann, H.; Zimprich, F.; Morgan, B.P.; Linington, C. The demyelinating potential of antibodies to myelin oligodendrocyte glycoprotein is related to their ability to fix complement. Am. J. Pathol. 1993, 143, 555–564. [Google Scholar] [CrossRef]
- Bettelli, E.; Baeten, D.; Jager, A.; Sobel, R.A.; Kuchroo, V.K. Myelin oligodendrocyte glycoprotein-specific t and b cells cooperate to induce a devic-like disease in mice. J. Clin. Investig. 2006, 116, 2393–2402. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, G.; Lassmann, H.; Wekerle, H.; Holz, A. Spontaneous opticospinal encephalomyelitis in a double-transgenic mouse model of autoimmune t cell/b cell cooperation. J. Clin. Investig. 2006, 116, 2385–2392. [Google Scholar] [CrossRef]
- Saadoun, S.; Waters, P.; Owens, G.P.; Bennett, J.L.; Vincent, A.; Papadopoulos, M.C. Neuromyelitis optica mog-igg causes reversible lesions in mouse brain. Acta Neuropathol. Commun. 2014, 2, 35. [Google Scholar] [CrossRef]
- Sato, D.K.; Callegaro, D.; Lana-Peixoto, M.A.; Waters, P.J.; de Haidar Jorge, F.M.; Takahashi, T.; Nakashima, I.; Apostolos-Pereira, S.L.; Talim, N.; Simm, R.F.; et al. Distinction between mog antibody-positive and aqp4 antibody-positive nmo spectrum disorders. Neurology 2014, 82, 474–481. [Google Scholar] [CrossRef]
- Jarius, S.; Ruprecht, K.; Kleiter, I.; Borisow, N.; Asgari, N.; Pitarokoili, K.; Pache, F.; Stich, O.; Beume, L.A.; Hummert, M.W.; et al. Mog-igg in nmo and related disorders: A multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J. Neuroinflamm. 2016, 13, 280. [Google Scholar] [CrossRef]
- Jarius, S.; Paul, F.; Aktas, O.; Asgari, N.; Dale, R.C.; de Seze, J.; Franciotta, D.; Fujihara, K.; Jacob, A.; Kim, H.J.; et al. Mog encephalomyelitis: International recommendations on diagnosis and antibody testing. J. Neuroinflamm. 2018, 15, 134. [Google Scholar] [CrossRef] [PubMed]
- Chalmoukou, K.; Alexopoulos, H.; Akrivou, S.; Stathopoulos, P.; Reindl, M.; Dalakas, M.C. Anti-mog antibodies are frequently associated with steroid-sensitive recurrent optic neuritis. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e131. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Authors, Year | Country | Number of Cases | Incidence (95% CI) (per Million per Year) | Prevalence (95% CI) (per 100,000) |
|---|---|---|---|---|
| Rivera et al., 2008 [54] | Mexico | 34 | 0.20 (0.05–0.35) | 1 |
| Cabrera-Gómez et al., 2009 [56] | Cuba | 58 | 0.44 (0.3–0.62) | 0.43 (0.29–0.61) |
| Asgari et al., 2011 [55] | Denmark | 42 | 4 (3.0–5.4) | 4.41 (3.1–5.7) |
| Aboul Enein et al., 2011 [57] | Austria | 71 | 0.54 (0.01–0.03) | 0.71 (0.17–0.96) |
| Cossburn et al., 2012 [58] | UK | 14 | NA | 1.96 (1.22–2.97) |
| Houzen et al., 2012 [59] | Japan | 3 | 0.8 (0.3–1.6) | 0.72 (0.31–1.42) |
| Jacob et al., 2013 [60] | UK | 13 | 0.8 (0.3–1.6) | 0.72 (0.31–1.42) |
| Etemadifar et al., 2014 [61] | Iran | 95 | NA | 1.95 (1.62–2.23) |
| Pandit et al., 2014 [62] | India | 11 | NA | 2.6 |
| Kashipazha et al., 2015 [63] | Iran | 51 | NA | 0.8 (0.54–1.06) |
| Flanagan et al., 2016 [53] | USA | 6 | 0.7 (0.0–2.1) | 3.9 (0.8–7.1) |
| Martinique | 39 | 7.3 (4.1–10.1) | 10.0 (6.8–13.2) | |
| van Pelt et al., 2016 [64] | Netherlands | 1.2 | NA | |
| Houzen et al., 2017 [65] | Japan | 14 | NA | 4.1 (2.2–6.9) |
| Hor et al., 2017 [66] | Malaysia | 14 | NA | 1.99 (1.09–3.35) |
| Bukhari et al., 2017 [67] | ANZ | 81 | 0.37 (0.36–0.38) | 0.7 (0.66–0.74) |
| Sepulveda et al., 2017 [68] | Spain | 74 | 0.63 (0.45–0.8) | 0.89 (0.87–0.91) |
| Holroyd et al., 2018 [69] | United Arab Emirates | 10 | 0.59 | 0.34 |
| Site | Symtoms |
|---|---|
| Optic nerve/chiasm | Eye pain or headache |
| Blurred vision | |
| Disturbance of color vision | |
| Amaurosis | |
| Optic disc edema | |
| Optic atrophy | |
| Scotomas and other visual field defects | |
| Spinal cord | Limb weakness |
| Lower limb spasticity | |
| Gait abnormalities | |
| Sensory disturbances | |
| Radicular pain | |
| Pruritus | |
| Painful tonic spasms | |
| Trunk and limb ataxia | |
| Sphincter disturbances | |
| Respiratory weakness | |
| Lhermitte phenomenon | |
| Brainstem | Motor and sensory disturbances |
| Incoercible nausea, vomiting and hiccups | |
| Intractable cough | |
| Weight loss | |
| Anorexia | |
| Diplopia/ocular movement disorders | |
| Facial dysesthesia and trigeminal neuralgia | |
| Dysgeusia | |
| Facial paralysis | |
| Hearing loss, tinnitus | |
| Vertigo | |
| Dysarthria/dysphagia | |
| Diencephalon | Narcolepsy |
| Hypophyseal abnormalities | |
| Antidiuretic hormone syndrome | |
| Pre-syncopal symptoms | |
| Disturbances of body temperature | |
| Anhydrosis/excessive sweating | |
| Hyperphagia | |
| Cerebrum | Posterior reversible encephalopathy syndrome (PRES) |
| Mental confusion | |
| Seizures | |
| Aphasia | |
| Apraxia | |
| Cognitive dysfunction | |
| Psychiatric symptoms |
| 1. Diagnostic criteria for NMOSD with AQP4-IgG | |||
| 1. At least one core clinical characteristic | |||
| 2. Exclusion of alternative diagnoses | |||
| 2. Diagnostic criteria for NMOSD without AQP4-IgG or NMOSD with unknown AQP4-IgG status | |||
| 1. At least two core clinical characteristics meeting all of the following requirements: | |||
| a. At least one core clinical characteristic must be optic neuritis, acute myelitis with LETM, or area postrema syndrome | |||
| b. Dissemination in space (two or more different core clinical characteristics) | |||
| c. Core clinical syndromes must be associated with respective MRI findings: | |||
| i. Optic neuritis: | |||
| 1. Brain MRI is normal or with nonspecific lesions; OR | |||
| 2. Optic nerve lesion extending over ½ of the optic nerve length; or chiasmal lesion | |||
| ii. Acute myelitis: MRI with lesion or spinal atrophy extending over ≥3 contiguous segments | |||
| iii. Area postrema syndrome: MRI with dorsal medulla/area postrema lesions | |||
| iv. Acute brainstem syndrome: MRI with periependymal brainstem lesions | |||
| v. Narcolepsy or acute diencephalic clinical syndrome: MRI with NMOSD-typical diencephalic lesions | |||
| 2. Exclusion of alternative diagnoses | |||
| Distinctive characteristics of MS | |
| Progressive course | |
| Partial transverse myelitis | |
| Brain MRI features | |
| Perpendicular periventricular lesions (Dawson fingers) | |
| Periventricular lesions in the inferior temporal lobe | |
| Juxtacortical lesions involving subcortical U-fibers | |
| Cortical lesions | |
| More severe brain atrophy | |
| Spinal cord MRI features | |
| Lesions <3 complete vertebral segments | |
| Lesions located predominantly in the peripheral cord | |
| Diffuse, indistinct signal change on T2-weighted sequences | |
| Cerebrospinal fluid analysis | |
| Presence of oligoclonal bands | |
| Optic coherence tomography features | |
| Predominant atrophy of temporal RNFL | |
| Distinctive characteristics of NMOSD | |
| Complete transverse myelitis | |
| Brain MRI features | |
| Multiple patchy enhancement with blurred margin in adjacent regions (cloud-like enhancement) | |
| Large and edematous callosal lesions | |
| Large and confluent white matter lesions (as in PRES) | |
| Predominantly posterior brainstem lesions (around the fourth ventricle lesions and periaqueductal lesions) | |
| Hypothalamic lesions | |
| Extensive optic nerve lesions and chiasmal lesions | |
| Spinal cord MRI features | |
| Longitudinally extensive transverse myelitis lesions (≥3 contiguous segments) | |
| Longitudinally extensive spinal cord atrophy (≥3 contiguous segments) | |
| Centrally-located or holomedullary spinal cord lesions | |
| Cerebrospinal fluid analysis | |
| Moderate or marked pleocytosis | |
| Presence of neutrophils and eosinophils | |
| Optic coherence tomography features | |
| Predominant atrophy of superior and inferior RNFL | |
| Multiple Sclerosis | |
|---|---|
| Acute disseminated encephalomyelitis | |
| MOG-related disorders | |
| Sarcoidosis | |
| Lymphoma | |
| Paraneoplastic disease | |
| Central nervous system infections | |
| Syphilis | |
| Tuberculosis | |
| Human T-lymphotropic virus-I (HTLV-I) infection | |
| Herpes virus infection | |
| Dengue-virus infection | |
| Lyme disease | |
| Schistosomiasis | |
| Sjogren syndrome | |
| Systemic lupus erythematosus | |
| Neuro-Behçet’s disease | |
| Spinal dural arteriovenous fistula | |
| Drugs | Route | Regimen | Comments |
|---|---|---|---|
| Prednisone | Oral | ≥30 mg/d | Keep until until azathioprine or mycophenolate fully effective, then taper over six months |
| Azathioprine | Oral | 2-3 mg/kg/d in 2 doses | First line treatment; latency four to six months; target dose guided by ALC and MCV; monitor liver function |
| Mycophenolate mofetil | Oral | 1500–3000 mg/d in 2 doses | Target dose guided by ALC and blood concentration (1–2 μg/mL) |
| Rituximab | IV | 1000 mg given twice, 14 d apart.Repeat every 6 mo or based on reemergence of CD19 B cells | First-line therapy; CD19 B cells as a marker |
| Methotrexate | Oral | 15–25.0 mg weekly | Supplement with folic acid 1 mg/d, monitor liver function |
| Ciclosporin A | Oral | 2–5 mg/kg/day in 2 doses | Nephrotoxic, target dose guided by blood concentration (70–100 ng/mL) |
| Tacrolimus | Oral | 1–6 mg/day in 2 doses | Nephrotoxic, target dose guided by blood concentration (5–10 ng/mL) |
| Mitoxantrone | IV | 12 mg/m2 every 1–3 months | Cardiac monitoring (LVEF), target dose guided by leukocyte count; total cumulative dose 100 mg/m2 |
| Tocilizumab | IV | 8 mg/kg every 4 weeks | 8 mg/kg every four weeks; monitoring for infections; CRP no reliable biomarker for infection |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lana-Peixoto, M.A.; Talim, N. Neuromyelitis Optica Spectrum Disorder and Anti-MOG Syndromes. Biomedicines 2019, 7, 42. https://doi.org/10.3390/biomedicines7020042
Lana-Peixoto MA, Talim N. Neuromyelitis Optica Spectrum Disorder and Anti-MOG Syndromes. Biomedicines. 2019; 7(2):42. https://doi.org/10.3390/biomedicines7020042
Chicago/Turabian StyleLana-Peixoto, Marco A., and Natália Talim. 2019. "Neuromyelitis Optica Spectrum Disorder and Anti-MOG Syndromes" Biomedicines 7, no. 2: 42. https://doi.org/10.3390/biomedicines7020042
APA StyleLana-Peixoto, M. A., & Talim, N. (2019). Neuromyelitis Optica Spectrum Disorder and Anti-MOG Syndromes. Biomedicines, 7(2), 42. https://doi.org/10.3390/biomedicines7020042