Zebrafish: A Powerful Model for Understanding the Functional Relevance of Noncoding Region Mutations in Human Genetic Diseases

Abstract
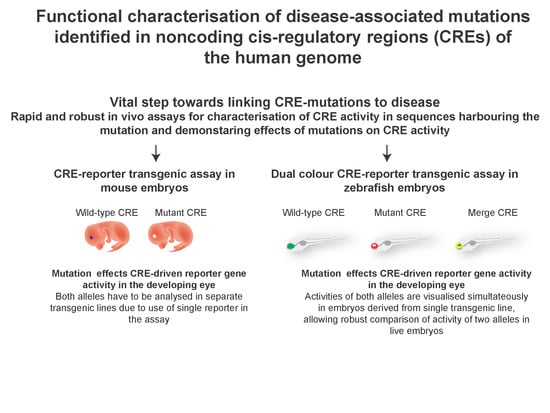
1. Introduction
2. Prediction of Cis-Regulatory or Enhancer Activity in Noncoding Regions of Human Genome
3. Testing the Predicted CRE Activity in CRE-Reporter Assays
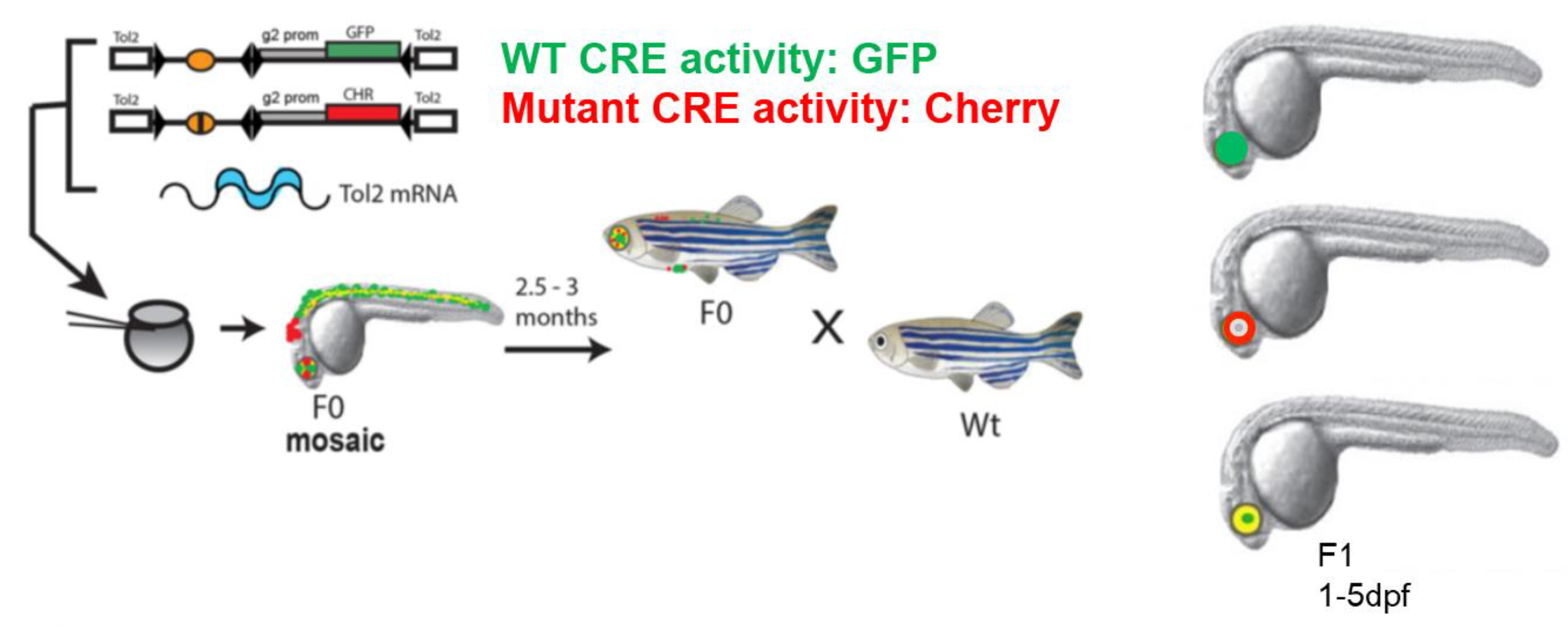
4. Zebrafish Dual-Colour CRE-Reporter Assay for Assessing Effects of Mutations on CRE Function
Author Contributions
Funding
Conflicts of Interest
References
- Lee, T.I.; Young, R.A. Transcriptional Regulation and its Misregulation in Disease. Cell 2013, 152, 1237–1251. [Google Scholar] [CrossRef] [PubMed]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef] [PubMed]
- Schoenfelder, S.; Fraser, P. Long-range enhancer–promoter contacts in gene expression control. Nat. Rev. Genet. 2019, 20, 437–455. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Kleinjan, D.A. Disruption of long-range gene regulation in human genetic disease: A kaleidoscope of general principles, diverse mechanisms and unique phenotypic consequences. Qual. Life Res. 2014, 133, 815–845. [Google Scholar] [CrossRef] [PubMed]
- Loots, G.G. Genomic Identification of Regulatory Elements by Evolutionary Sequence Comparison and Functional Analysis. Nonviral Vectors Gene Ther. Phys. Methods Med Transl. 2008, 61, 269–293. [Google Scholar]
- Naville, M.; Ishibashi, M.; Ferg, M.; Bengani, H.; Rinkwitz, S.; Krecsmarik, M.; Hawkins, T.A.; Wilson, S.W.; Manning, E.; Chilamakuri, C.S.R.; et al. Long-range evolutionary constraints reveal cis-regulatory interactions on the human X chromosome. Nat. Commun. 2015, 6, 6904. [Google Scholar] [CrossRef]
- Newburger, D.E.; Bulyk, M.L. UniPROBE: An online database of protein binding microarray data on protein-DNA interactions. Nucleic Acids Res. 2009, 37, D77–D82. [Google Scholar] [CrossRef]
- Zinzen, R.P.; Girardot, C.; Gagneur, J.; Braun, M.; Furlong, E.E.M. Combinatorial binding predicts spatio-temporal cis-regulatory activity. Nature 2009, 462, 65–70. [Google Scholar] [CrossRef]
- John, S.; Sabo, P.J.; Canfield, T.K.; Lee, K.; Vong, S.; Weaver, M.; Wang, H.; Vierstra, J.; Reynolds, A.P.; Thurman, R.E.; et al. Genome-scale Mapping of DNaseI Hypersensitivity. Curr. Protoc. Mol. Boil. 2013, 103, 21–27. [Google Scholar]
- Giresi, P.G.; Kim, J.; McDaniell, R.M.; Iyer, V.R.; Lieb, J.D. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2007, 17, 877–885. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Wu, B.; Litzenburger, U.M.; Ruff, D.; Gonzales, M.L.; Snyder, M.P.; Chang, H.Y.; Greenleaf, W.J. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 2015, 523, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Heintzman, N.D.; Ren, B. The gateway to transcription: Identifying, characterizing and understanding promoters in the eukaryotic genome. Cell Mol. Life Sci. 2007, 64, 386–400. [Google Scholar] [CrossRef] [PubMed]
- Rada-Iglesias, A.; Bajpai, R.; Swigut, T.; Brugmann, S.A.; Flynn, R.A.; Wysocka, J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011, 470, 279–283. [Google Scholar] [CrossRef] [PubMed]
- De Wit, E.; de Laat, W. A decade of 3C technologies: Insights into nuclear organization. Genes Dev. 2012, 26, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Van Steensel, B.; Dekker, J. Genomics tools for unraveling chromosome architecture. Nat. Biotechnol 2010, 28, 1089–1095. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, K.R.; Dreszer, T.R.; Long, J.C.; Malladi, V.S.; Sloan, C.A.; Raney, B.J.; Cline, M.S.; Karolchik, D.; Barber, G.P.; Clawson, H.; et al. ENCODE whole-genome data in the UCSC Genome Browser: Update 2012. Nucleic Acids Res. 2012, 40, D912–D917. [Google Scholar] [CrossRef] [PubMed]
- Kwasnieski, J.C.; Mogno, I.; Myers, C.A.; Corbo, J.C.; Cohen, B.A. Complex effects of nucleotide variants in a mammalian cis-regulatory element. Proc. Natl. Acad. Sci. USA 2012, 109, 19498–19503. [Google Scholar] [CrossRef] [PubMed]
- Arnold, C.D.; Gerlach, D.; Stelzer, C.; Boryn, L.M.; Rath, M.; Stark, A. Genome-Wide Quantitative Enhancer Activity Maps Identified by STARR-seq. Science 2013, 339, 1074–1077. [Google Scholar] [CrossRef]
- Goode, D.K.; Elgar, G. Capturing the regulatory interactions of eukaryote genomes. Brief Funct. Genom. 2013, 12, 142–160. [Google Scholar] [CrossRef]
- Bhatia, S.; Gordon, C.T.; Foster, R.G.; Melin, L.; Abadie, V.; Baujat, G.; Vazquez, M.-P.; Amiel, J.; Lyonnet, S.; Van Heyningen, V.; et al. Functional Assessment of Disease-Associated Regulatory Variants In Vivo Using a Versatile Dual Colour Transgenesis Strategy in Zebrafish. PLoS Genet. 2015, 11, e1005193. [Google Scholar] [CrossRef]
- Walker, S.L.; Ariga, J.; Mathias, J.R.; Coothankandaswamy, V.; Xie, X.; Distel, M.; Köster, R.W.; Parsons, M.J.; Bhalla, K.N.; Saxena, M.T.; et al. Automated Reporter Quantification In Vivo: High-Throughput Screening Method for Reporter-Based Assays in Zebrafish. PLoS ONE 2012, 7, e29916. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.A.; Miguel-Escalada, I.; Slovik, K.J.; Walsh, K.T.; Hadzhiev, Y.; Sanges, R.; Stupka, E.; Marsh, E.K.; Balciuniene, J.; Balciunas, D.; et al. Targeted transgene integration overcomes variability of position effects in zebrafish. Development 2014, 141, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Hadzhiev, Y.; Miguel-Escalada, I.; Balciunas, D.; Müller, F. Testing of Cis-Regulatory Elements by Targeted Transgene Integration in Zebrafish Using PhiC31 Integrase. Adv. Struct. Saf. Stud. 2016, 1451, 81–91. [Google Scholar]
- Kleinjan, D.A.; Seawright, A.; Schedl, A.; A Quinlan, R.; Danes, S.; Van Heyningen, V. Aniridia-associated translocations, DNase hypersensitivity, sequence comparison and transgenic analysis redefine the functional domain of PAX6. Hum. Mol. Genet. 2001, 10, 2049–2059. [Google Scholar] [CrossRef] [PubMed]
- Kokubu, C.; Horie, K.; Abe, K.; Ikeda, R.; Mizuno, S.; Uno, Y.; Ogiwara, S.; Ohtsuka, M.; Isotani, A.; Okabe, M.; et al. A transposon-based chromosomal engineering method to survey a large cis-regulatory landscape in mice. Nat. Genet. 2009, 41, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Ruf, S.; Symmons, O.; Uslu, V.V.; Dolle, D.; Hot, C.; Ettwiller, L.; Spitz, F. Large-scale analysis of the regulatory architecture of the mouse genome with a transposon-associated sensor. Nat. Genet. 2011, 43, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.B.; Westerfield, M. Zebrafish models in translational research: Tipping the scales toward advancements in human health. Dis. Model. Mech. 2014, 7, 739–743. [Google Scholar] [CrossRef]
- Bhatia, S.; Monahan, J.M.; Ravi, V.; Gautier, P.; Murdoch, E.; Brenner, S.; Van Heyningen, V.; Venkatesh, B.; Kleinjan, D.A. A survey of ancient conserved non-coding elements in the PAX6 locus reveals a landscape of interdigitated cis-regulatory archipelagos. Dev. Boil. 2014, 387, 214–228. [Google Scholar] [CrossRef]
- Bhatia, S.; Bengani, H.; Fish, M.; Brown, A.; Divizia, M.T.; De Marco, R.; Damante, G.; Grainger, R.; Van Heyningen, V.; Kleinjan, D.A. Disruption of Autoregulatory Feedback by a Mutation in a Remote, Ultraconserved PAX6 Enhancer Causes Aniridia. Am. J. Hum. Genet. 2013, 93, 1126–1134. [Google Scholar] [CrossRef]
- Ravi, V.; Bhatia, S.; Gautier, P.; Loosli, F.; Tay, B.-H.; Tay, A.; Murdoch, E.; Coutinho, P.; Van Heyningen, V.; Brenner, S.; et al. Sequencing of Pax6 Loci from the Elephant Shark Reveals a Family of Pax6 Genes in Vertebrate Genomes, Forged by Ancient Duplications and Divergences. PLoS Genet. 2013, 9, e1003177. [Google Scholar] [CrossRef]
- Rainger, J.K.; Bhatia, S.; Bengani, H.; Gautier, P.; Rainger, J.; Pearson, M.; Ansari, M.; Crow, J.; Mehendale, F.; Palinkasova, B.; et al. Disruption of SATB2 or its long-range cis-regulation by SOX9 causes a syndromic form of Pierre Robin sequence. Hum. Mol. Genet. 2014, 23, 2569–2579. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Song, M.; Devine, P.; Bruneau, B.G.; Scott, I.C.; Wilson, M.D. Heart enhancers with deeply conserved regulatory activity are established early in zebrafish development. Nat. Commun. 2018, 9, 4977. [Google Scholar] [CrossRef] [PubMed]
- Chahal, G.; Tyagi, S.; Ramialison, M. Navigating the non-coding genome in heart development and Congenital Heart Disease. Differentiation. 2019, 107, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, M.; Mechaly, A.S.; Becker, T.S.; Rinkwitz, S. Using zebrafish transgenesis to test human genomic sequences for specific enhancer activity. Methods 2013, 62, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, T.J.; Lancman, J.J.; Berenguer, M.; Dong, P.D.S.; Duester, G. Genomic Knockout of Two Presumed Forelimb Tbx5 Enhancers Reveals They Are Nonessential for Limb Development. Cell Rep. 2018, 23, 3146–3151. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| CRE-reporter assays in in vitro cultured cell lines Description:
|
| CRE-reporter assays in zebrafish Description:
|
| CRE-reporter assays in mouse Description:
|
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mann, A.; Bhatia, S. Zebrafish: A Powerful Model for Understanding the Functional Relevance of Noncoding Region Mutations in Human Genetic Diseases. Biomedicines 2019, 7, 71. https://doi.org/10.3390/biomedicines7030071
Mann A, Bhatia S. Zebrafish: A Powerful Model for Understanding the Functional Relevance of Noncoding Region Mutations in Human Genetic Diseases. Biomedicines. 2019; 7(3):71. https://doi.org/10.3390/biomedicines7030071
Chicago/Turabian StyleMann, Anita, and Shipra Bhatia. 2019. "Zebrafish: A Powerful Model for Understanding the Functional Relevance of Noncoding Region Mutations in Human Genetic Diseases" Biomedicines 7, no. 3: 71. https://doi.org/10.3390/biomedicines7030071
APA StyleMann, A., & Bhatia, S. (2019). Zebrafish: A Powerful Model for Understanding the Functional Relevance of Noncoding Region Mutations in Human Genetic Diseases. Biomedicines, 7(3), 71. https://doi.org/10.3390/biomedicines7030071