Simplified Admix Archaeal Glycolipid Adjuvanted Vaccine and Checkpoint Inhibitor Therapy Combination Enhances Protection from Murine Melanoma

 , , ,
, , ,
Abstract
:
1. Introduction
2. Experimental Section
2.1. Mouse Strains
2.2. Tumor Model (B16-OVA, B16 Melanoma)
2.3. Vaccine Preparation and Route of Immunization
2.4. Checkpoint Inhibitor Combination Therapy
2.5. Cellular Processing and Detection of OVA-Specific CD8+ T Cells
2.6. Statistical Analysis
2.7. Processing and Immunohistochemical Detection of Tumor-Infiltrating Immune Cells
2.8. Histological Scoring
3. Results
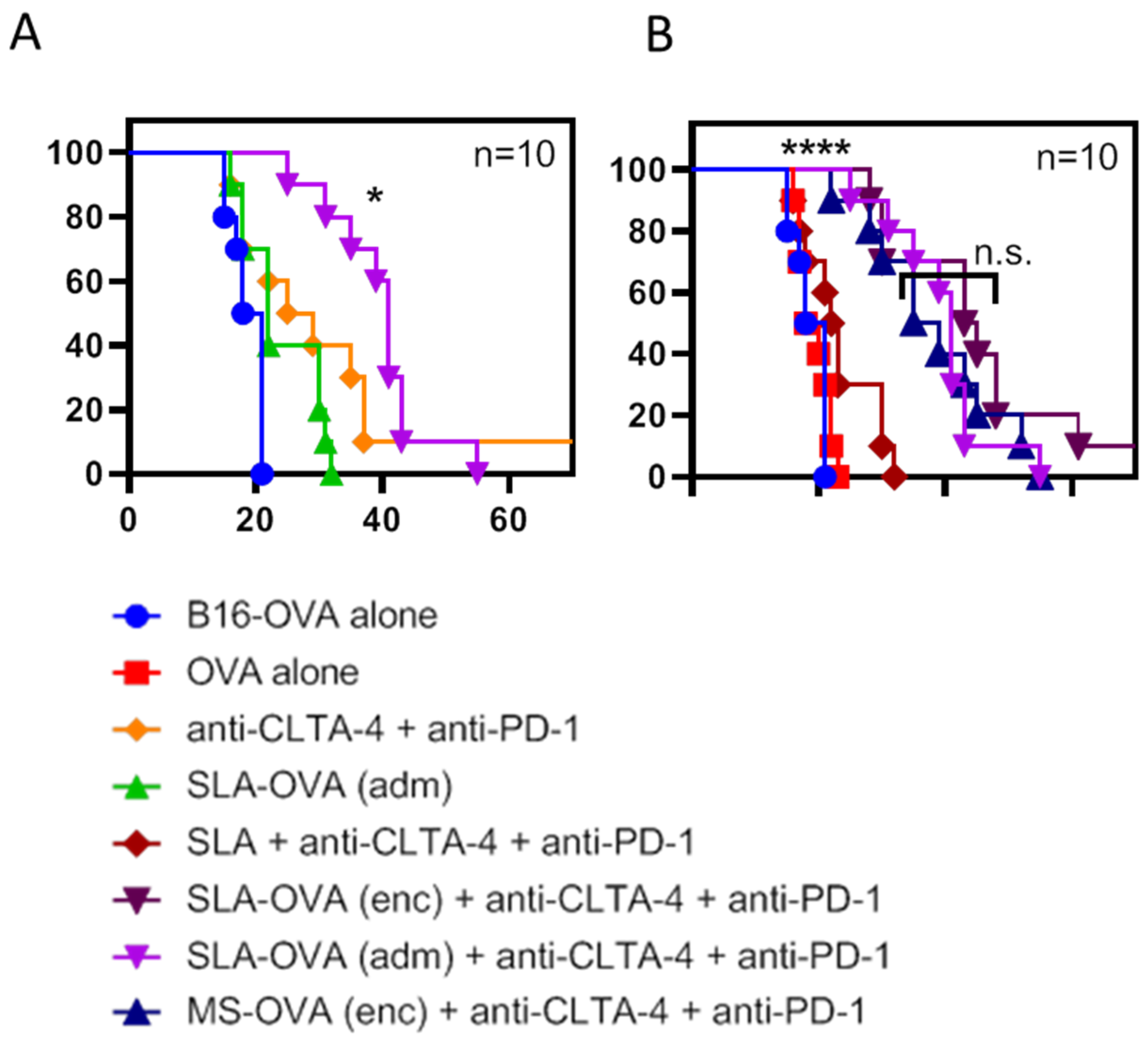
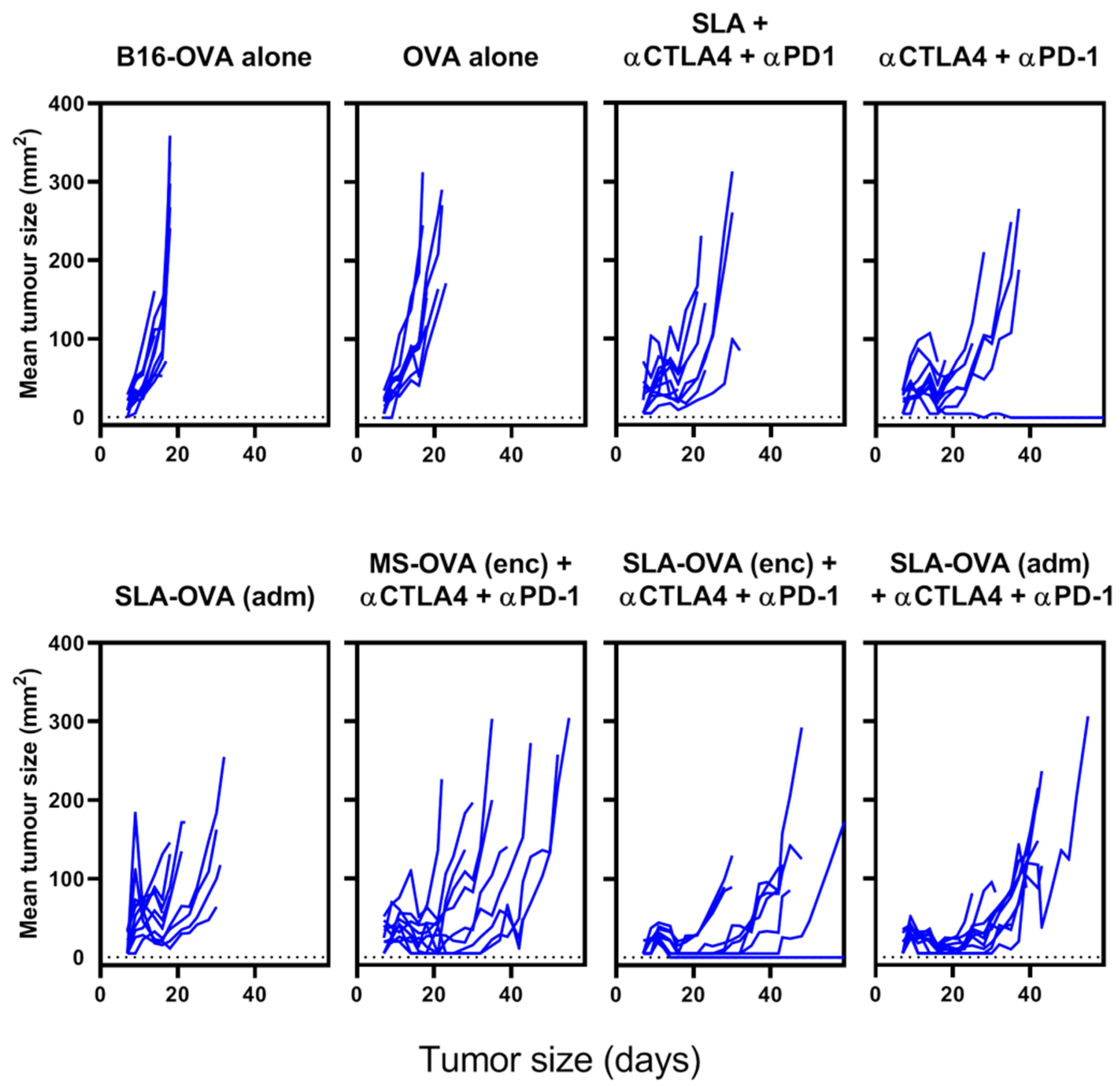
3.1. SLA–OVA (adm) Immunization Synergizes with Anti-CTLA-4 and Anti-PD-1 Therapy to Provide Protection in a B16-OVA Melanoma Model
3.2. SLA–OVA (adm) Therapy Increased the Frequencies of OVA-CD8+ T Cells in the Spleen and Tumor
3.3. Histological Properties of B16 Tumors
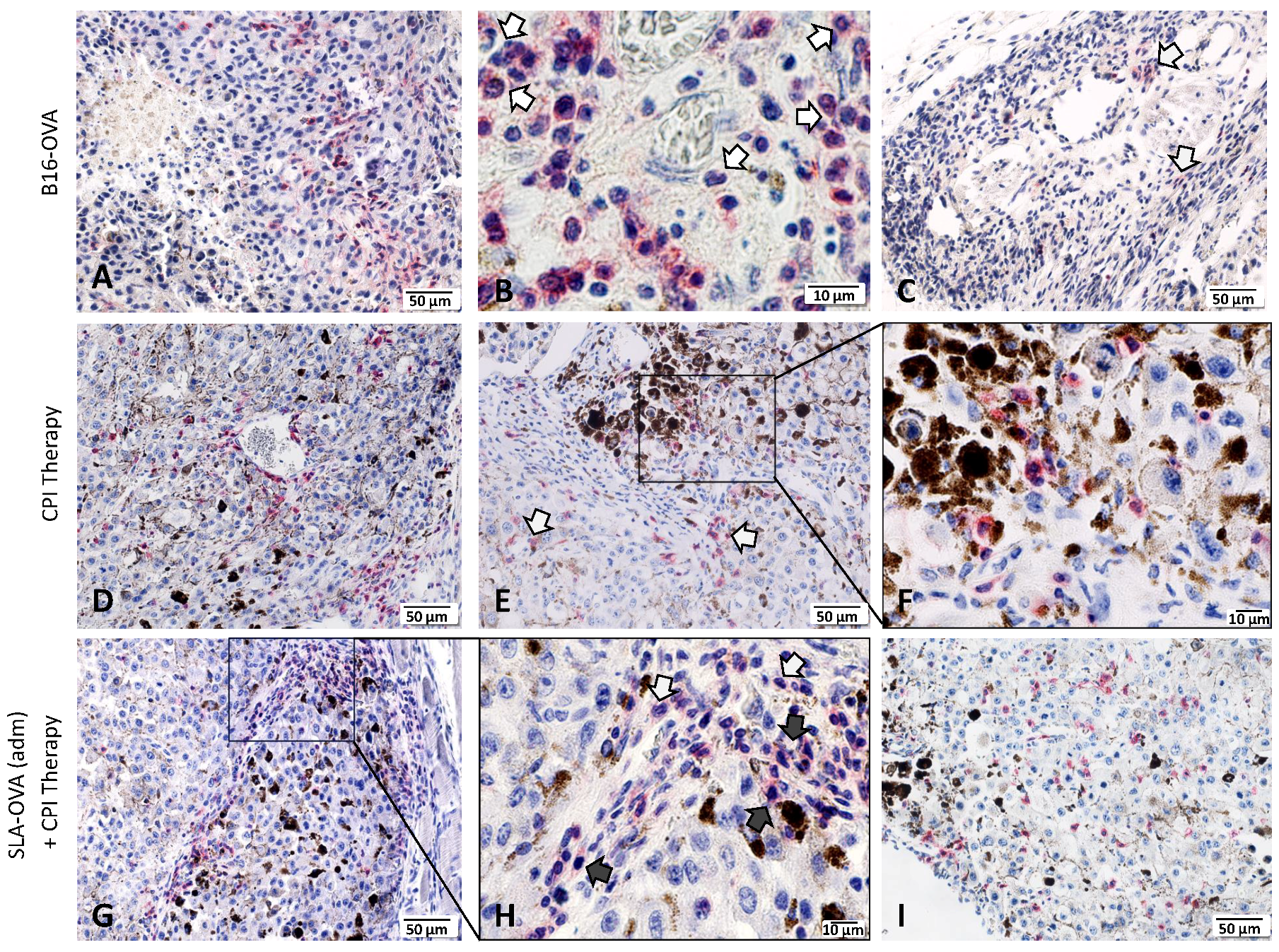
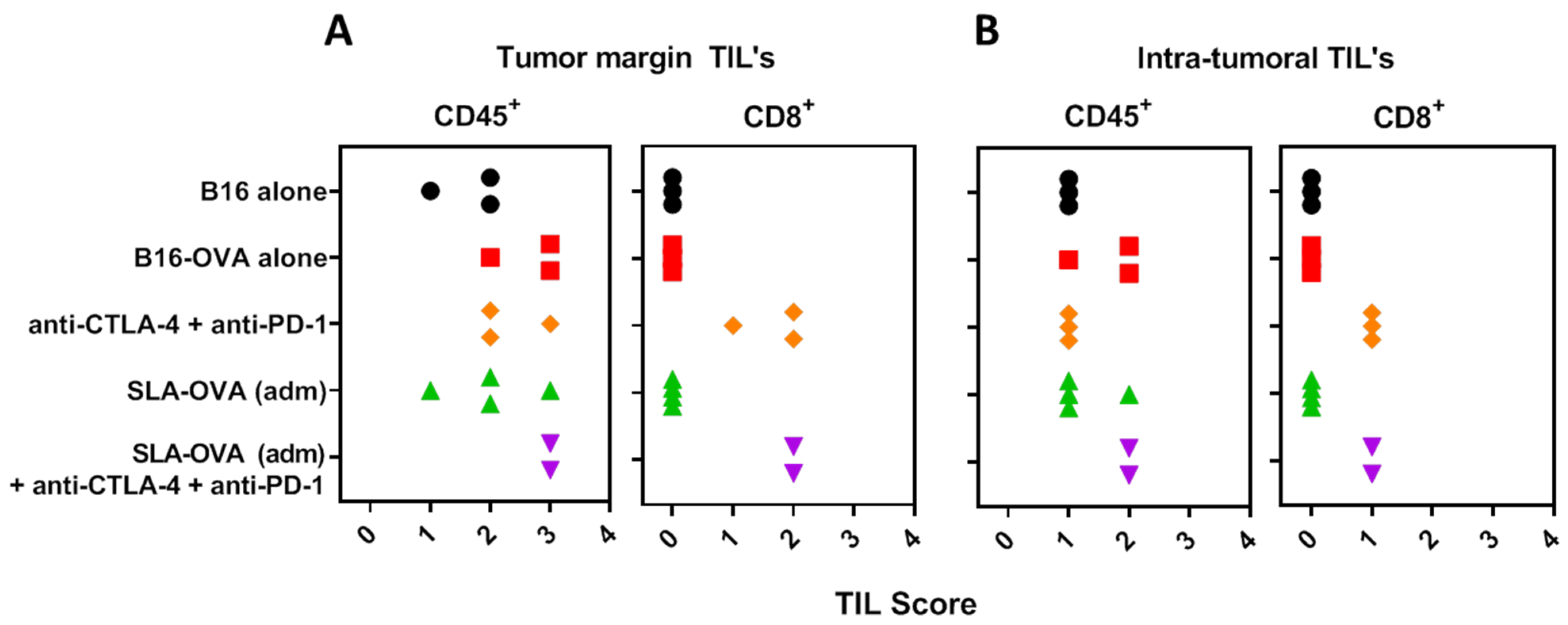
3.4. Immunohistochemical Evaluation of CD8+ T Cells in B16-OVA Tumor-Bearing Mice Receiving SLA–OVA and Checkpoint Therapy
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alexander, W. The Checkpoint Immunotherapy Revolution: what started as a trickle has become a flood, despite some daunting adverse effects; new drugs, indications, and combinations continue to emerge. Pharm. Ther. 2016, 41, 185–191. [Google Scholar]
- Barone, A.; Hazarika, M.; Theoret, M.R.; Mishra-Kalyani, P.; Chen, H.; He, K.; Sridhara, R.; Subramaniam, S.; Pfuma, E.; Wang, Y.; et al. FDA Approval Summary: Pembrolizumab for the Treatment of Patients with Unresectable or Metastatic Melanoma. Clin. Cancer Res. 2017, 23, 5661–5665. [Google Scholar] [CrossRef] [PubMed]
- Pai-Scherf, L.; Blumenthal, G.M.; Li, H.; Subramaniam, S.; Mishra-Kalyani, P.S.; He, K.; Zhao, H.; Yu, J.; Paciga, M.; Goldberg, K.B.; et al. FDA Approval Summary: Pembrolizumab for Treatment of Metastatic Non-Small Cell Lung Cancer: First-Line Therapy and Beyond. Oncologist 2017, 22, 1392–1399. [Google Scholar] [CrossRef] [PubMed]
- Lugowska, I.; Teterycz, P.; Rutkowski, P. Immunotherapy of melanoma. Contemp. Oncol. 2018, 22, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Zhao, Z.; Barber, B.; Farr, A.M.; Ivanov, B.; Novich, M. Overall survival in patients with metastatic melanoma. Curr. Med. Res. Opin. 2015, 31, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Callahan, M.K.; Kluger, H.; Postow, M.A.; Segal, N.H.; Lesokhin, A.; Atkins, M.B.; Kirkwood, J.M.; Krishnan, S.; Bhore, R.; Horak, C.; et al. Nivolumab Plus Ipilimumab in Patients with Advanced Melanoma: Updated Survival, Response, and Safety Data in a Phase I Dose-Escalation Study. J. Clin. Oncol. 2017, 36, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Conlan, J.W.; Krishnan, L.; Willick, G.E.; Patel, G.B.; Sprott, G.D. Immunization of mice with lipopeptide antigens encapsulated in novel liposomes prepared from the polar lipids of various Archaeobacteria elicits rapid and prolonged specific protective immunity against infection with the facultative intracellular pathogen, Listeria monocytogenes. Vaccine 2001, 19, 3509–3517. [Google Scholar]
- Krishnan, L.; Deschatelets, L.; Stark, F.C.; Gurnani, K.; Sprott, G.D. Archaeosome adjuvant overcomes tolerance to tumor-associated melanoma antigens inducing protective CD8 T cell responses. Clin. Dev. Immunol. 2010, 2010, 578432. [Google Scholar] [CrossRef]
- Landi, A.; Law, J.; Hockman, D.; Logan, M.; Crawford, K.; Chen, C.; Kundu, J.; Ebensen, T.; Guzman, C.A.; Deschatelets, L.; et al. Superior immunogenicity of HCV envelope glycoproteins when adjuvanted with cyclic-di-AMP, a STING activator or archaeosomes. Vaccine 2017, 35, 6949–6956. [Google Scholar] [CrossRef]
- Akache, B.; Stark, F.C.; Jia, Y.; Deschatelets, L.; Dudani, R.; Harrison, B.A.; Agbayani, G.; Williams, D.; Jamshidi, M.P.; Krishnan, L.; et al. Sulfated archaeol glycolipids: Comparison with other immunological adjuvants in mice. PLoS ONE 2018, 13, e0208067. [Google Scholar] [CrossRef]
- Stark, F.C.; Akache, B.; Ponce, A.; Dudani, R.; Deschatelets, L.; Jia, Y.; Sauvageau, J.; Williams, D.; Jamshidi, M.P.; Agbayani, G.; et al. Archaeal glycolipid adjuvanted vaccines induce strong influenza-specific immune responses through direct immunization in young and aged mice or through passive maternal immunization. Vaccine 2019, 37, 7108–7116. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Zubair, S.; Tufail, S.; Ahmad, E.; Khan, M.R.; Quadri, Z.; Owais, M. Ether lipid vesicle-based antigens impart protection against experimental listeriosis. Int. J. Nanomed. 2012, 7, 2433–2447. [Google Scholar]
- Higa, L.H.; Corral, R.S.; Morilla, M.J.; Romero, E.L.; Petray, P.B. Archaeosomes display immunoadjuvant potential for a vaccine against Chagas disease. Hum. Vaccin Immunother. 2013, 9, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Zubair, S.; Mahmood, A.; Gupta, P.; Khan, A.A.; Gupta, U.D.; Arora, A.; Owais, M. RD antigen based nanovaccine imparts long term protection by inducing memory response against experimental murine tuberculosis. PLoS ONE 2011, 6, e22889. [Google Scholar] [CrossRef] [PubMed]
- Stark, F.C.; Weeratna, R.D.; Deschatelets, L.; Gurnani, K.; Dudani, R.; McCluskie, M.J.; Krishnan, L. An Archaeosome-Adjuvanted Vaccine and Checkpoint Inhibitor Therapy Combination Significantly Enhances Protection from Murine Melanoma. Vaccines 2017, 5, 38. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, L.; Dicaire, C.J.; Patel, G.B.; Sprott, G.D. Archaeosome vaccine adjuvants induce strong humoral, cell-mediated, and memory responses: Comparison to conventional liposomes and alum. Infect. Immun. 2000, 68, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Sprott, G.D.; Patel, G.B.; Krishnan, L. Archaeobacterial ether lipid liposomes as vaccine adjuvants. Meth. Enzymol 2003, 373, 155–172. [Google Scholar]
- McCluskie, M.J.; Deschatelets, L.; Krishnan, L. Sulfated archaeal glycolipid archaeosomes as a safe and effective vaccine adjuvant for induction of cell-mediated immunity. Hum. Vaccin. Immunother. 2017, 13, 2772–2779. [Google Scholar] [CrossRef]
- Akache, B.; Stark, F.C.; Iqbal, U.; Chen, W.; Jia, Y.; Krishnan, L.; McCluskie, M.J. Safety and biodistribution of sulfated archaeal glycolipid archaeosomes as vaccine adjuvants. Hum. Vaccin. Immunother. 2018, 14, 1746–1759. [Google Scholar] [CrossRef]
- Brown, D.M.; Fisher, T.L.; Wei, C.; Frelinger, J.G.; Lord, E.M. Tumours can act as adjuvants for humoral immunity. Immunology 2001, 102, 486–497. [Google Scholar] [CrossRef]
- Dudani, R.; Chapdelaine, Y.; van Faassen, H.; Smith, D.K.; Shen, H.; Krishnan, L.; Sad, S. Multiple mechanisms compensate to enhance tumor-protective CD8+ T cell response in the long-term despite poor CD8(+) T cell priming initially: Comparison between an acute versus a chronic intracellular bacterium expressing a model antigen. J. Immunol. 2002, 168, 5737–5745. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, D.M.; Yu, S.H.; Dicaire, C.J.; Sprott, G.D. Development of new glycosylation methodologies for the synthesis of archaeal-derived glycolipid adjuvants. Carbohydr. Res. 2010, 345, 214–229. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, D.M.; Sprott, D.G.; Krishnan, L. Sulfated-Glycolipids as Adjuvants for Vaccines. WO2016004512A1, 14 January 2016. [Google Scholar]
- Krishnan, L.; Gurnani, K.; Dicaire, C.J.; van Faassen, H.; Zafer, A.; Kirschning, C.J.; Sad, S.; Sprott, G.D. Rapid clonal expansion and prolonged maintenance of memory CD8+ T cells of the effector (CD44highCD62Llow) and central (CD44highCD62Lhigh) phenotype by an archaeosome adjuvant independent of TLR2. J. Immunol. 2007, 178, 2396–2406. [Google Scholar] [CrossRef] [PubMed]
- Sprott, G.D.; Yeung, A.; Dicaire, C.J.; Yu, S.H.; Whitfield, D.M. Synthetic archaeosome vaccines containing triglycosylarchaeols can provide additive and long-lasting immune responses that are enhanced by archaetidylserine. Archaea 2012, 2012, 513231. [Google Scholar] [CrossRef]
- Jia, Y.; Akache, B.; Deschatelets, L.; Qian, H.; Dudani, R.; Harrison, B.A.; Stark, F.C.; Chandan, V.; Jamshidi, M.P.; Krishnan, L.; et al. A comparison of the immune responses induced by antigens in three different archaeosome-based vaccine formulations. Int. J. Pharm. 2019, 561, 187–196. [Google Scholar] [CrossRef]
- Stark, F.C.; McCluskie, M.J.; Krishnan, L. Homologous Prime-Boost Vaccination with OVA Entrapped in Self-Adjuvanting Archaeosomes Induces High Numbers of OVA-Specific CD8+ T Cells that Protect Against Subcutaneous B16-OVA Melanoma. Vaccines 2016, 4, 44. [Google Scholar] [CrossRef]
- Azimi, F.; Scolyer, R.A.; Rumcheva, P.; Moncrieff, M.; Murali, R.; McCarthy, S.W.; Saw, R.P.; Thompson, J.F. Tumor-infiltrating lymphocyte grade is an independent predictor of sentinel lymph node status and survival in patients with cutaneous melanoma. J. Clin. Oncol. 2012, 30, 2678–2683. [Google Scholar] [CrossRef]
- Hendry, S.; Salgado, R.; Gevaert, T.; Russell, P.A.; John, T.; Thapa, B.; Christie, M.; van de Vijver, K.; Estrada, M.V.; Gonzalez-Ericsson, P.I.; et al. Assessing Tumor-Infiltrating Lymphocytes in Solid Tumors: A Practical Review for Pathologists and Proposal for a Standardized Method from the International Immuno-Oncology Biomarkers Working Group: Part 2: TILs in Melanoma, Gastrointestinal Tract Carcinomas, Non-Small Cell Lung Carcinoma and Mesothelioma, Endometrial and Ovarian Carcinomas, Squamous Cell Carcinoma of the Head and Neck, Genitourinary Carcinomas, and Primary Brain Tumors. Adv. Anat. Pathol. 2017, 24, 311. [Google Scholar]
- Stark, F.; McCluskie, M. National Research Council of Canada, Ottawa, ON. In a repeat experiment, all treatment groups were repeated with the addition of a separate group that included anti-PD-L1 therapy. The addition of anti-PD-L1 did not further enhance survival in combination therapy with anti-PD-1, anti-CTLA-4 and the adjuvant SLA admixed with ovalbumin protein in a therapeutic B16-OVA melanoma model. 2019; unpublished work. [Google Scholar]
- Halse, H.; Colebatch, A.J.; Petrone, P.; Henderson, M.A.; Mills, J.K.; Snow, H.; Westwood, J.A.; Sandhu, S.; Raleigh, J.M.; Behren, A.; et al. Multiplex immunohistochemistry accurately defines the immune context of metastatic melanoma. Sci. Rep. 2018, 8, 11158. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer–immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Kyi, C.; Postow, M.A. Immune checkpoint inhibitor combinations in solid tumors: Opportunities and challenges. Immunotherapy 2016, 8, 821–837. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Immunotherapy Study for Patients with Stage IV Melanoma. Available online: https://clinicaltrials.gov/ct2/show/NCT02054520 (accessed on 23 July 2019).
- Pilot Study of VigilTM + Pembrolizumab for Advanced Melanoma. Available online: https://clinicaltrials.gov/ct2/show/NCT02574533 (accessed on 23 July 2019).
- A Study of RO7198457 as a Single Agent and in Combination with Atezolizumab in Participants with Locally Advanced or Metastatic Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT03289962 (accessed on 23 July 2019).
- A Personal Cancer Vaccine (NEO-PV-01) and APX005M or Ipilimumab with Nivolumab in Patients with Advanced Melanoma. Available online: https://clinicaltrials.gov/ct2/show/NCT03597282 (accessed on 23 July 2019).
- A Personal Cancer Vaccine (NEO-PV-01) w/ Nivolumab for Patients with Melanoma, Lung Cancer or Bladder Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT02897765 (accessed on 23 July 2019).
- A Study to Evaluate Safety, Feasibility, Efficacy of Multiple Dosing with VB10.NEO Immunotherapy in Patients with Locally Advanced or Metastatic Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03548467 (accessed on 23 July 2019).
- A Phase I/II Trial to Evaluate a Peptide Vaccine plus Ipilimumab in Patients with Melanoma. Available online: https://clinicaltrials.gov/ct2/show/NCT02385669 (accessed on 23 July 2019).
- Safety, Tolerability, Immunogenicity, and Antitumor Activity of GEN-009 Adjuvanted Vaccine. Available online: https://clinicaltrials.gov/ct2/show/NCT03633110 (accessed on 23 July 2019).
- A Phase 1/2 Study of in Situ Vaccination with Tremelimumab and IV Durvalumab plus PolyICLC in Subjects with Advanced, Measurable, Biopsy-Accessible Cancers. Available online: https://clinicaltrials.gov/ct2/show/NCT02643303 (accessed on 23 July 2019).
- Hu-Lieskovan, S.; Ribas, A. New combination strategies using PD-1/ L1 checkpoint inhibitors as a backbone. Cancer J. 2017, 23, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Haq, K.; Jia, Y.; Krishnan, L. Archaeal lipid vaccine adjuvants for induction of cell-mediated immunity. Expert Rev. Vaccines 2016, 15, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [Green Version]
- Five-Year Outcomes for Opdivo (nivolumab) in Combination with Yervoy (ipilimumab) Demonstrate Durable Long-Term Survival Benefits in Patients with Advanced Melanoma | BMS Newsroom. Available online: https://news.bms.com/press-release/corporatefinancial-news/five-year-outcomes-opdivo-nivolumab-combination-yervoy-ipilimu (accessed on 6 November 2019).
- Wei, S.C.; Anang, N.-A.A.S.; Sharma, R.; Andrews, M.C.; Reuben, A.; Levine, J.H.; Cogdill, A.P.; Mancuso, J.J.; Wargo, J.A.; Pe’er, D.; et al. Combination anti-CTLA-4 plus anti-PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies. Proc. Natl. Acad. Sci. USA 2019, 116, 22699–22709. [Google Scholar] [CrossRef] [Green Version]
- Boutros, C.; Tarhini, A.; Routier, E.; Lambotte, O.; Ladurie, F.L.; Carbonnel, F.; Izzeddine, H.; Marabelle, A.; Champiat, S.; Berdelou, A.; et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat. Rev. Clin. Oncol. 2016, 13, 473–486. [Google Scholar] [CrossRef]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.-L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti–PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [Green Version]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.M.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, J.; Chehrazi-Raffle, A.; Placencio-Hickok, V.; Guan, M.; Hendifar, A.; Salgia, R. The gut microbiome and response to immune checkpoint inhibitors: Preclinical and clinical strategies. Clin. Transl. Med. 2019, 8, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunnhoelzl, D.; Wang, J. Acute tumor lysis syndrome after anti-pd-1 immunotherapy nivolumab for metastatic melanoma. J. Mol. Oncol. Res. 2017, 1, 5–6. [Google Scholar] [CrossRef]
- Brunnhoelzl, D.; Weed, M.; Trepet, R.; Wang, J. Tumor Lysis Syndrome Following a Single Atezolizumab Infusion for Metastatic Urothelial Carcinoma Involving Both Upper and Lower Tract. Arch. Can. Res. 2017, 5. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Frequency of OVA-CD8+ T Cells of All CD8+ T Cells in the Spleen and Tumor at the Humane Endpoint | ||||
|---|---|---|---|---|
| Spleen | Tumour | |||
| Geometric Mean | %95 CI (Lower + Upper) | Geometric Mean | %95 CI (Lower + Upper) | |
| SLA-OVA (adm) | 1.13 | 0.38 and 1.79 | 0.55 | 0.23 and 1.35 |
| Anti- CTLA4 + anti-PD-1 | 0.17 | −0.03 and 0.56 | 0.56 | 0.17 and 3.12 |
| SLA-OVA (adm) + anti-CTLA4 + anti-PD-1 | 0.27 | 0.14 and 1.38 | 1.85 | 0.18 and 5.50 |
| SLA-OVA (enc) + anti-CTLA4 + anti-PD-1 | 1.49 | 0.44 and 3.07 | 8.02 | 0.41 and 21.97 |
| MS-OVA (enc) + anti-CTLA4 + anti-PD-1 | 1.27 | 0.55 and 1.37 | 1.74 | 0.67 and 3.04 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stark, F.C.; Agbayani, G.; Sandhu, J.K.; Akache, B.; McPherson, C.; Deschatelets, L.; Dudani, R.; Hewitt, M.; Jia, Y.; Krishnan, L.; et al. Simplified Admix Archaeal Glycolipid Adjuvanted Vaccine and Checkpoint Inhibitor Therapy Combination Enhances Protection from Murine Melanoma. Biomedicines 2019, 7, 91. https://doi.org/10.3390/biomedicines7040091
Stark FC, Agbayani G, Sandhu JK, Akache B, McPherson C, Deschatelets L, Dudani R, Hewitt M, Jia Y, Krishnan L, et al. Simplified Admix Archaeal Glycolipid Adjuvanted Vaccine and Checkpoint Inhibitor Therapy Combination Enhances Protection from Murine Melanoma. Biomedicines. 2019; 7(4):91. https://doi.org/10.3390/biomedicines7040091
Chicago/Turabian StyleStark, Felicity C., Gerard Agbayani, Jagdeep K. Sandhu, Bassel Akache, Charis McPherson, Lise Deschatelets, Renu Dudani, Melissa Hewitt, Yimei Jia, Lakshmi Krishnan, and et al. 2019. "Simplified Admix Archaeal Glycolipid Adjuvanted Vaccine and Checkpoint Inhibitor Therapy Combination Enhances Protection from Murine Melanoma" Biomedicines 7, no. 4: 91. https://doi.org/10.3390/biomedicines7040091
APA StyleStark, F. C., Agbayani, G., Sandhu, J. K., Akache, B., McPherson, C., Deschatelets, L., Dudani, R., Hewitt, M., Jia, Y., Krishnan, L., & McCluskie, M. J. (2019). Simplified Admix Archaeal Glycolipid Adjuvanted Vaccine and Checkpoint Inhibitor Therapy Combination Enhances Protection from Murine Melanoma. Biomedicines, 7(4), 91. https://doi.org/10.3390/biomedicines7040091