Oncostatin M in the Regulation of Connective Tissue Cells and Macrophages in Pulmonary Disease



{kind=link}
Abstract
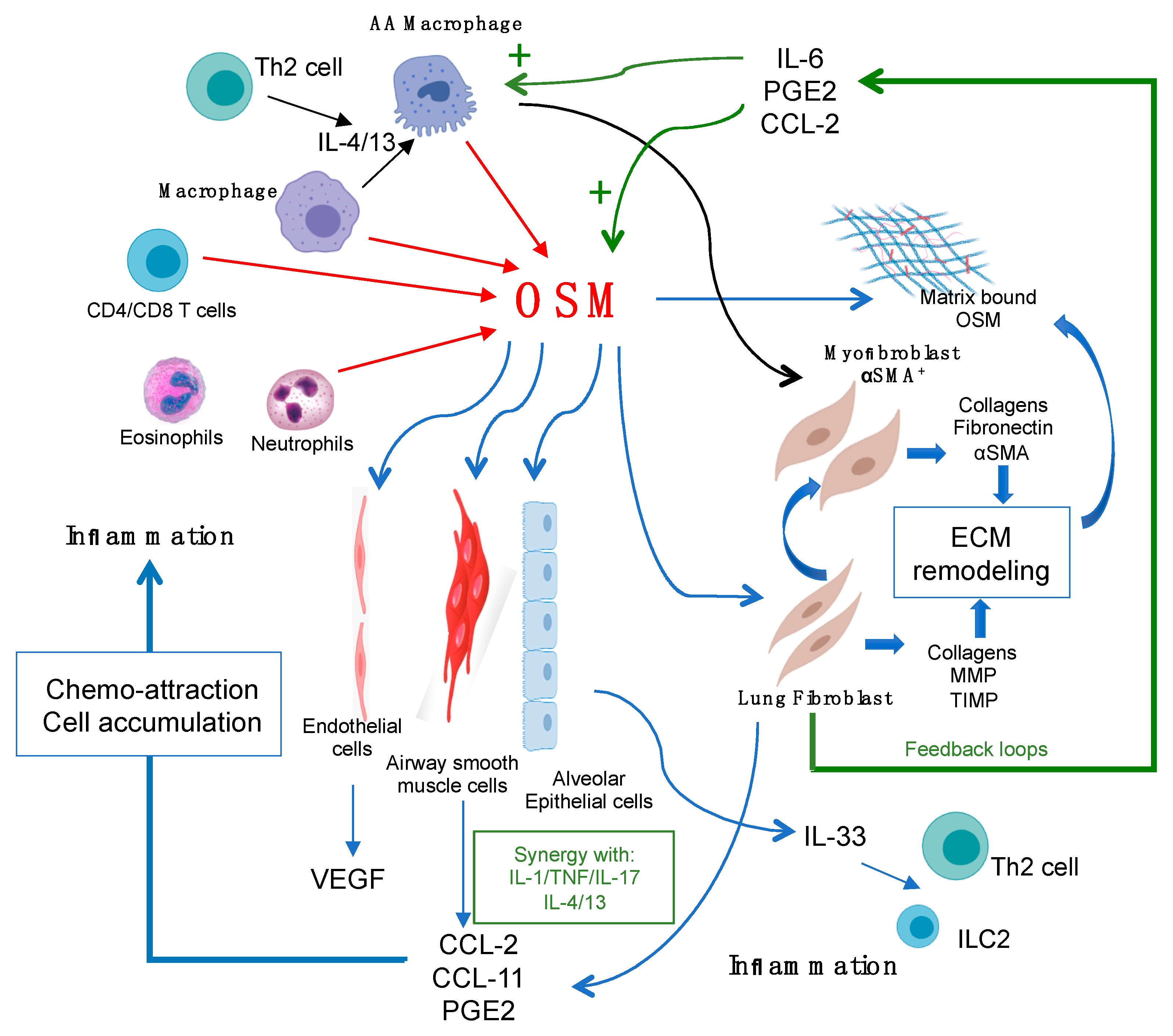
:1. Introduction
2. Oncostatin M Receptors and Cell Responses
3. Extracellular Matrix Remodeling
3.1. Transforming Growth Factor and Oncostatin M in Extracellular Matrix Remodeling In Vivo
3.2. Oncostatin M Regulation of Connective Tissue Cells In Vitro
4. Regulation of Inflammatory Mediators in Lung Connective Tissue Cells
4.1. Chemokines
4.2. Interleukin-6
4.3. Vascular Endothelial Growth Factor (VEGF) and Prostaglandin E
4.4. Lung Epithelial Cells and IL-33
5. Sources of Oncostatin M Ligands and Macrophages
Other Sources of Oncostatin M
6. Feedback Loops between Connective Tissue Cells and Macrophages
7. Targeting the Oncostatin M Pathway as a Potential Therapeutic
8. Conclusions
Funding
Conflicts of Interest
References
- Jones, S.A.; Jenkins, B.J. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat. Rev. Immunol. 2018, 18, 773–789. [Google Scholar] [CrossRef] [PubMed]
- Silver, J.S.; Hunter, C.A. Gp130 at the nexus of inflammation, autoimmunity, and cancer. J. Leukoc. Biol. 2010, 88, 1145–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaper, F.; Rose-John, S. Interleukin-6: Biology, signaling and strategies of blockade. Cytokine Growth Factor Rev. 2015, 26, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Wang, Y.; Zhou, G.; Wang, Y.; Li, X. Review: The Roles and Mechanisms of Glycoprotein 130 Cytokines in the Regulation of Adipocyte Biological Function. Inflammation 2019, 42, 790–798. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Miyahima, A. Onconstatin M, A Multifunctional Cytokine BT-Reviews of Physiology, Biochemistry and Pharmacology; Springer: Berlin/Heidelberg, Germany, 2004; pp. 39–52. [Google Scholar]
- Hermanns, H.M. Oncostatin M and interleukin-31: Cytokines, receptors, signal transduction and physiology. Cytokine Growth Factor Rev. 2015, 26, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Richards, C.D. The enigmatic cytokine oncostatin m and roles in disease. ISRN Inflamm. 2013, 2013, 512103. [Google Scholar] [CrossRef] [Green Version]
- West, N.R.; Owens, B.M.J.; Hegazy, A.N. The oncostatin M-stromal cell axis in health and disease. Scand. J. Immunol. 2018, 88, 1–18. [Google Scholar] [CrossRef]
- West, N.R. Coordination of immune-stroma crosstalk by IL-6 family cytokines. Front. Immunol. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Houben, E.; Hellings, N.; Broux, B. Oncostatin M, an Underestimated Player in the Central Nervous System. Front. Immunol. 2019, 10, 1165. [Google Scholar] [CrossRef]
- Sims, N.A.; Quinn, J.M.W. Osteoimmunology: Oncostatin M as a pleiotropic regulator of bone formation and resorption in health and disease. Bonekey Rep. 2014, 3, 527. [Google Scholar] [CrossRef]
- Mozaffarian, A.W.A.; Brewer, E.S.; Trueblood, I.G.; Luzina, N.W.; Todd, S.P.; Atamas, H.; Arnett, A. Mechanisms of Oncostatin M-Induced Pulmonary Inflammation and Fibrosis. J. Immunol. 2008, 181, 7243–7253. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.L.; Baines, K.J.; Boyle, M.J.; Scott, R.J.; Gibson, P.G. Oncostatin m (osm) is increased in asthma with incompletely reversible airflow obstruction. Exp. Lung Res. 2009, 35, 781–794. [Google Scholar] [CrossRef] [PubMed]
- Pothoven, K.L.; Norton, J.E.; Hulse, K.E.; Suh, L.A.; Carter, R.G.; Rocci, E.; Harris, K.E.; Shintani-Smith, S.; Conley, D.B.; Chandra, R.K.; et al. Oncostatin M promotes mucosal epithelial barrier dysfunction, and its expression is increased in patients with eosinophilic mucosal disease. J. Allergy Clin. Immunol. 2015, 136, 737–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baines, K.J.; Simpson, J.L.; Gibson, P.G. Innate immune responses are increased in chronic obstructive pulmonary disease. PLoS ONE 2011, 6, e18426. [Google Scholar] [CrossRef]
- Langdon, C.; Kerr, C.; Hassen, M.; Hara, T.; Arsenault, A.L.; Richards, C.D. Murine oncostatin M stimulates mouse synovial fibroblasts in vitro and induces inflammation and destruction in mouse joints in vivo. Am. J. Pathol. 2000, 157, 1187–1196. [Google Scholar] [CrossRef]
- Fritz, D.K.; Kerr, C.; Fattouh, R.; Llop-Guevara, A.; Khan, W.I.; Jordana, M.; Richards, C.D. A Mouse Model of Airway Disease: Oncostatin M-Induced Pulmonary Eosinophilia, Goblet Cell Hyperplasia, and Airway Hyperresponsiveness Are STAT6 Dependent, and Interstitial Pulmonary Fibrosis Is STAT6 Independent. J. Immunol. 2011, 186, 1107–1118. [Google Scholar] [CrossRef]
- Wong, S.; Botelho, F.M.; Rodrigues, R.M.; Richards, C.D. Oncostatin M overexpression induces matrix deposition, STAT3 activation, and SMAD1 dysregulation in lungs of fibrosis-resistant BALB/c mice. Lab. Investig. 2014, 94, 1003–1016. [Google Scholar] [CrossRef] [Green Version]
- Pohin, M.; Guesdon, W.; Mekouo, A.A.; Rabeony, H.; Paris, I.; Atanassov, H.; Favot, L.; Mcheik, J.; Bernard, F.X.; Richards, C.D.; et al. Oncostatin M overexpression induces skin inflammation but is not required in the mouse model of imiquimod-induced psoriasis-like inflammation. Eur. J. Immunol. 2016, 46, 1737–1751. [Google Scholar] [CrossRef]
- Vincenti, M.P. The Matrix Metalloproteinase (MMP) and Tissue Inhibitor of Metalloproteinase (TIMP) Genes. In Matrix Metalloproteinase Protocols; Clark, I.M., Ed.; Humana Press: Totowa, NJ, USA, 2001; pp. 121–148. [Google Scholar]
- White, J.M. ADAMs: Modulators of cell-cell and cell-matrix interactions. Curr. Opin. Cell Biol. 2003, 15, 598–606. [Google Scholar] [CrossRef]
- Willis, B.C.; Liebler, J.M.; Luby-Phelps, K.; Nicholson, A.G.; Crandall, E.D.; Bois, R.M.; Borok, Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: Potential role in idiopathic pulmonary fibrosis. Am. J. Pathol. 2005, 166, 1321–1332. [Google Scholar] [CrossRef]
- Chapman, H.A. Epithelial-Mesenchymal Interactions in Pulmonary Fibrosis. Annu. Rev. Physiol. 2011, 73, 413–435. [Google Scholar] [CrossRef] [PubMed]
- Sime, P.J.; Xing, Z.; Graham, F.L.; Csaky, K.G.; Gauldie, J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J. Clin. Invest. 1997, 100, 768–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sime, P.J.; O’reilly, K.M.A. Fibrosis of the lung and other tissues: New concepts in pathogenesis and treatment. Clin. Immunol. 2001, 99, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and Safety of Nintedanib in Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [Green Version]
- Wollin, L.; Wex, E.; Pautsch, A.; Schnapp, G.; Hostettler, K.E.; Stowasser, S.; Kolb, M. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1434–1445. [Google Scholar] [CrossRef]
- Lancaster, L.; Albera, C.; Bradford, W.Z.; Costabel, U.; Du Bois, R.M.; Fagan, E.A.; Fishman, R.S.; Glaspole, I.; Glassberg, M.K.; King, T.E.; et al. Safety of pirfenidone in patients with idiopathic pulmonary fibrosis: Integrated analysis of cumulative data from 5 clinical trials. BMJ Open Respir. Res. 2016, 3, e000105. [Google Scholar] [CrossRef] [Green Version]
- Fattouh, R.; Midence, N.G.; Arias, K.; Johnson, J.R.; Walker, T.D.; Goncharova, S.; Souza, K.P.; Gregory, R.C., Jr.; Lonning, S.; Gauldie, J.; et al. Transforming Growth Factor-β Regulates House Dust Mite–induced Allergic Airway Inflammation but Not Airway Remodeling. Am. J. Respir. Crit. Care Med. 2008, 177, 593–603. [Google Scholar] [CrossRef]
- O’Donoghue, R.J.; Knight, D.A.; Richards, C.D.; Prêle, C.M.; Lau, H.L.; Jarnicki, A.G.; Jones, J.; Bozinovski, S.; Vlahos, R.; Thiem, S.; et al. Genetic partitioning of interleukin-6 signalling in mice dissociates Stat3 from Smad3-mediated lung fibrosis. EMBO Mol. Med. 2012, 4, 939–951. [Google Scholar] [CrossRef]
- Eivers, E.; Demagny, H.; de Robertis, E.M. Integration of BMP and Wnt signaling via vertebrate Smad1/5/8 and Drosophila Mad. Cytokine Growth Factor Rev. 2009, 20, 357–365. [Google Scholar] [CrossRef] [Green Version]
- Myllärniemi, M.; Lindholm, P.; Ryynänen, M.J.; Kliment, C.R.; Salmenkivi, K.; Keski-Oja, J.; Kinnula, V.L.; Oury, T.D.; Koli, K. Gremlin-mediated decrease in bone morphogenetic protein signaling promotes pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008, 177, 321–329. [Google Scholar] [CrossRef]
- Duncan, M.R.; Hasan, A.; Berman, B. Oncostatin M stimulates collagen and glycosaminoglycan production by cultured normal dermal fibroblasts: Insensitivity of sclerodermal and keloidal fibroblasts. J. Invest. Dermatol. 1995, 104, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, A.K.; Mutsaers, S.E.; Moodley, Y.P.; McAnulty, R.J.; Laurent, G.J.; Thompson, P.J.; Knight, D.A. Oncostatin M stimulates proliferation, induces collagen production and inhibits apoptosis of human lung fibroblasts. Br. J. Pharmacol. 2002, 136, 793–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, C.D.; Shoyab, M.; Brown, T.J.; Gauldie, J. Selective regulation of metalloproteinase inhibitor (TIMP-1) by oncostatin M in fibroblasts in culture. J. Immunol. 1993, 150, 5596–5603. [Google Scholar] [PubMed]
- O’Kane, C.M.; Elkington, P.T.; Friedland, J.S. Monocyte-dependent oncostatin M and TNF-α synergize to stimulate unopposed matrix metalloproteinase-1/3 secretion from human lung fibroblasts in tuberculosis. Eur. J. Immunol. 2008, 38, 1321–1330. [Google Scholar] [CrossRef]
- Nagahama, K.Y.; Togo, S.; Holz, O.; Magnussen, H.; Liu, X.; Seyama, K.; Takahashi, K.; Rennard, S.I. Oncostatin M modulates fibroblast function via signal transducers and activators of transcription proteins-3. Am. J. Respir. Cell Mol. Biol. 2013, 49, 582–591. [Google Scholar] [CrossRef]
- Cohen, M.; Marchand-Adam, S.; Lecon-Malas, V.; Marchal-Somme, J.; Boutten, A.; Durand, G.; Crestani, B.; Dehoux, M. HGF synthesis in human lung fibroblasts is regulated by oncostatin M. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, 1097–1103. [Google Scholar] [CrossRef] [Green Version]
- Dally, J.; Khan, J.S.; Voisey, A.; Charalambous, C.; John, H.L.; Woods, E.L.; Steadman, R.; Moseley, R.; Midgley, A.C. Hepatocyte growth factor mediates enhanced wound healing responses and resistance to transforming growth factor-β1-driven myofibroblast differentiation in oral mucosal fibroblasts. Int. J. Mol. Sci. 2017, 18, 1843. [Google Scholar] [CrossRef]
- Fritz, D.K.; Kerr, C.; Botelho, F.; Stampfli, M.; Richards, C.D. Oncostatin M (OSM) primes IL-13- and IL-4-induced eotaxin responses in fibroblasts: Regulation of the type-II IL-4 receptor chains IL-4Rα and IL-13Rα1. Exp. Cell Res. 2009, 315, 3486–3499. [Google Scholar] [CrossRef]
- Fritz, D.K.; Kerr, C.; Tong, L.; Smyth, D.; Richards, C.D. Oncostatin-M up-regulates VCAM-1 and synergizes with IL-4 in eotaxin expression: involvement of STAT6. J. Immunol. 2006, 176, 4352–4360. [Google Scholar] [CrossRef] [Green Version]
- Waters, D.W.; Blokland, K.E.C.; Pathinayake, P.S.; Burgess, J.K.; Mutsaers, S.E.; Prele, C.M.; Schuliga, M.; Grainge, C.L.; Knight, D.A. Fibroblast senescence in the pathology of idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 315, L162–L172. [Google Scholar] [CrossRef] [Green Version]
- Waters, D.W.; Blokland, K.E.; Pathinayake, P.S.; Wei, L.; Schuliga, M.; Jaffar, J.; Westall, G.P.; Hansbro, P.M.; Prele, C.M.; Mutsaers, S.E.; et al. STAT3 regulates the onset of oxidant-induced senescence in lung fibroblasts. Am. J. Respir. Cell Mol. Biol. 2019, 61, 61–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huguier, V.; Giot, J.P.; Simonneau, M.; Levillain, P.; Charreau, S.; Garcia, M.; Jégou, J.F.; Bodet, C.; Morel, F.; Lecron, J.C.; et al. Oncostatin M exerts a protective effect against excessive scarring by counteracting the inductive effect of TGFβ1 on fibrosis markers. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.J.; Rowe, J.M.; Liu, J.W.; Shoyab, M. Regulation of IL-6 expression by oncostatin M. J. Immunol. 2017, 147, 2175–2180. [Google Scholar]
- Lieschke, G.J.; Burgess, A.W. Granulocyte Colony-Stimulating Factor and Granulocyte-Macrophage Colony-Stimulating Factor Expression by Oncostatin M. Blood 1993, 82, 33–38. [Google Scholar]
- Richards, C.D.; Brown, T.J.; Shoyab, M.; Baumann, H.; Gauldie, J. Recombinant oncostatin M stimulates the production of acute phase proteins in HepG2 cells and rat primary hepatocytes in vitro. J. Immunol. 1992, 148, 1731–1736. [Google Scholar]
- Langdon, C.; Kerr, C.; Tong, L.; Richards, C.D. Oncostatin M Regulates Eotaxin Expression in Fibroblasts and Eosinophilic Inflammation in C57BL/6 Mice. J. Immunol. 2003, 170, 548–555. [Google Scholar] [CrossRef]
- Kwofie, K.; Scott, M.; Rodrigues, R.; Guerette, J.; Radford, K.; Nair, P.; Richards, C.D. Regulation of IL-17A responses in human airway smooth muscle cells by Oncostatin M. Respir. Res. 2015, 16, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Traber, K.E.; Hilliard, K.L.; Allen, E.; Wasserman, G.A.; Yamamoto, K.; Jones, M.R.; Mizgerd, J.P.; Quinton, L.J. Induction of STAT3-dependent CXCL5 expression and neutrophil recruitment by oncostatin-M during pneumonia. Am. J. Respir. Cell Mol. Biol. 2015, 53, 479–488. [Google Scholar] [CrossRef]
- Faffe, D.S.; Flynt, L.; Mellema, M.; Moore, P.E.; Silverman, E.S.; Subramaniam, V.; Jones, M.R.; Mizgerd, J.P.; Whitehead, T.; Imrich, A.; et al. Oncostatin M causes eotaxin-1 release from airway smooth muscle: Synergy with IL-4 and IL-13. J. Allergy Clin. Immunol. 2005, 115, 514–520. [Google Scholar] [CrossRef]
- Richards, C.D.; Langdon, C.; Botelho, F.; Brown, T.J.; Agro, A. Oncostatin M inhibits IL-1-induced expression of IL-8 and granulocyte-macrophage colony-stimulating factor by synovial and lung fibroblasts. J. Immunol. 1996, 156, 343–349. [Google Scholar]
- Botelho, F.M.; Rangel-Moreno, J.; Fritz, D.; Randall, T.D.; Xing, Z.; Richards, C.D. Pulmonary Expression of Oncostatin M (OSM) Promotes Inducible BALT Formation Independently of IL-6, Despite a Role for IL-6 in OSM-Driven Pulmonary Inflammation. J. Immunol. 2013, 191, 1453–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubey, A.; Izakelian, L.; Ayaub, E.A.; Ho, L.; Stephenson, K.; Wong, S.; Kwofie, K.; Austin, R.C.; Botelho, F.; Ask, K.; et al. Separate roles of IL-6 and oncostatin M in mouse macrophage polarization in vitro and in vivo. Immunol. Cell Biol. 2018, 96, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Ayaub, E.A.; Dubey, A.; Imani, J.; Botelho, F.; Kolb, M.R.J.; Richards, C.D.; Ask, K. Overexpression of OSM and IL-6 impacts the polarization of pro-fibrotic macrophages and the development of bleomycin-induced lung fibrosis. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauer, J.; Chaurasia, B.; Goldau, J.; Vogt, M.C.; Ruud, J.; Nguyen, K.D.; Theurich, S.; Hausen, A.C.; Schmitz, J.; Brönneke, H.S.; et al. Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat. Immunol. 2014, 15, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Heusinkveld, M.; van Steenwijk, P.J.; Goedemans, R.; Ramwadhdoebe, T.H.; Gorter, A.; Welters, M.J.; van Hall, T.; van der Burg, S.H. M2 Macrophages Induced by Prostaglandin E 2 and IL-6 from Cervical Carcinoma Are Switched to Activated M1 Macrophages by CD4 + Th1 Cells. J. Immunol. 2011, 187, 1157–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasse, M.; Pourtau, J.; Trochon, V.; Muraine, M.; Vannier, J.P.; Lu, H.; Soria, J.; Soria, C. Oncostatin M Induces Angiogenesis In Vitro and In Vivo. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1835–1842. [Google Scholar] [CrossRef] [Green Version]
- Modur, V.J.M.; Feldhaus, A.S.; Weyrich, D.L.; Jicha, S.M.; Prescott, G.A.Z.; McIntyre, T.M. Oncostatin M is a proinflammatory mediator. In vivo effects correlate with endothelial cell expression of inflammatory cytokines and adhesion molecules. J. Clin. Invest. 1997, 100, 158–168. [Google Scholar] [CrossRef] [Green Version]
- van Keulen, D.G.M.; Pouwer, G.; Pasterkamp, A.J.v.; Tempel, D. Inflammatory cytokine oncostatin M induces endothelial activation in macro-and microvascular endothelial cells and in APOE*3Leiden.CETP mice. PLoS ONE 2018, 13, e0204911. [Google Scholar] [CrossRef]
- Faffe, D.S.L.; Flynt, M.; Mellema, T.R.; Whitehead, K.; Bourgeois, R.A.; Panettieri, E.S. Silverman, and S. A. Shore. Oncostatin M causes VEGF release from human airway smooth muscle: Synergy with IL-1β. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 288, 1040–1048. [Google Scholar] [CrossRef] [Green Version]
- Bernard, C.; Merval, R.; Lebret, M.; Delerive, P.; Dusanter-Fourt, I.; Lehoux, S.; Créminon, C.; Staels, B.; Maclouf, J.; Tedgui, A. Oncostatin M Induces Interleukin-6 and Cyclooxygenase-2 Expression in Human Vascular Smooth Muscle Cells. Circ. Res. 1999, 85, 1124–1131. [Google Scholar] [CrossRef] [Green Version]
- Barksby, H.E.; Hui, W.; Wappler, I.; Peters, H.H.; Milner, J.M.; Richards, C.D.; Cawston, T.E.; Rowan, A.D. Interleukin-1 in combination with oncostatin M up-regulates multiple genes in chondrocytes: Implications for cartilage destruction and repair. Arthritis Rheum. 2006, 54, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Cawston, T.E.; Curry, V.A.; Summers, C.A.; Clark, I.M.; Riley, G.P.; Life, P.F.; Spaull, J.R.; Goldring, M.B.; Koshy, P.J.; Rowan, A.D. The role of oncostatin M in animal and human connective tissue collagen turnover and its localization within the rheumatoid joint. Arthritis Rheum. 1998, 41, 1760–1771. [Google Scholar] [CrossRef]
- Rowan, A.D.; Hui, W.; Cawston, T.E.; Richards, C.D. Adenoviral gene transfer of interleukin-1 in combination with oncostatin M induces significant joint damage in a murine model. Am. J. Pathol. 2003, 162, 1975–1984. [Google Scholar] [CrossRef] [Green Version]
- Cichy, J.; Rose-John, S.; Travis, J. Oncostatin M, leukaemia-inhibitory factor and interleukin 6 trigger different effects on α1-proteinase inhibitor synthesis in human lung-derived epithelial cells. Biochem. J. 1998, 329, 335–339. [Google Scholar] [CrossRef] [Green Version]
- Sallenave, J.M.; Tremblay, M.G.; Gauldie, J.; Richards, C.D. Oncostatin M, but not interleukin-6 or leukemia inhibitory factor, stimulates expression of alpha1-proteinase inhibitor in A549 human alveolar epithelial cells. J. Interferon Cytokine Res. 1997, 17, 337–346. [Google Scholar] [CrossRef]
- Richards, C.D.; Izakelian, L.; Dubey, A.; Zhang, G.; Wong, S.; Kwofie, K.; Qureshi, A.; Botelho, F. Regulation of IL-33 by Oncostatin M in Mouse Lung Epithelial Cells. Mediators Inflamm. 2016, 11, 1–12. [Google Scholar] [CrossRef]
- Arshad, M.I.; Guihard, P.; Danger, Y.; Noel, G.; Le Seyec, J.; Boutet, M.A.; Richards, C.D.; L’Helgoualc’h, A.; Genet, V.; Lucas-Clerc, C.; et al. Oncostatin M induces IL-33 expression in liver endothelial cells in mice and expands ST2+CD4+ lymphocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G542–G553. [Google Scholar] [CrossRef]
- Beigel, F.; Friedrich, M.; Probst, C.; Sotlar, K.; Göke, B.; Diegelmann, J.; Brand, S. Oncostatin M mediates STAT3-dependent intestinal epithelial restitution via increased cell proliferation, decreased apoptosis and upregulation of SERPIN family members. PLoS ONE 2014, 9, e93498. [Google Scholar] [CrossRef]
- Pothoven, K.L.; Schleimer, R.P. The barrier hypothesis and Oncostatin M: Restoration of epithelial barrier function as a novel therapeutic strategy for the treatment of type 2 inflammatory disease. Tissue Barriers 2017, 5, 1–18. [Google Scholar] [CrossRef]
- Liew, F.Y.; Pitman, N.I.; McInnes, I.B. Disease-associated functions of IL-33: The new kid in the IL-1 family. Nat. Rev. Immunol. 2010, 10, 103–110. [Google Scholar] [CrossRef]
- Cayrol, C.; Girard, J.P. Interleukin-33 (IL-33): A nuclear cytokine from the IL-1 family. Immunol. Rev. 2018, 281, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.T.; Martin, M.U. Interleukin 33 is a guardian of barriers and a local alarmin. Nat. Immunol. 2016, 17, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.S.; Scott, I.C.; Majithiya, J.B.; Rapley, L.; Kemp, B.P.; England, E.; Rees, D.G.; Overed-Sayer, C.L.; Woods, J.; Bond, N.J.; et al. Oxidation of the alarmin IL-33 regulates ST2-dependent inflammation. Nat. Commun. 2015, 6, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hams, E.; Fallon, P.G. Innate type 2 cells and asthma. Curr. Opin. Pharmacol. 2012, 12, 503–509. [Google Scholar] [CrossRef]
- Nakae, S.; Morita, H.; Ohno, T.; Arae, K.; Matsumoto, K.; Saito, H. Role of Interleukin-33 in Innate-Type immune cells in allergy. Allergol. Int. 2013, 62, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Luzina, I.G.; Kopach, P.; Lockatell, V.; Kang, P.H.; Nagarsekar, A.; Burke, A.P.; Hasday, J.D.; Todd, N.W.; Atamas, S.P. Interleukin-33 potentiates Bleomycin-induced lung injury. Am. J. Respir. Cell Mol. Biol. 2013, 49, 999–1008. [Google Scholar] [CrossRef]
- Li, D.; Guabiraba, R.; Besnard, A.G.; Komai-Koma, M.; Jabir, M.S.; Zhang, L.; Graham, G.J.; Kurowska-Stolarska, M.; Liew, F.Y.; McSharry, C.; et al. IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J. Allergy Clin. Immunol. 2014, 134, 1422–1432.e11. [Google Scholar] [CrossRef]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [Green Version]
- Schupp, J.C.; Binder, H.; Jäger, B.; Cillis, G.; Zissel, G.; Müller-Quernheim, J.; Prasse, A. Macrophage activation in acute exacerbation of idiopathic pulmonary fibrosis. PLoS ONE 2015, 10, e0116775. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.C.; Zou, X.B.; Chai, Y.F.; Yao, Y.M. Macrophage polarization in inflammatory diseases. Int. J. Biol. Sci. 2014, 10, 520–529. [Google Scholar] [CrossRef]
- Guihard, P.; Danger, Y.; Brounais, B.; David, E.; Brion, R.; Delecrin, J.; Richards, C.D.; Chevalier, S.; Rédini, F.; Heymann, D.; et al. Induction of osteogenesis in mesenchymal stem cells by activated monocytes/macrophages depends on oncostatin M signaling. Stem Cells 2012, 30, 762–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suda, T.; Chida, K.; Todate, A.; Ide, K.; Asada, K.; Nakamura, Y.; Suzuki, K.; Kuwata, H.; Nakamura, H. Oncostatin M production by human dendritic cells in response to bacterial products. Cytokine 2002, 17, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Torossian, F.; Guerton, B.; Anginot, A.; Alexander, K.A.; Desterke, C.; Soave, S.; Tseng, H.W.; Arouche, N.; Boutin, L.; Kulina, I.; et al. Macrophage-derived oncostatin M contributes to human and mouse neurogenic heterotopic ossifications. JCI Insight 2017, 2, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Repovic, P.; Benveniste, E.N. Prostaglandin E2 is a novel inducer of oncostatin-M expression in macrophages and microglia. J. Neurosci. 2002, 22, 5334–5343. [Google Scholar] [CrossRef] [Green Version]
- Henkel, J.; Gärtner, D.; Dorn, C.; Hellerbrand, C.; Schanze, N.; Elz, S.R.; Püschel, G.P. Oncostatin M produced in Kupffer cells in response to PGE2: Possible contributor to hepatic insulin resistance and steatosis. Lab. Investig. 2011, 91, 1107–1117. [Google Scholar] [CrossRef]
- Kastl, S.P.; Speidl, W.S.; Kaun, C.; Katsaros, K.M.; Rega, G.; Afonyushkin, T.; Bochkov, V.N.; Valent, P.; Assadian, A.; Hagmueller, G.W.; et al. In human macrophages the complement component C5a induces the expression of oncostatin M via AP-1 activation. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 498–503. [Google Scholar] [CrossRef]
- Kastl, S.P.; Seidl, W.S.; Katsaros, K.M.; Kaun, C.; Rega, G.; Assadian, A.; Hagmueller, G.W.; Hoeth, M.; de Martin, R.; Ma, Y.; et al. Thrombin induces the expression of oncostatin M via AP-1 activation in human macrophages: A link between coagulation and inflammation. Blood 2009, 114, 2812–2818. [Google Scholar] [CrossRef]
- Misharin, A.V.; Nebreda, L.M.; Reyfman, P.A.; Cuda, C.M.; Walter, J.M.; Pimentel, A.C.M.; Chen, C.I.; Anekalla, K.R.; Joshi, N.; Williams, K.J.N.; et al. 2017. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 2017, 214, 2387–2404. [Google Scholar] [CrossRef] [Green Version]
- Hendriks, J.J.A.; Slaets, H.; Carmans, S.; de Vries, H.E.; Dijkstra, C.D.; Stinissen, P.; Hellings, N. Leukemia inhibitory factor modulates production of inflammatory mediators and myelin phagocytosis by macrophages. J. Neuroimmunol. 2008, 204, 52–57. [Google Scholar] [CrossRef]
- Biswas, S.K.; Sica, A.; Lewis, C.E. Plasticity of Macrophage Function during Tumor Progression: Regulation by Distinct Molecular Mechanisms. J. Immunol. 2008, 180, 2011–2017. [Google Scholar] [CrossRef]
- Tripathi, C.; Tewari, B.N.; Kanchan, R.K.; Baghel, K.S.; Nautiyal, N.; Shrivastava, R.; Kaur, H.; Bhatt, M.L.B.; Bhadauria, S. Macrophages are recruited to hypoxic tumor areas and acquire a Pro-Angiogenic M2-Polarized phenotype via hypoxic cancer cell derived cytokines Oncostatin M and Eotaxin. Oncotarget 2014, 5, 5350–5368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komori, T.; Tewari, B.N.; Kanchan, R.K.; Baghel, K.S.; Nautiyal, N.; Shrivastava, R.; Kaur, H.; Bhatt, M.L.B.; Bhadauria, S. Lack of oncostatin M receptor β leads to adipose tissue inflammation and insulin resistance by switching macrophage phenotype. J. Biol. Chem. 2013, 288, 21861–21875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broxmeyer, H.E.; Bruns, H.A.; Zhang, S.; Cooper, S.; Hangoc, G.; McKenzie, A.N.; Dent, A.L.; Schindler, U.; Naeger, L.K.; Hoey, T.; et al. Th1 cells regulate hematopoietic progenitor cell homeostasis by production of Oncostatin M. Immunity 2002, 16, 815–825. [Google Scholar] [CrossRef] [Green Version]
- Boniface, K.; Tewari, B.N.; Kanchan, R.K.; Baghel, K.S.; Nautiyal, N.; Shrivastava, R.; Kaur, H.; Bhatt, M.L.B.; Bhadauria, S. Oncostatin M Secreted by Skin Infiltrating T Lymphocytes Is a Potent Keratinocyte Activator Involved in Skin Inflammation. J. Immunol. 2007, 178, 4615–4622. [Google Scholar] [CrossRef] [Green Version]
- Luzina, I.G.; Atamas, S.P.; Wise, R.; Wigley, F.M.; Choi, J.; Xiao, H.Q.; White, B. Occurrence of an activated, profibrotic pattern of gene expression in lung CD8+ T cells from scleroderma patients. Arthritis Rheum. 2003, 48, 2262–2274. [Google Scholar] [CrossRef]
- Grenier, A.; Tewari, B.N.; Kanchan, R.K.; Baghel, K.S.; Nautiyal, N.; Shrivastava, R.; Kaur, H.; Bhatt, M.L.B.; Bhadauria, S. Oncostatin M production and regulation by human polymorphonuclear neutrophils. Blood 1999, 93, 1413–1421. [Google Scholar] [CrossRef]
- Cross, A.; Edwards, S.W.; Bucknall, R.C.; Moots, R.J. Secretion of Oncostatin M by Neutrophils in Rheumatoid arthritis. Arthritis Rheum. 2004, 50, 1430–1436. [Google Scholar] [CrossRef]
- Goren, I.; Kämpfer, H.; Müller, E.; Schiefelbein, D.; Pfeilschifter, J.; Frank, S. Oncostatin M expression is functionally connected to neutrophils in the early inflammatory phase of skin repair: Implications for normal and diabetes-impaired wounds. J. Invest. Dermatol. 2006, 126, 628–637. [Google Scholar] [CrossRef] [Green Version]
- Quinton, L.J.; Jones, M.R.; Robson, B.E.; Simms, B.T.; Whitsett, J.A.; Mizgerd, J.P. Alveolar Epithelial STAT3, IL-6 Family Cytokines, and Host Defense during Escherichia coli Pneumonia. Am. J. Respir. Cell Mol. Biol. 2008, 38, 699–706. [Google Scholar] [CrossRef] [Green Version]
- Pothoven, K.L.; Norton, J.E.; Suh, L.A.; Carter, R.G.; Harris, K.E.; Biyasheva, A.; Welch, K.; Shintani-Smith, S.; Conley, B.D.; Liu, M.C.; et al. Neutrophils are a major source of the epithelial barrier disrupting cytokine oncostatin M in patients with mucosal airways disease. J. Allergy Clin. Immunol. 2017, 139, 1966–1978. [Google Scholar] [CrossRef]
- Hurst, S.M.; McLoughlin, R.M.; Monslow, J.; Owens, S.; Morgan, L.; Fuller, G.M.; Topley, N.; Jones, S.A. Secretion of Oncostatin M by Infiltrating Neutrophils: Regulation of IL-6 and Chemokine Expression in Human Mesothelial Cells. J. Immunol. 2002, 169, 5244–5251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lodyga, M.; Cambridge, E.; Karvonen, H.M.; Pakshir, P.; Wu, B.; Boo, S.; Kiebalo, M.; Kaarteenaho, R.; Glogauer, M.; Kapoor, M.; et al. Cadherin-11–mediated adhesion of macrophages to myofibroblasts establishes a profibrotic niche of active TGF-β. Sci. Signal. 2019, 12, eaao3469. [Google Scholar] [CrossRef] [PubMed]
- Ryan, R.E.; Martin, B.; Mellor, L.; Jacob, R.B.; Tawara, K.; McDougal, O.M.; Oxford, J.T.; Jorcyk, C.L. Oncostatin M binds to extracellular matrix in a bioactive conformation: Implications for inflammation and metastasis. Cytokine 2015, 72, 71–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choy, E.H.; Bendit, M.; McAleer, D.; Liu, F.; Feeney, M.; Brett, S.; Zamuner, S.; Campanile, A.; Toso, J. Safety, tolerability, pharmacokinetics and pharmacodynamics of an anti- oncostatin M monoclonal antibody in rheumatoid arthritis: Results from phase II randomized, placebo-controlled trials. Arthritis Res. Ther. 2013, 15, R132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, N.R.; Hegazy, A.N.; Owens, B.M.; Bullers, S.J.; Linggi, B.; Buonocore, S.; Coccia, M.; Görtz, D.; This, S.; Stockenhuber, K.; et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor–neutralizing therapy in patients with inflammatory bowel disease. Nat. Med. 2017, 23, 579–589. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Richards, C.D.; Botelho, F. Oncostatin M in the Regulation of Connective Tissue Cells and Macrophages in Pulmonary Disease. Biomedicines 2019, 7, 95. https://doi.org/10.3390/biomedicines7040095
Richards CD, Botelho F. Oncostatin M in the Regulation of Connective Tissue Cells and Macrophages in Pulmonary Disease. Biomedicines. 2019; 7(4):95. https://doi.org/10.3390/biomedicines7040095
Chicago/Turabian StyleRichards, Carl D., and Fernando Botelho. 2019. "Oncostatin M in the Regulation of Connective Tissue Cells and Macrophages in Pulmonary Disease" Biomedicines 7, no. 4: 95. https://doi.org/10.3390/biomedicines7040095
APA StyleRichards, C. D., & Botelho, F. (2019). Oncostatin M in the Regulation of Connective Tissue Cells and Macrophages in Pulmonary Disease. Biomedicines, 7(4), 95. https://doi.org/10.3390/biomedicines7040095