Obesity Augments Glucocorticoid-Dependent Muscle Atrophy in Male C57BL/6J Mice



Abstract
:1. Introduction
2. Results
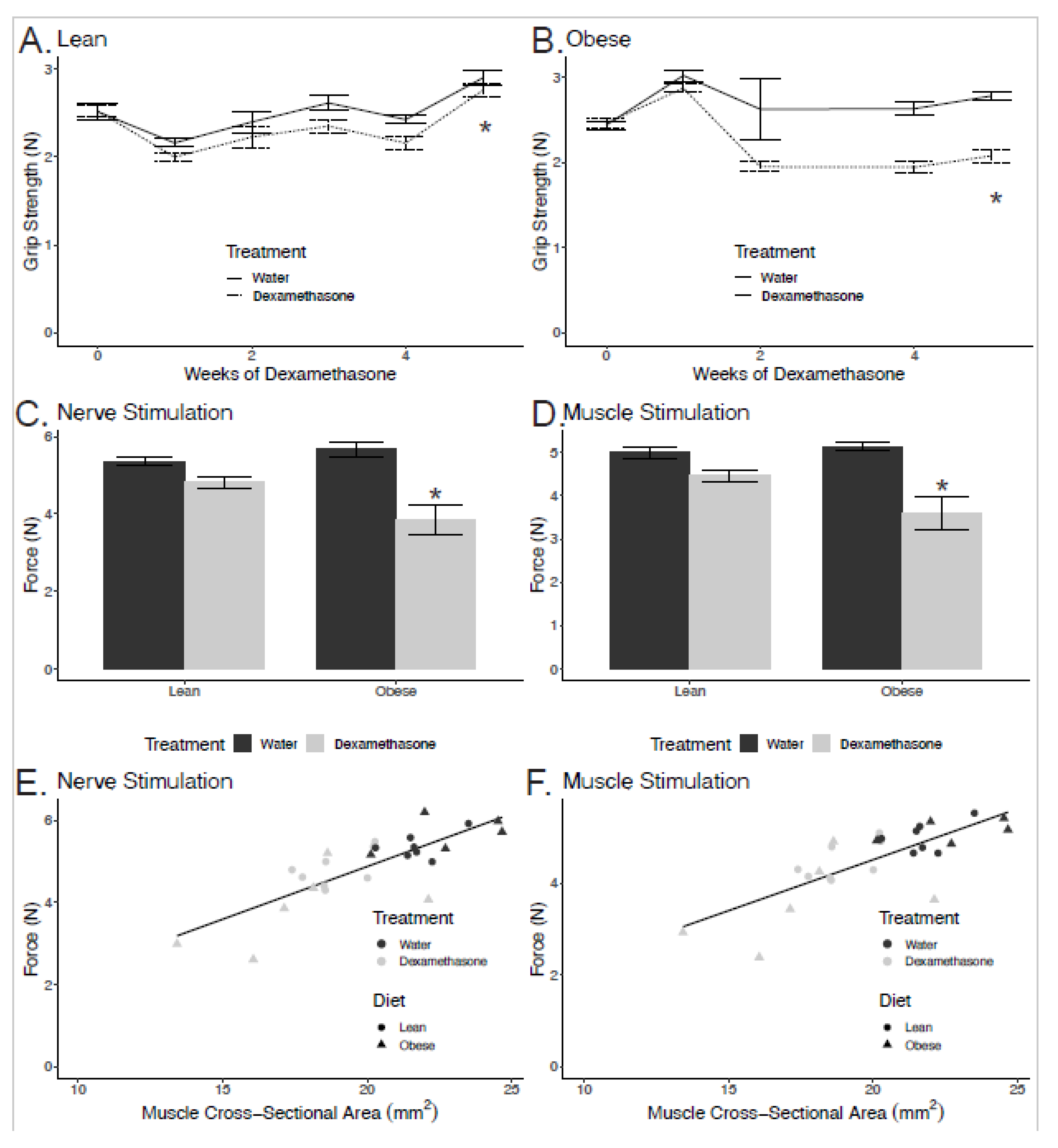
2.1. Greater Losses in Grip Strength in Obese Dexamethasone-Treated Mice
2.2. Reductions in Strength are Related to Smaller Cross-Sectional Area
2.3. Enhanced Muscle Atrophy in Obese Mice
2.4. Obesity with Dexamethasone Treatment Resulted in Smaller Type II Muscle Fibers
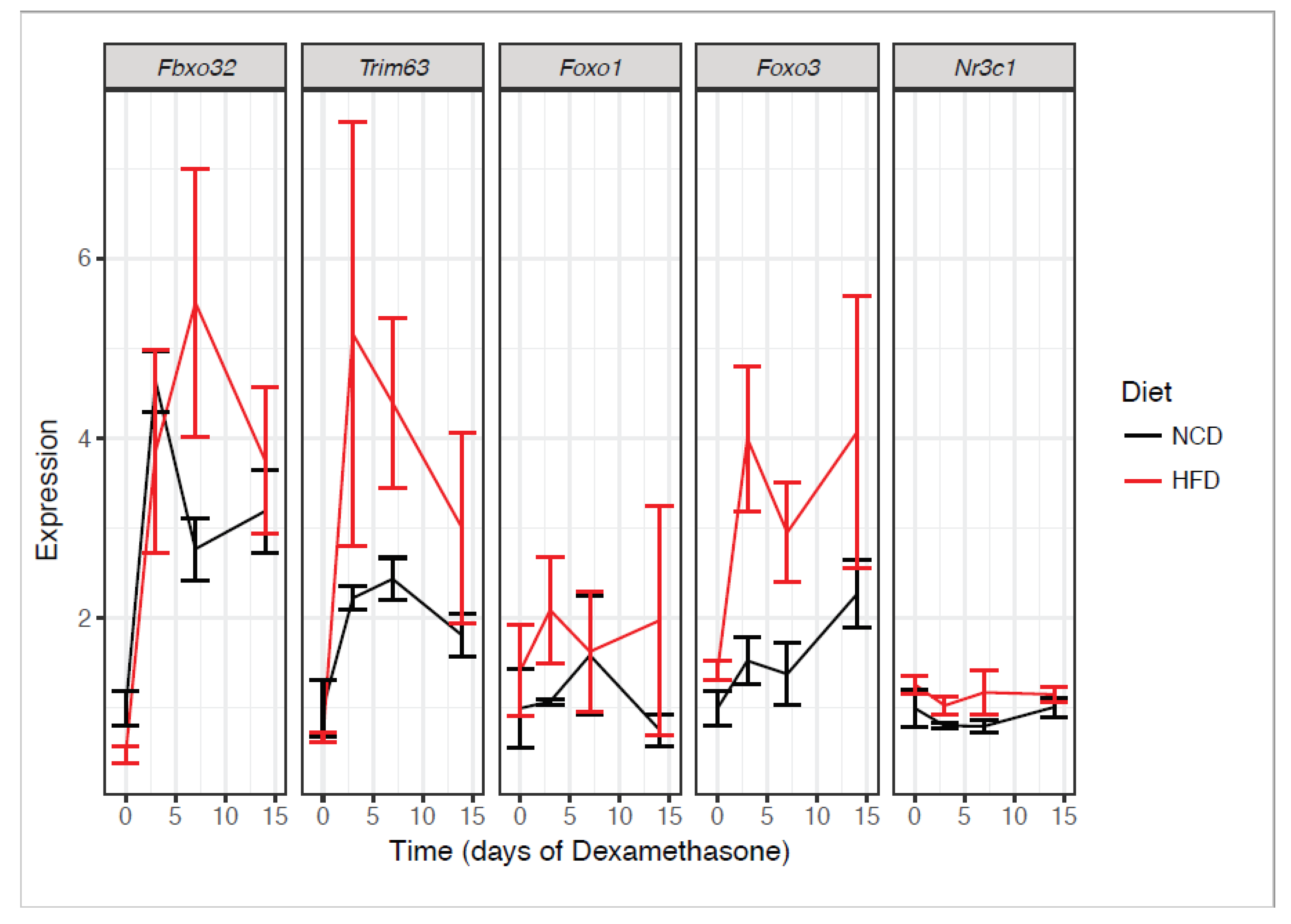
2.5. Obesity and Dexamethasone Cause Elevated Atrogene Expression
2.6. Obese Dexamethasone-Treated Mice Are Insulin Resistant After Adjusting for Muscle Mass
3. Discussion
4. Materials and Methods
4.1. Animal Husbandry
4.2. Grip Strength
4.3. In situ Contractile Measurements
4.4. Histology and Fiber Type Quantifications
4.5. mRNA Quantification
4.6. Protein Quantification
4.7. Assessment of Insulin Tolerance
4.8. Body, Fat, and Lean Mass Determination
5. Statistics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wolfe, R.R. The underappreciated role of muscle in health and disease. Am. J. Clin. Nutr. 2018, 84, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Lecker, S.H.; Jagoe, R.T.; Gilbert, A.; Gomes, M.; Baracos, V.E.; Bailey, J.; Price, S.R.; Mitch, W.E.; Goldberg, A.L. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004, 18, 39–51. [Google Scholar] [CrossRef]
- Staa, T.P.V.A.N.; Leufkens, H.G.M.; Abenhaim, L.; Begaud, B.; Zhang, B.; Cooper, C.; van Staa, T.P.; Leufkens, H.G.M.; Abenhaim, L.; Begaud, B.; et al. Use of oral corticosteroids in the United Kingdom. QJM 2000, 93, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overman, R.A.; Yeh, J.Y.; Deal, C.L. Prevalence of oral glucocorticoid usage in the United States: A general population perspective. Arthritis Care Res. 2013, 65, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, R.M.; Labad, J.; Strachan, M.W.J.; Braun, A.; Fowkes, F.G.R.; Lee, A.J.; Frier, B.M.; Seckl, J.R.; Walker, B.R.; Price, J.F. Elevated Fasting Plasma Cortisol Is Associated with Ischemic Heart Disease and Its Risk Factors in People with Type 2 Diabetes: The Edinburgh Type 2 Diabetes Study. J. Clin. Endocrinol. Metab. 2010, 95, 1602–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackett, R.A.; Kivimäki, M.; Kumari, M.; Steptoe, A. Diurnal Cortisol Patterns, Future Diabetes, and Impaired Glucose Metabolism in the Whitehall II Cohort Study. J. Clin. Endocrinol. Metab. 2016, 101, 619–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flegal, K.M.; Kruszon-Moran, D.; Carroll, M.D.; Fryar, C.D.; Ogden, C.L. Trends in Obesity Among Adults in the United States, 2005 to 2014. JAMA 2016, 315, 2284. [Google Scholar] [CrossRef] [Green Version]
- Heymsfield, S.B.; Wadden, T.A. Mechanisms, Pathophysiology, and Management of Obesity. N. Engl. J. Med. 2017, 376, 254–266. [Google Scholar] [CrossRef]
- Kelly, F.J.; Goldspink, D.F. The differing responses of four muscle types to dexamethasone treatment in the the Rat. Biochem 1982, 175, 147–151. [Google Scholar] [CrossRef] [Green Version]
- Bhasin, S.; Artaza, J.; Mahabadi, V.; Mallidis, C.; Ma, K.; Gonzalez-Cadavid, N.; Arias, J.; Salehian, B. Glucocorticoid-induced skeletal muscle atrophy is associated with upregulation of myostatin gene expression. Am. J. Physiol. Metab. 2015, 285, E363–E371. [Google Scholar] [CrossRef]
- Peeters, G.M.E.E.; Van Schoor, N.M.; Van Rossum, E.F.C.; Visser, M.; Lips, P. The relationship between cortisol, muscle mass and muscle strength in older persons and the role of genetic variations in the glucocorticoid receptor. Clin. Endocrinol. (Oxf.) 2008, 69, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Schakman, O.; Kalista, S.; Barbé, C.; Loumaye, A.; Thissen, J.P.P. Glucocorticoid-induced skeletal muscle atrophy. Int. J. Biochem. Cell Biol. 2013, 45, 2163–2172. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Liao, Q.; Liu, J.; Pan, R.; Lee, S.M.Y.; Lin, L. Myricanol rescues dexamethasone-induced muscle dysfunction via a sirtuin 1-dependent mechanism. J. Cachexia Sarcopenia Muscle 2019, 10, 429–444. [Google Scholar] [CrossRef]
- Hochberg, I.; Harvey, I.; Tran, Q.T.; Stephenson, E.J.; Barkan, A.L.; Saltiel, A.R.; Chandler, W.F.; Bridges, D. Gene expression changes in subcutaneous adipose tissue due to Cushing’s disease. J. Mol. Endocrinol. 2015, 55, 81–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, R.M.R.; Freire de Carvalho, J. Glucocorticoid-induced myopathy. J. Bone Spine 2011, 78, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef] [Green Version]
- Burke, S.J.; Batdorf, H.M.; Huang, T.-Y.; Jackson, J.W.; Jones, K.A.; Martin, T.M.; Rohli, K.E.; Karlstad, M.D.; Sparer, T.E.; Burk, D.H.; et al. One week of continuous corticosterone exposure impairs hepatic metabolic flexibility, promotes islet β-cell proliferation, and reduces physical activity in male C57BL/6 J mice. J. Steroid Biochem. Mol. Biol. 2019, 195, 105468. [Google Scholar] [CrossRef] [PubMed]
- Bentzinger, C.F.; Lin, S.; Romanino, K.; Castets, P.; Guridi, M.; Summermatter, S.; Handschin, C.; Tintignac, L.A.; Hall, M.N.; Rüegg, M.A. Differential response of skeletal muscles to mTORC1 signaling during atrophy and hypertrophy. Skelet. Muscle 2013, 3, 6. [Google Scholar] [CrossRef] [Green Version]
- Bentzinger, C.F.; Romanino, K.; Cloëtta, D.; Lin, S.; Mascarenhas, J.B.; Oliveri, F.; Xia, J.; Casanova, E.; Costa, C.F.; Brink, M.; et al. Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab. 2008, 8, 411–424. [Google Scholar] [CrossRef] [Green Version]
- Bodine, S.C.; Stitt, T.N.; Gonzalez, M.; Kline, W.O.; Stover, G.L.; Bauerlein, R.; Zlotchenko, E.; Scrimgeour, A.; Lawrence, J.C.; Glass, D.J.; et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat. Cell Biol. 2001, 3, 1014–1019. [Google Scholar] [CrossRef]
- Harvey, I.; Stephenson, E.J.; Redd, J.R.; Tran, Q.T.; Hochberg, I.; Qi, N.; Bridges, D. Glucocorticoid-Induced Metabolic Disturbances Are Exacerbated in Obese Male Mice. Endocrinology 2018, 159, 2275–2287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shpilberg, Y.; Beaudry, J.L.; D’Souza, A.; Campbell, J.E.; Peckett, A.; Riddell, M.C. A rodent model of rapid-onset diabetes induced by glucocorticoids and high-fat feeding. Dis. Model. Mech. 2012, 5, 671–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaudry, J.L.; D’souza, A.M.; Teich, T.; Tsushima, R.; Riddell, M.C. Exogenous glucocorticoids and a high-fat diet cause severe hyperglycemia and hyperinsulinemia and limit islet glucose responsiveness in young male Sprague-Dawley rats. Endocrinology 2013, 154, 3197–3208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adhikary, S.; Kothari, P.; Choudhary, D.; Tripathi, A.K.; Trivedi, R. Glucocorticoid aggravates bone micro-architecture deterioration and skeletal muscle atrophy in mice fed on high-fat diet. Steroids 2019. [Google Scholar] [CrossRef] [PubMed]
- Sher, J.; Cardasis, C. Skeletal Muscle Fiber Types in the Adult Mouse. Acta Neurol. Scand. 1976, 54, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [Green Version]
- DeFronzo, R.A. Lilly lecture 1987. The triumvirate: Beta-cell, muscle, liver. A collusion responsible for NIDDM. Diabetes 1988, 37, 667–687. [Google Scholar] [CrossRef]
- Son, Y.H.; Jang, E.J.; Kim, Y.W.; Lee, J.H. Sulforaphane prevents dexamethasone-induced muscle atrophy via regulation of the Akt/Foxo1 axis in C2C12 myotubes. Biomed. Pharmacother. 2017, 95, 1486–1492. [Google Scholar] [CrossRef]
- Shimizu, N.; Yoshikawa, N.; Ito, N.; Maruyama, T.; Suzuki, Y.; Takeda, S.I.; Nakae, J.; Tagata, Y.; Nishitani, S.; Takehana, K.; et al. Crosstalk between glucocorticoid receptor and nutritional sensor mTOR in skeletal muscle. Cell Metab. 2011, 13, 170–182. [Google Scholar] [CrossRef] [Green Version]
- Falduto, M.T.; Czerwinski, S.M.; Hickson, R.C. Glucocorticoid-induced muscle atrophy prevention by exercise in fast-twitch fibers. J. Appl. Physiol. 2017, 69, 1058–1062. [Google Scholar] [CrossRef]
- Maffiuletti, N.A.; Jubeau, M.; Munzinger, U.; Bizzini, M.; Agosti, F.; De Col, A.; Lafortuna, C.L.; Sartorio, A. Differences in quadriceps muscle strength and fatigue between lean and obese subjects. Eur. J. Appl. Physiol. 2007, 101, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Abdelmoula, A.; Martin, V.; Bouchant, A.; Walrand, S.; Lavet, C.; Taillardat, M.; Maffiuletti, N.A.; Boisseau, N.; Duché, P.; Ratel, S. Knee extension strength in obese and nonobese male adolescents. Appl. Physiol. Nutr. Metab. 2012, 37, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Hulens, M.; Vansant, G.; Lysens, R.; Claessens, A.L.; Muls, E. Exercise capacity in lean versus obese women. Scand. J. Med. Sci. Sport. 2001, 11, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Zoico, E.; Di Francesco, V.; Guralnik, J.M.; Mazzali, G.; Bortolani, A.; Guariento, S.; Sergi, G.; Bosello, O.; Zamboni, M. Physical disability and muscular strength in relation to obesity and different body composition indexes in a sample of healthy elderly women. Int. J. Obes. 2004, 28, 234–241. [Google Scholar] [CrossRef] [Green Version]
- Wester, V.L.; Staufenbiel, S.M.; Veldhorst, M.A.B.; Visser, J.A.; Manenschijn, L.; Koper, J.W.; Klessens-Godfroy, F.J.M.; van den Akker, E.L.T.; van Rossum, E.F.C. Long-term cortisol levels measured in scalp hair of obese patients. Obesity 2014, 22, 1–3. [Google Scholar] [CrossRef]
- Rosmond, R.; Chagnon, Y.C.C.; Chagnon, M.; Pe, L.; Chagnon, M.; Russe, L.P.E.; Carlsson, R.N.; Lindell, K.; Holm, G.; Chagnon, M.; et al. A glucocorticoid receptor gene marker is associated with abdominal obesity, leptin, and dysregulation of the hypothalamic-pituitary-adrenal axis. Obes. Res. 2000, 8, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levitan, R.D.; Vaccarino, F.J.; Brown, G.M.; Kennedy, S.H. Low-dose dexamethasone challenge in women with atypical major depression: Pilot study. J. Psychiatry Neurosci. 2002, 27, 47–51. [Google Scholar]
- Lopes, M.W.; Leal, R.B.; Guarnieri, R.; Schwarzbold, M.L.; Hoeller, A.; Diaz, A.P.; Boos, G.L.; Lin, K.; Linhares, M.N.; Nunes, J.C.; et al. A single high dose of dexamethasone affects the phosphorylation state of glutamate AMPA receptors in the human limbic system. Transl. Psychiatry 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27. [Google Scholar] [CrossRef] [Green Version]
- Becker, D.E. Basic and Clinical Pharmacology of Glucocorticosteroids. Anesth. Prog. 2013, 60, 25–32. [Google Scholar] [CrossRef]
- Rouleau, G.; Karpati, G.; Carpenter, S.; Soza, M.; Prescott, S.; Holland, P. Glucocorticoid excess induces preferential depletion of myosin in denervated skeletal muscle fibers. Muscle Nerve 1987, 10, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.M.; Loenneke, J.P.; Jo, E.; Wilson, G.J.; Zourdos, M.C.; Kim, J.-S. The Effects of Endurance, Strength, and Power Training on Muscle Fiber Type Shifting. J. Strength Cond. Res. 2012, 26, 1724–1729. [Google Scholar] [CrossRef]
- Kang, S.; Tsai, L.T.-Y.; Rosen, E.D. Nuclear Mechanisms of Insulin Resistance. Trends Cell Biol. 2016, 26, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Heller, E.A.; Cates, H.M.; Peña, C.J.; Herman, J.P.; Walsh, J.J. Mechanisms of Glucocorticoid-Induced Insulin Resistance: Focus on Adipose Tissue Function and Lipid Metabolism. Endocrinol Metab Clin. N. Am. 2015, 17, 1720–1727. [Google Scholar] [CrossRef]
- Hebling, A.; Scabora, J.E.; Esquisatto, M.A.M. Muscle Fibre Types and Connective Tissue Morphometry in Frontal Muscle of Norfolk Rabbits (Oryctolagus cuniculus). Int. J. Morphol. 2009, 27, 187–191. [Google Scholar] [CrossRef] [Green Version]
- Bergmeister, K.D.; Gröger, M.; Aman, M.; Willensdorfer, A.; Manzano-Szalai, K.; Salminger, S.; Aszmann, O.C. Automated muscle fiber type population analysis with ImageJ of whole rat muscles using rapid myosin heavy chain immunohistochemistry. Muscle Nerve 2016, 54, 292–299. [Google Scholar] [CrossRef]
- Harvey, C.J.d.C.; Schofield, G.M.; Zinn, C.; Thornley, S.J.; Crofts, C.; Merien, F.L.R. Low-carbohydrate diets differing in carbohydrate restriction improve cardiometabolic and anthropometric markers in healthy adults: A randomised clinical trial. PeerJ 2019, 7, e6273. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing; RC Team: Vienna, Austria, 2019. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NCD, Water | NCD, Dexamethasone | HFD, Water | HFD, Dexamethasone | |
|---|---|---|---|---|
| Body weight at sacrifice (g) | 31.5 ± 0.7 | 29.2 ± 1.5 | 46.5 ± 1.0 | 34.2 ± 1.6 * |
| Lean mass at sacrifice (g) | 26.2 ± 0.4 | 23.8 ± 0.6 | 27.8 ± 1.1 | 23.7 ± 0.3 |
| Fat mass at sacrifice (g) | 3.1 ± 0.6 | 3.6 ± 0.5 | 16.0 ± 1.3 | 11.6 ± 1.6 * |
| Percent fat mass at sacrifice | 9.8 ± 1.7 | 12 ± 1.2 | 34 ± 2.0 | 33 ± 3.2 |
| Food intake per mouse per day during dexamethasone treatment (g) | 3.5 ± 0.09 | 3.7 ± 0.21 | 2.1 ± 1.0 | 3.6 ± 0.31 |
| Calorie intake per mouse per day during dexamethasone treatment (kcal) | 10.1 ± 0.26 | 10.8 ± 0.61 | 9.9 ± 4.7 | 17.0 ± 1.5 * |
| Fluid intake per mouse per day during dexamethasone treatment (mL) | 11.7 ± 3.0 | 9.3 ± 3.0 | 15.9 ± 1.0 | 8.6 ± 1.7 |
| Gene | Forward 5′-3′ Sequence | Reverse 5′-3′ Sequence |
|---|---|---|
| Fbxo32 | CTTCTCGACTGCCATCCTGG | GTTCTTTTGGGCGATGCCAC |
| Trim63 | GAGGGCCATTGACTTTGGGA | TTTACCCTCTGTGGTCACGC |
| Foxo1 | AGTGGATGGTGAAGAGCGTG | GAAGGGACAGATTGTGGCGA |
| Foxo3 | AAACGGCTCACTTTGTCCCA | ATTCTGAACGCGCATGAAGC |
| Nr3c1 | CAAGGGTCTGGAGAGGACAAC | GCTGGACGGAGGAGAACTCA |
| Pgk1 | CAAGCTACTGTGGCCTCTGG | CCCACAGCCTCGGCATATTT |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gunder, L.C.; Harvey, I.; Redd, J.R.; Davis, C.S.; AL-Tamimi, A.; Brooks, S.V.; Bridges, D. Obesity Augments Glucocorticoid-Dependent Muscle Atrophy in Male C57BL/6J Mice. Biomedicines 2020, 8, 420. https://doi.org/10.3390/biomedicines8100420
Gunder LC, Harvey I, Redd JR, Davis CS, AL-Tamimi A, Brooks SV, Bridges D. Obesity Augments Glucocorticoid-Dependent Muscle Atrophy in Male C57BL/6J Mice. Biomedicines. 2020; 8(10):420. https://doi.org/10.3390/biomedicines8100420
Chicago/Turabian StyleGunder, Laura C., Innocence Harvey, JeAnna R. Redd, Carol S. Davis, Ayat AL-Tamimi, Susan V. Brooks, and Dave Bridges. 2020. "Obesity Augments Glucocorticoid-Dependent Muscle Atrophy in Male C57BL/6J Mice" Biomedicines 8, no. 10: 420. https://doi.org/10.3390/biomedicines8100420