Osmotic Adaptation by Na+-Dependent Transporters and ACE2: Correlation with Hemostatic Crisis in COVID-19




Abstract
:
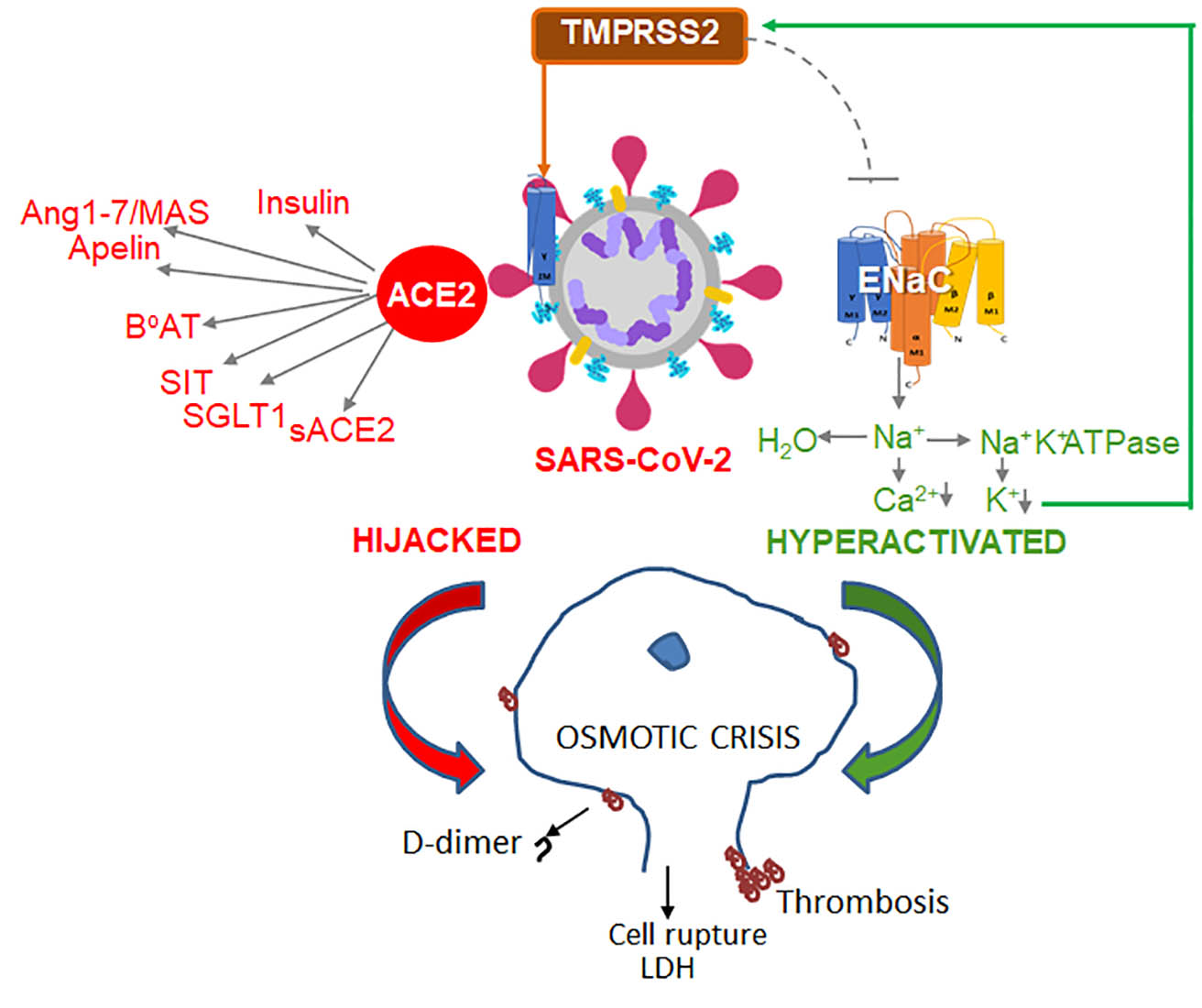{kind=link}
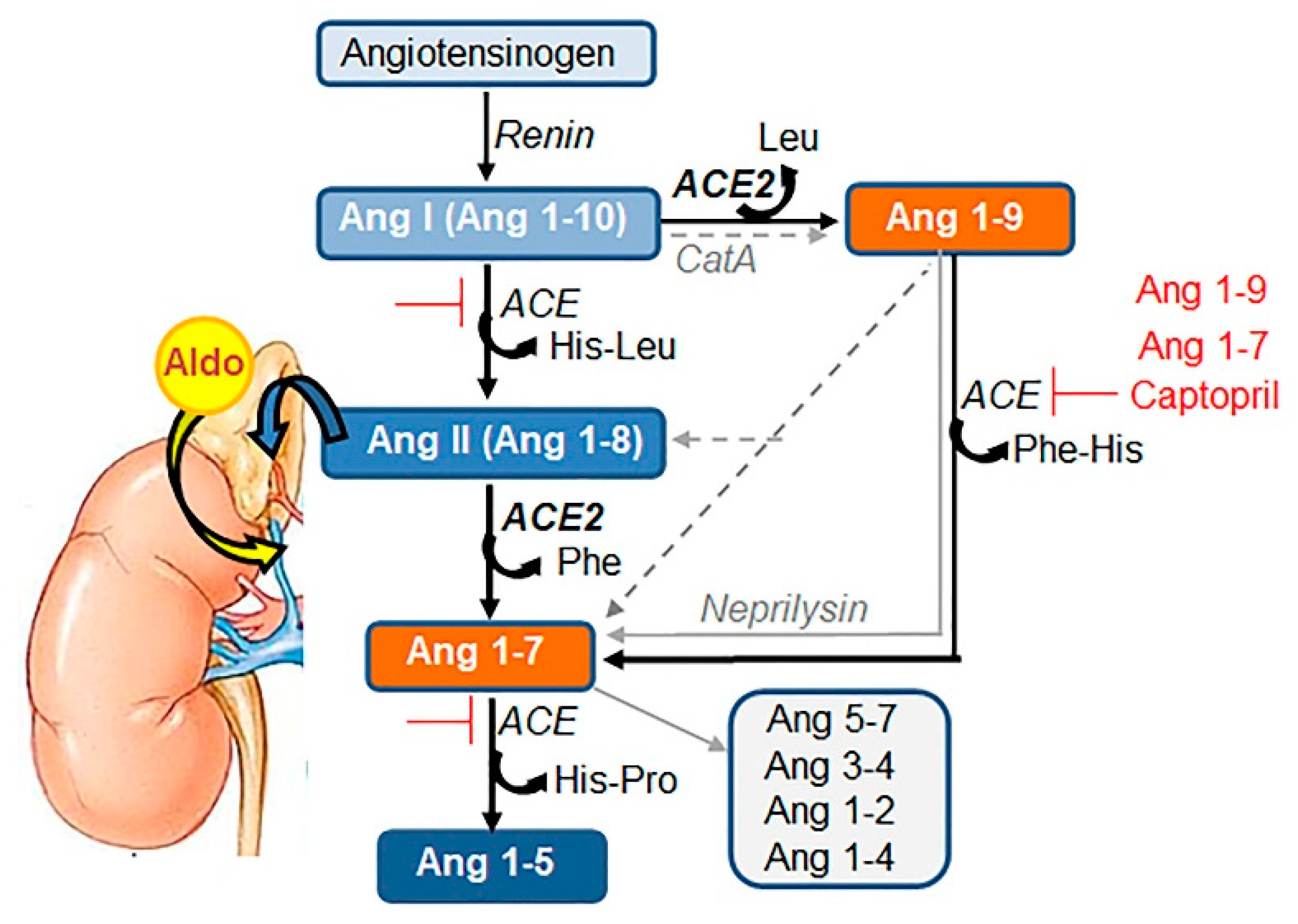{kind=link}
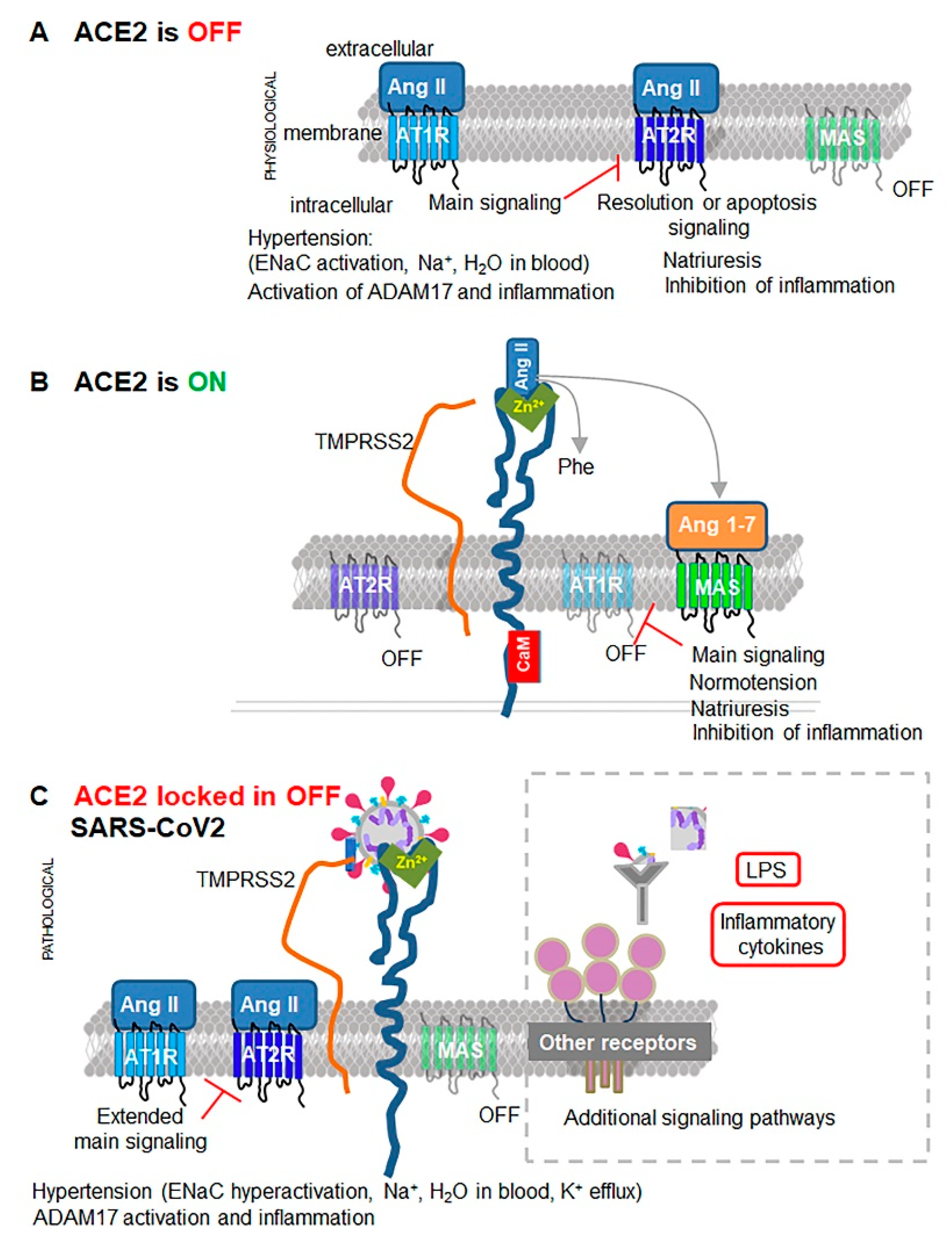{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
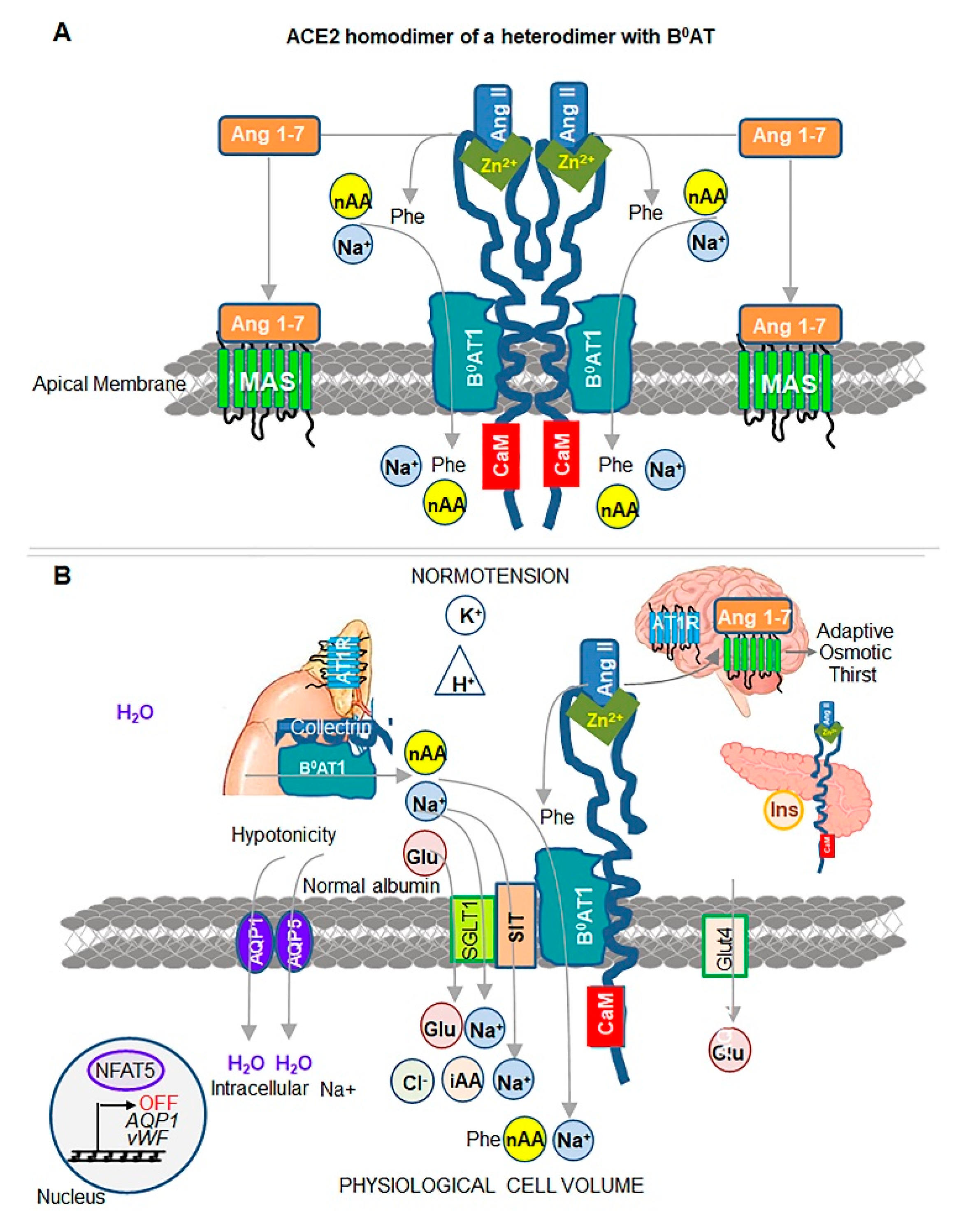
2. ACE2 Structure and Catalytic Site
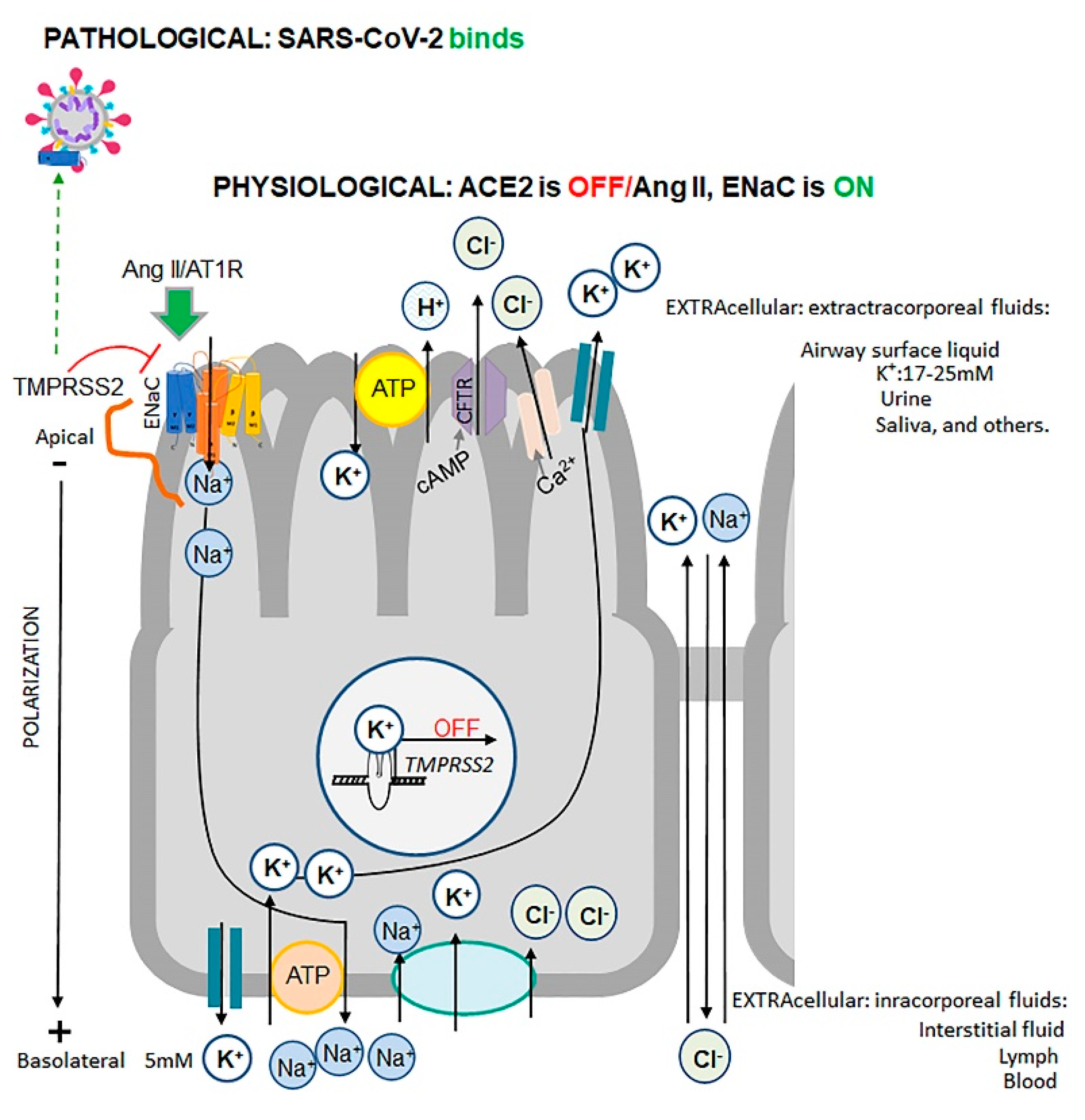
3. Effects of Ang II and Its Downstream Target ENaC on Osmosis, Hypertension, and Swelling
4. ACE2 Partnership with Neutral aa Transporter B0AT1
5. Evidence of ACE2-Dependent Regulation of Osmolality, Cell Volume, and Susceptibility to Thrombosis
6. Disruptive Viral Partners ACE2 Catalysis
- Extracellular: Host’s principal protease TMPRSS2 cleaves S1-S2 proteins of SARS-CoV-2 [102,104,106].Relevant physiological functions of TMPRSS2: (1) ACE2 cleavage and inactivation [25],(2) ENaC inhibitor [46].
- Relevant physiological functions of furin: (1) ENaC cleavage and activation [108].
- Alternative proteases: Cathepsins B/L (Cat B/L) [103].
6.1. Structural Site of SARS-CoV-2 and Hyperactivation of ENaC
6.2. Evidence of Osmotic Crisis in COVID-19
6.3. Osmotic Instability as a Risk Factor for Severe COVID-19 Pathogenesis
6.4. Sensory and Neuronal Interplay of ENaC, ACE2, and TMPRSS2 in COVID-19
7. SARS-CoV-2 Furin Cleavage Site and Infectivity
8. Conclusions
9. Therapeutic Perspectives
10. Patents
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AA | Amino Acids |
| ACE | Angiotensin-converting enzyme |
| ACE2 | Angiotensin-converting enzyme 2 |
| ADAM17 | Disintegrin and metalloproteinase 17 (Alias: TACE) |
| AMPK | Adenosine monophosphate-activated protein kinase) |
| Ang II | Angiotensin II (Alias: Ang 1–8) |
| Ang (1–10) | Angiotensin I |
| AP1 | Activator protein 1 transcription factor |
| AQP1 | Aquaporin 1, a water channel protein |
| AT1R | Angiotensin II Type 1 Receptor |
| APJ | Apelin receptor |
| ARDS | Acute Respiratory Distress Syndrome |
| ASCT2 | Alanine, serine, cysteine-preferring transporter 2 (gene SLC1A5) |
| AVP | Arginin vasopressin |
| B0AT1 | B degrees amino acid transporter 1 (gene SLC6A19) |
| CALHM1/3 | Calcium homeostasis modulator 1 and 3 |
| CAM | Calmodulin |
| CAMKII | CaM-dependent kinase |
| CAP1 | Channel-activating protease |
| CatA | Cathepsin A |
| CK2 | Casein kinase 2 |
| CNS | Central nervous system |
| COX2 | Cyclooxygenase 2 |
| COVID-19 | Coronavirus Disease of 2019 |
| CRP | C-reactive protein |
| CT | Chorda tympani |
| DAG | Diacylglycerol |
| ECM | Extracellular matrix |
| EDTA | Ethylenediaminetetraacetic acid |
| ENaC | Epithelial sodium channel |
| ER | Endoplasmic reticulum |
| Gαq/11 | Guanine nucleotide binding protein (G protein), alpha 11 (Gq class) |
| GABA | Gamma-aminobutyric acid |
| GDP | Guanosine diphosphate |
| GI | Gastrointestinal tract |
| Glu | Glucose |
| GLUT1 | Glucose transporter 1 |
| GTP | Guanosine triphosphate |
| HAT | Transmembrane serine protease 11D (Alias: TMPRSS11D) |
| H1N1 | Influenza A |
| HCoV-NL63 | Human coronavirus NL63 |
| HIF1 | Hypoxia Induced Transcription Factor |
| HMOX1 | Heme Oxygenase-1 |
| HPA | Hypothalamic-pituitary-adrenal |
| iAA | Imino acids, e.g., proline |
| IP3 | Inositol 3 phosphate |
| LDH | Lactate dehydrogenase (Alias: Lactic acid dehydrogenase) |
| LPS | Lipopolysaccharide |
| MAS | MAS1 proto-oncogene, G protein-coupled receptor |
| mTORC1 | Mammalian target of rapamycin complex 1 (Alias: Mechanistic target of rapamycin complex 1) |
| nAA | Neutral amino acids |
| NFAT5 | Nuclear Factor of Activated T cells (Alias: TonEBP, tonicity-responsive enhancer binding protein) |
| NHE3 | Sodium–hydrogen exchanger 3 |
| NO/iNOS | Nitric Oxide |
| ORF8 | Envelope glycoprotein B (Gene: ORF8) |
| PAI-1 | Plasminogen Activator Inhibitor-1 |
| PKC | Protein kinase C |
| PLC | Phospholipase C |
| RAS | Renin-Angiotensin System |
| RBD | Receptor binding domain |
| ROS | Reactive Oxygen Species |
| sACE2 | Soluble angiotensin-converting enzyme 2 |
| SARS-CoV | Severe Acute Respiratory Syndrome Coronavirus |
| SARS-CoV-2 | Severe Acute Respiratory Syndrome Coronavirus 2 |
| SGK1.1 | Glucocorticoid-induced kinase 1 |
| SGLT1 | Sodium-dependent Glucose Transporter 1 |
| SIT1 | Sodium/Imino-acid Transporter 1 (gene: SLC6A20) |
| SLC6A19 | Solute Carrier Family 6 Member 19 (protein BoAT1) |
| TACE | Tumor Necrosis Factor-α Converting Enzyme (Alias: ADAM 17) |
| TAPI2 | TNF Protease Inhibitor 2 |
| TMEM27 | Transmembrane protein 27 (Alias: Collectrin, Gene: CLTRN) |
| TMPRSS2 | Transmembrane Serine Protease 2 |
| VP | Vasopressin |
| vWF | von Willebrand factor |
| WT | Wild type |
References
- Hartenian, E.; Nandakumar, D.; Lari, A.; Ly, M.; Tucker, J.M.; Glaunsinger, B.A. The molecular virology of Coronaviruses. J. Biol. Chem. 2020, 295, 12910–12934. [Google Scholar] [CrossRef]
- Dell’Italia, L.J. Translational success stories: Angiotensin receptor 1 antagonists in heart failure. Circ. Res. 2011, 109, 437–452. [Google Scholar] [CrossRef] [Green Version]
- Kai, H.; Kai, M. Interactions of coronaviruses with ACE2, angiotensin II, and RAS inhibitors-lessons from available evidence and insights into COVID-19. Hypertens. Res. 2020, 43, 648–654. [Google Scholar] [CrossRef]
- Dostal, D.E.; Baker, K.M. The cardiac renin-angiotensin system: Conceptual, or a regulator of cardiac function? Circ. Res. 1999, 85, 643–650. [Google Scholar] [CrossRef]
- Bader, M.; Alenina, N.; Young, D.; Santos, R.A.S.; Touyz, R.M. The Meaning of Mas. Hypertension 2018, 72, 1072–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- South, A.M.; Shaltout, H.A.; Washburn, L.K.; Hendricks, A.S.; Diz, D.I.; Chappell, M.C. Fetal programming and the angiotensin-(1-7) axis: A review of the experimental and clinical data. Clin. Sci. 2019, 133, 55–74. [Google Scholar] [CrossRef]
- Ferrario, C.M. Angiotensin-converting enzyme 2 and angiotensin-(1-7): An evolving story in cardiovascular regulation. Hypertension 2006, 47, 515–521. [Google Scholar] [CrossRef]
- Crackower, M.A.; Sarao, R.; Oudit, G.Y.; Yagil, C.; Kozieradzki, I.; Scanga, S.E.; Oliveira-dos-Santos, A.J.; da Costa, J.; Zhang, L.; Pei, Y.; et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002, 417, 822–828. [Google Scholar] [CrossRef]
- Singer, D.; Camargo, S.M.; Ramadan, T.; Schafer, M.; Mariotta, L.; Herzog, B.; Huggel, K.; Wolfer, D.; Werner, S.; Penninger, J.M.; et al. Defective intestinal amino acid absorption in Ace2 null mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G686–G695. [Google Scholar] [CrossRef]
- Camargo, S.M.; Singer, D.; Makrides, V.; Huggel, K.; Pos, K.M.; Wagner, C.A.; Kuba, K.; Danilczyk, U.; Skovby, F.; Kleta, R.; et al. Tissue-specific amino acid transporter partners ACE2 and collectrin differentially interact with hartnup mutations. Gastroenterology 2009, 136, 872–882. [Google Scholar] [CrossRef] [Green Version]
- Kowalczuk, S.; Broer, A.; Tietze, N.; Vanslambrouck, J.M.; Rasko, J.E.; Broer, S. A protein complex in the brush-border membrane explains a Hartnup disorder allele. FASEB J. 2008, 22, 2880–2887. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Lambert, D.W.; Yarski, M.; Warner, F.J.; Thornhill, P.; Parkin, E.T.; Smith, A.I.; Hooper, N.M.; Turner, A.J. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J. Biol. Chem. 2005, 280, 30113–30119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, N.E.; Fisher, M.J.; Porter, K.E.; Lambert, D.W.; Turner, A.J. Angiotensin converting enzyme (ACE) and ACE2 bind integrins and ACE2 regulates integrin signalling. PLoS ONE 2012, 7, e34747. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sui, J.; Li, W.; Murakami, A.; Tamin, A.; Matthews, L.J.; Wong, S.K.; Moore, M.J.; Tallarico, A.S.; Olurinde, M.; Choe, H.; et al. Potent neutralization of severe acute respiratory syndrome (SARS) coronavirus by a human mAb to S1 protein that blocks receptor association. Proc. Natl. Acad. Sci. USA 2004, 101, 2536–2541. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://coronavirus.jhu.edu/map.html (accessed on 1 October 2020).
- Verdecchia, P.; Cavallini, C.; Spanevello, A.; Angeli, F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 2020, 76, 14–20. [Google Scholar] [CrossRef]
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 324, 749–751. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Anand, P.; Puranik, A.; Aravamudan, M.; Venkatakrishnan, A.J.; Soundararajan, V. SARS-CoV-2 strategically mimics proteolytic activation of human ENaC. Elife 2020, 9, e58603. [Google Scholar] [CrossRef]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef] [Green Version]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef]
- Vickers, C.; Hales, P.; Kaushik, V.; Dick, L.; Gavin, J.; Tang, J.; Godbout, K.; Parsons, T.; Baronas, E.; Hsieh, F.; et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J. Biol. Chem. 2002, 277, 14838–14843. [Google Scholar] [CrossRef] [Green Version]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pohlmann, S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef] [Green Version]
- Freis, E.D. Salt, volume and the prevention of hypertension. Circulation 1976, 53, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Butterworth, M.B. Regulation of the epithelial sodium channel (ENaC) by membrane trafficking. Biochim. Biophys. Acta 2010, 1802, 1166–1177. [Google Scholar] [CrossRef] [Green Version]
- Veiras, L.C.; McFarlin, B.E.; Ralph, D.L.; Buncha, V.; Prescott, J.; Shirvani, B.S.; McDonough, J.C.; Ha, D.; Giani, J.; Gurley, S.B.; et al. Electrolyte and transporter responses to angiotensin II induced hypertension in female and male rats and mice. Acta Physiol. 2020, 229, e13448. [Google Scholar] [CrossRef]
- Li, X.C.; Shao, Y.; Zhuo, J.L. AT1a receptor signaling is required for basal and water deprivation-induced urine concentration in AT1a receptor-deficient mice. Am. J. Physiol. Ren. Physiol. 2012, 303, F746–F756. [Google Scholar] [CrossRef] [Green Version]
- Mamenko, M.; Zaika, O.; Prieto, M.C.; Jensen, V.B.; Doris, P.A.; Navar, L.G.; Pochynyuk, O. Chronic angiotensin II infusion drives extensive aldosterone-independent epithelial Na+ channel activation. Hypertension 2013, 62, 1111–1122. [Google Scholar] [CrossRef] [Green Version]
- Peti-Peterdi, J.; Warnock, D.G.; Bell, P.D. Angiotensin II directly stimulates ENaC activity in the cortical collecting duct via AT(1) receptors. J. Am. Soc. Nephrol. 2002, 13, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Boscardin, E.; Alijevic, O.; Hummler, E.; Frateschi, S.; Kellenberger, S. The function and regulation of acid-sensing ion channels (ASICs) and the epithelial Na(+) channel (ENaC): IUPHAR Review 19. Br. J. Pharmacol. 2016, 173, 2671–2701. [Google Scholar] [CrossRef]
- Mobasheri, A.; Wray, S.; Marples, D. Distribution of AQP2 and AQP3 water channels in human tissue microarrays. J. Mol. Histol. 2005, 36, 1–14. [Google Scholar] [CrossRef]
- Miteva, D.O.; Rutkowski, J.M.; Dixon, J.B.; Kilarski, W.; Shields, J.D.; Swartz, M.A. Transmural flow modulates cell and fluid transport functions of lymphatic endothelium. Circ. Res. 2010, 106, 920–931. [Google Scholar] [CrossRef]
- Wittekindt, O.H.; Dietl, P. Aquaporins in the lung. Pflugers Arch. 2019, 471, 519–532. [Google Scholar] [CrossRef]
- Zhang, W.; Zitron, E.; Homme, M.; Kihm, L.; Morath, C.; Scherer, D.; Hegge, S.; Thomas, D.; Schmitt, C.P.; Zeier, M.; et al. Aquaporin-1 channel function is positively regulated by protein kinase C. J. Biol. Chem. 2007, 282, 20933–20940. [Google Scholar] [CrossRef] [Green Version]
- Weber, J.; Rajan, S.; Schremmer, C.; Chao, Y.K.; Krasteva-Christ, G.; Kannler, M.; Yildirim, A.O.; Brosien, M.; Schredelseker, J.; Weissmann, N.; et al. TRPV4 channels are essential for alveolar epithelial barrier function as protection from lung edema. JCI Insight 2020. [Google Scholar] [CrossRef]
- Li, H.; Kan, H.; He, C.; Zhang, X.; Yang, Z.; Jin, J.; Zhang, P.; Ma, X. TRPV4 activates cytosolic phospholipase A2 via Ca(2+) -dependent PKC/ERK1/2 signalling in controlling hypertensive contraction. Clin. Exp. Pharmacol. Physiol. 2018. [Google Scholar] [CrossRef]
- Namkung, W.; Song, Y.; Mills, A.D.; Padmawar, P.; Finkbeiner, W.E.; Verkman, A.S. In situ measurement of airway surface liquid [K+] using a ratioable K+-sensitive fluorescent dye. J. Biol. Chem. 2009, 284, 15916–15926. [Google Scholar] [CrossRef] [Green Version]
- Cook, D.I.; Dinudom, A.; Komwatana, P.; Kumar, S.; Young, J.A. Patch-clamp studies on epithelial sodium channels in salivary duct cells. Cell Biochem. Biophys. 2002, 36, 105–113. [Google Scholar] [CrossRef]
- Knoepp, F.; Ashley, Z.; Barth, D.; Baldin, J.P.; Jennings, M.; Kazantseva, M.; Saw, E.L.; Katare, R.; Alvarez de la Rosa, D.; Weissmann, N.; et al. Shear force sensing of epithelial Na(+) channel (ENaC) relies on N-glycosylated asparagines in the palm and knuckle domains of alphaENaC. Proc. Natl. Acad. Sci. USA 2020, 117, 717–726. [Google Scholar] [CrossRef] [Green Version]
- Biffi, G.; Tannahill, D.; McCafferty, J.; Balasubramanian, S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 2013, 5, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Seenisamy, J.; Rezler, E.M.; Powell, T.J.; Tye, D.; Gokhale, V.; Joshi, C.S.; Siddiqui-Jain, A.; Hurley, L.H. The dynamic character of the G-quadruplex element in the c-MYC promoter and modification by TMPyP4. J. Am. Chem. Soc. 2004, 126, 8702–8709. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.W.; Qian, M.Q.; Yu, K.; Narva, S.; Yu, F.; Wu, Y.L.; Zhang, W. Inhibition of Influenza A virus propagation by benzoselenoxanthenes stabilizing TMPRSS2 Gene G-quadruplex and hence down-regulating TMPRSS2 expression. Sci. Rep. 2020, 10, 7635. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Neidle, S. G-quadruplex nucleic acids as therapeutic targets. Curr. Opin. Chem. Biol. 2009, 13, 345–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donaldson, S.H.; Hirsh, A.; Li, D.C.; Holloway, G.; Chao, J.; Boucher, R.C.; Gabriel, S.E. Regulation of the epithelial sodium channel by serine proteases in human airways. J. Biol. Chem. 2002, 277, 8338–8345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afar, D.E.; Vivanco, I.; Hubert, R.S.; Kuo, J.; Chen, E.; Saffran, D.C.; Raitano, A.B.; Jakobovits, A. Catalytic cleavage of the androgen-regulated TMPRSS2 protease results in its secretion by prostate and prostate cancer epithelia. Cancer Res. 2001, 61, 1686–1692. [Google Scholar]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, 1016–1035. [Google Scholar] [CrossRef]
- Liu, Y.; Jiang, B.J.; Zhao, R.Z.; Ji, H.L. Epithelial Sodium Channels in Pulmonary Epithelial Progenitor and Stem Cells. Int. J. Biol. Sci. 2016, 12, 1150–1154. [Google Scholar] [CrossRef] [Green Version]
- Carsana, L.; Sonzogni, A.; Nasr, A.; Rossi, R.S.; Pellegrinelli, A.; Zerbi, P.; Rech, R.; Colombo, R.; Antinori, S.; Corbellino, M.; et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: A two-centre descriptive study. Lancet Infect. Dis. 2020, 20, 1135–1140. [Google Scholar] [CrossRef]
- Damiani, S.; Fiorentino, M.; De Palma, A.; Foschini, M.P.; Lazzarotto, T.; Gabrielli, L.; Viale, P.L.; Attard, L.; Riefolo, M.; D’Errico, A. Pathological Post Mortem Findings in Lungs Infected With Sars-Cov 2. J. Pathol. 2020. [Google Scholar] [CrossRef]
- Solymosi, E.A.; Kaestle-Gembardt, S.M.; Vadasz, I.; Wang, L.; Neye, N.; Chupin, C.J.; Rozowsky, S.; Ruehl, R.; Tabuchi, A.; Schulz, H.; et al. Chloride transport-driven alveolar fluid secretion is a major contributor to cardiogenic lung edema. Proc. Natl. Acad. Sci. USA 2013, 110, E2308–E2316. [Google Scholar] [CrossRef] [Green Version]
- Veiras, L.C.; Han, J.; Ralph, D.L.; McDonough, A.A. Potassium Supplementation Prevents Sodium Chloride Cotransporter Stimulation During Angiotensin II Hypertension. Hypertension 2016, 68, 904–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, P.; Gao, Z.X.; Duan, X.P.; Su, X.T.; Wang, M.X.; Lin, D.H.; Gu, R.; Wang, W.H. AT2R (Angiotensin II Type 2 Receptor)-Mediated Regulation of NCC (Na-Cl Cotransporter) and Renal K Excretion Depends on the K Channel, Kir4.1. Hypertension 2018, 71, 622–630. [Google Scholar] [CrossRef]
- Planes, C.; Caughey, G.H. Regulation of the epithelial Na+ channel by peptidases. Curr. Top. Dev. Biol. 2007, 78, 23–46. [Google Scholar]
- Kimura, T.; Kawabe, H.; Jiang, C.; Zhang, W.; Xiang, Y.Y.; Lu, C.; Salter, M.W.; Brose, N.; Lu, W.Y.; Rotin, D. Deletion of the ubiquitin ligase Nedd4L in lung epithelia causes cystic fibrosis-like disease. Proc. Natl. Acad. Sci. USA 2011, 108, 3216–3221. [Google Scholar] [CrossRef] [Green Version]
- Goel, P.; Manning, J.A.; Kumar, S. NEDD4-2 (NEDD4L): The ubiquitin ligase for multiple membrane proteins. Gene 2015, 557, 1–10. [Google Scholar] [CrossRef]
- Abriel, H.; Loffing, J.; Rebhun, J.F.; Pratt, J.H.; Schild, L.; Horisberger, J.D.; Rotin, D.; Staub, O. Defective regulation of the epithelial Na+ channel by Nedd4 in Liddle’s syndrome. J. Clin. Investig. 1999, 103, 667–673. [Google Scholar] [CrossRef] [Green Version]
- Wynne, B.M.; Zou, L.; Linck, V.; Hoover, R.S.; Ma, H.P.; Eaton, D.C. Regulation of Lung Epithelial Sodium Channels by Cytokines and Chemokines. Front. Immunol. 2017, 8, 766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussolati, O.; Dall’Asta, V.; Franchi-Gazzola, R.; Sala, R.; Rotoli, B.M.; Visigalli, R.; Casado, J.; Lopez-Fontanals, M.; Pastor-Anglada, M.; Gazzola, G.C. The role of system A for neutral amino acid transport in the regulation of cell volume. Mol. Membr. Biol. 2001, 18, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.P.; Debnam, E.S.; Leung, P.S. Involvement of an enterocyte renin-angiotensin system in the local control of SGLT1-dependent glucose uptake across the rat small intestinal brush border membrane. J. Physiol. 2007, 584, 613–623. [Google Scholar] [CrossRef]
- Tanemoto, M. Effect of serum albumin on serum sodium: Necessity to consider the Donnan effect. QJM 2008, 101, 827–828. [Google Scholar] [CrossRef]
- Dunn, M.J. The roles of angiotensin II and prostaglandins in the regulation of the glomerular filtration of albumin. J. Hypertens. Suppl. 1990, 8, S47–S51. [Google Scholar] [CrossRef]
- Lim, S.C.; Liu, J.J.; Subramaniam, T.; Sum, C.F. Elevated circulating alpha-klotho by angiotensin II receptor blocker losartan is associated with reduction of albuminuria in type 2 diabetic patients. J. Renin Angiotensin Aldosterone Syst. 2014, 15, 487–490. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Sato, T.; Rodriguez-Iturbe, B.; Vaziri, N.D. Role of intrarenal angiotensin system activation, oxidative stress, inflammation, and impaired nuclear factor-erythroid-2-related factor 2 activity in the progression of focal glomerulosclerosis. J. Pharmacol. Exp. Ther. 2011, 337, 583–590. [Google Scholar] [CrossRef] [Green Version]
- Dandona, P.; Fonseca, V.; Baron, D.N. Hypoalbuminaemic hyponatraemia: A new syndrome? Br. Med. J. 1985, 291, 1253–1255. [Google Scholar] [CrossRef] [Green Version]
- Namkung, Y.; LeGouill, C.; Kumar, S.; Cao, Y.; Teixeira, L.B.; Lukasheva, V.; Giubilaro, J.; Simoes, S.C.; Longpre, J.M.; Devost, D.; et al. Functional selectivity profiling of the angiotensin II type 1 receptor using pathway-wide BRET signaling sensors. Sci. Signal. 2018, 11, eaat1631. [Google Scholar] [CrossRef] [Green Version]
- Redig, A.J.; Platanias, L.C. The protein kinase C (PKC) family of proteins in cytokine signaling in hematopoiesis. J. Interferon Cytokine Res. 2007, 27, 623–636. [Google Scholar] [CrossRef]
- Conner, M.T.; Conner, A.C.; Brown, J.E.; Bill, R.M. Membrane trafficking of aquaporin 1 is mediated by protein kinase C via microtubules and regulated by tonicity. Biochemistry 2010, 49, 821–823. [Google Scholar] [CrossRef]
- Kitchen, P.; Oberg, F.; Sjohamn, J.; Hedfalk, K.; Bill, R.M.; Conner, A.C.; Conner, M.T.; Tornroth-Horsefield, S. Plasma Membrane Abundance of Human Aquaporin 5 Is Dynamically Regulated by Multiple Pathways. PLoS ONE 2015, 10, e0143027. [Google Scholar] [CrossRef]
- Li, C.; Wang, W.; Rivard, C.J.; Lanaspa, M.A.; Summer, S.; Schrier, R.W. Molecular mechanisms of angiotensin II stimulation on aquaporin-2 expression and trafficking. Am. J. Physiol. Ren. Physiol. 2011, 300, F1255–F1261. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, S.; Terris, J.; Andersen, D.; Ecelbarger, C.; Frokiaer, J.; Jonassen, T.; Marples, D.; Knepper, M.A.; Petersen, J.S. Congestive heart failure in rats is associated with increased expression and targeting of aquaporin-2 water channel in collecting duct. Proc. Natl. Acad. Sci. USA 1997, 94, 5450–5455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrier, R.W.; Martin, P.Y. Recent advances in the understanding of water metabolism in heart failure. Adv. Exp. Med. Biol. 1998, 449, 415–426. [Google Scholar] [PubMed]
- Feng, Y.; Hans, C.; McIlwain, E.; Varner, K.J.; Lazartigues, E. Angiotensin-converting enzyme 2 over-expression in the central nervous system reduces angiotensin-II-mediated cardiac hypertrophy. PLoS ONE 2012, 7, e48910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broer, A.; Juelich, T.; Vanslambrouck, J.M.; Tietze, N.; Solomon, P.S.; Holst, J.; Bailey, C.G.; Rasko, J.E.; Broer, S. Impaired nutrient signaling and body weight control in a Na+ neutral amino acid cotransporter (Slc6a19)-deficient mouse. J. Biol. Chem. 2011, 286, 26638–26651. [Google Scholar] [CrossRef] [Green Version]
- Dos-Santos, R.C.; Monteiro, L.; Paes-Leme, B.; Lustrino, D.; Antunes-Rodrigues, J.; Mecawi, A.S.; Reis, L.C. Central angiotensin-(1-7) increases osmotic thirst. Exp. Physiol. 2017, 102, 1397–1404. [Google Scholar] [CrossRef]
- Danilczyk, U.; Sarao, R.; Remy, C.; Benabbas, C.; Stange, G.; Richter, A.; Arya, S.; Pospisilik, J.A.; Singer, D.; Camargo, S.M.; et al. Essential role for collectrin in renal amino acid transport. Nature 2006, 444, 1088–1091. [Google Scholar] [CrossRef]
- Seow, H.F.; Broer, S.; Broer, A.; Bailey, C.G.; Potter, S.J.; Cavanaugh, J.A.; Rasko, J.E. Hartnup disorder is caused by mutations in the gene encoding the neutral amino acid transporter SLC6A19. Nat. Genet. 2004, 36, 1003–1007. [Google Scholar] [CrossRef]
- Baron, D.N.; Dent, C.E.; Harris, H.; Hart, E.W.; Jepson, J.B. Hereditary pellagra-like skin rash with temporary cerebellar ataxia, constant renal amino-aciduria, and other bizarre biochemical features. Lancet 1956, 271, 421–428. [Google Scholar] [CrossRef]
- Vuille-dit-Bille, R.N.; Camargo, S.M.; Emmenegger, L.; Sasse, T.; Kummer, E.; Jando, J.; Hamie, Q.M.; Meier, C.F.; Hunziker, S.; Forras-Kaufmann, Z.; et al. Human intestine luminal ACE2 and amino acid transporter expression increased by ACE-inhibitors. Amino Acids 2015, 47, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, S.; Tikellis, C.; Candido, R.; Tsorotes, D.; Pickering, R.J.; Bossi, F.; Carretta, R.; Fabris, B.; Cooper, M.E.; Thomas, M.C. ACE2 deficiency shifts energy metabolism towards glucose utilization. Metabolism 2015, 64, 406–415. [Google Scholar] [CrossRef]
- Javed, K.; Broer, S. Mice Lacking the Intestinal and Renal Neutral Amino Acid Transporter SLC6A19 Demonstrate the Relationship between Dietary Protein Intake and Amino Acid Malabsorption. Nutrients 2019, 11, 2024. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, K.B.; Chhabra, K.H.; Nguyen, V.K.; Xia, H.; Lazartigues, E. The transcription factor HNF1alpha induces expression of angiotensin-converting enzyme 2 (ACE2) in pancreatic islets from evolutionarily conserved promoter motifs. Biochim. Biophys. Acta 2013, 1829, 1225–1235. [Google Scholar] [CrossRef] [Green Version]
- Niu, M.J.; Yang, J.K.; Lin, S.S.; Ji, X.J.; Guo, L.M. Loss of angiotensin-converting enzyme 2 leads to impaired glucose homeostasis in mice. Endocrine 2008, 34, 56–61. [Google Scholar] [CrossRef] [PubMed]
- van Twist, D.J.; Kroon, A.A.; de Leeuw, P.W. Angiotensin-(1-7) as a strategy in the treatment of hypertension? Curr. Opin. Nephrol. Hypertens. 2014, 23, 480–486. [Google Scholar] [CrossRef]
- Zhang, F.; Tang, H.; Sun, S.; Luo, Y.; Ren, X.; Chen, A.; Xu, Y.; Li, P.; Han, Y. Angiotensin-(1-7) induced vascular relaxation in spontaneously hypertensive rats. Nitric Oxide 2019, 88, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ocaranza, M.P.; Moya, J.; Barrientos, V.; Alzamora, R.; Hevia, D.; Morales, C.; Pinto, M.; Escudero, N.; Garcia, L.; Novoa, U.; et al. Angiotensin-(1-9) reverses experimental hypertension and cardiovascular damage by inhibition of the angiotensin converting enzyme/Ang II axis. J. Hypertens. 2014, 32, 771–783. [Google Scholar] [CrossRef]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000147003-CLTRN/tissue (accessed on 31 July 2020).
- Moritani, T.; Iwai, M.; Kanno, H.; Nakaoka, H.; Iwanami, J.; Higaki, T.; Ishii, E.; Horiuchi, M. ACE2 deficiency induced perivascular fibrosis and cardiac hypertrophy during postnatal development in mice. J. Am. Soc. Hypertens. 2013, 7, 259–266. [Google Scholar] [CrossRef]
- Giani, J.F.; Gironacci, M.M.; Munoz, M.C.; Pena, C.; Turyn, D.; Dominici, F.P. Angiotensin-(1 7) stimulates the phosphorylation of JAK2, IRS-1 and Akt in rat heart in vivo: Role of the AT1 and Mas receptors. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1154–H1163. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.D.; Fuchs, S.; Bernstein, E.A.; Li, P.; Campbell, D.J.; Bernstein, K.E. Mice expressing ACE only in the heart show that increased cardiac angiotensin II is not associated with cardiac hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H659–H667. [Google Scholar] [CrossRef]
- Wilson, C.S.; Mongin, A.A. Cell Volume Control in Healthy Brain and Neuropathologies. Curr. Top. Membr. 2018, 81, 385–455. [Google Scholar] [PubMed]
- Avissar, N.E.; Ryan, C.K.; Ganapathy, V.; Sax, H.C. Na(+)-dependent neutral amino acid transporter ATB(0) is a rabbit epithelial cell brush-border protein. Am. J. Physiol. Cell Physiol. 2001, 281, C963–C971. [Google Scholar] [CrossRef] [PubMed]
- Ranek, M.J.; Kokkonen-Simon, K.M.; Chen, A.; Dunkerly-Eyring, B.L.; Vera, M.P.; Oeing, C.U.; Patel, C.H.; Nakamura, T.; Zhu, G.; Bedja, D.; et al. PKG1-modified TSC2 regulates mTORC1 activity to counter adverse cardiac stress. Nature 2019, 566, 264–269. [Google Scholar] [CrossRef]
- Seol, S.Y.; Lee, S.Y.; Kim, Y.D.; Do, E.J.; Kwon, J.A.; Kim, S.I.; Chu, I.S.; Leem, S.H. Minisatellite polymorphisms of the SLC6A19: Susceptibility in hypertension. Biochem. Biophys. Res. Commun. 2008, 374, 714–719. [Google Scholar] [CrossRef]
- Franchi-Gazzola, R.; Visigalli, R.; Dall’Asta, V.; Sala, R.; Woo, S.K.; Kwon, H.M.; Gazzola, G.C.; Bussolati, O. Amino acid depletion activates TonEBP and sodium-coupled inositol transport. Am. J. Physiol. Cell Physiol. 2001, 280, C1465–C1474. [Google Scholar] [CrossRef] [PubMed]
- Dmitrieva, N.I.; Burg, M.B. Secretion of von Willebrand factor by endothelial cells links sodium to hypercoagulability and thrombosis. Proc. Natl. Acad. Sci. USA 2014, 111, 6485–6490. [Google Scholar] [CrossRef] [Green Version]
- Madonna, R.; Giovannelli, G.; Confalone, P.; Renna, F.V.; Geng, Y.J.; De Caterina, R. High glucose-induced hyperosmolarity contributes to COX-2 expression and angiogenesis: Implications for diabetic retinopathy. Cardiovasc. Diabetol. 2016, 15, 18. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Li, W.; Peng, G.; Li, F. Crystal structure of NL63 respiratory coronavirus receptor-binding domain complexed with its human receptor. Proc. Natl. Acad. Sci. USA 2009, 106, 19970–19974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Simmons, G.; Gosalia, D.N.; Rennekamp, A.J.; Reeves, J.D.; Diamond, S.L.; Bates, P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. USA 2005, 102, 11876–11881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Pohlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784. [Google Scholar] [CrossRef]
- Brielle, E.S.; Schneidman-Duhovny, D.; Linial, M. The SARS-CoV-2 Exerts a Distinctive Strategy for Interacting with the ACE2 Human Receptor. Viruses 2020, 12, 497. [Google Scholar] [CrossRef]
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res. 2020, 176, 104742. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Kota, P.; Gentzsch, M.; Dang, Y.L.; Boucher, R.C.; Stutts, M.J. The N terminus of alpha-ENaC mediates ENaC cleavage and activation by furin. J. Gen. Physiol. 2018, 150, 1179–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C.; et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020, 63, 364–374. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.; Fan, Y.; Chen, M.; Wu, X.; Zhang, L.; He, T.; Wang, H.; Wan, J.; Wang, X.; Lu, Z. Cardiovascular Implications of Fatal Outcomes of Patients With Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fosbol, E.L.; Butt, J.H.; Ostergaard, L.; Andersson, C.; Selmer, C.; Kragholm, K.; Schou, M.; Phelps, M.; Gislason, G.H.; Gerds, T.A.; et al. Association of Angiotensin-Converting Enzyme Inhibitor or Angiotensin Receptor Blocker Use With COVID-19 Diagnosis and Mortality. JAMA 2020, 323, 1769–1770. [Google Scholar] [CrossRef]
- Jiang, R.D.; Liu, M.Q.; Chen, Y.; Shan, C.; Zhou, Y.W.; Shen, X.R.; Li, Q.; Zhang, L.; Zhu, Y.; Si, H.R.; et al. Pathogenesis of SARS-CoV-2 in Transgenic Mice Expressing Human Angiotensin-Converting Enzyme 2. Cell 2020, 182, 50–58. [Google Scholar] [CrossRef]
- Gualtieri, P.; Falcone, C.; Romano, L.; Macheda, S.; Correale, P.; Arciello, P.; Polimeni, N.; Lorenzo, A. Body Composition Findings by Computed Tomography in SARS-CoV-2 Patients: Increased Risk of Muscle Wasting in Obesity. Int. J. Mol. Sci. 2020, 21, 4670. [Google Scholar] [CrossRef] [PubMed]
- Mao, R.; Qiu, Y.; He, J.S.; Tan, J.Y.; Li, X.H.; Liang, J.; Shen, J.; Zhu, L.R.; Chen, Y.; Iacucci, M.; et al. Manifestations and prognosis of gastrointestinal and liver involvement in patients with COVID-19: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2020, 5, 667–678. [Google Scholar] [CrossRef]
- Kiekens, C.; Boldrini, P.; Andreoli, A.; Avesani, R.; Gamna, F.; Grandi, M.; Lombardi, F.; Lusuardi, M.; Molteni, F.; Perboni, A.; et al. Rehabilitation and respiratory management in the acute and early post-acute phase. “Instant paper from the field” on rehabilitation answers to the Covid-19 emergency. Eur. J. Phys. Rehabil. Med. 2020, 56, 323–326. [Google Scholar] [CrossRef] [PubMed]
- South, A.M.; Tomlinson, L.; Edmonston, D.; Hiremath, S.; Sparks, M.A. Controversies of renin-angiotensin system inhibition during the COVID-19 pandemic. Nat. Rev. Nephrol. 2020, 16, 305–307. [Google Scholar] [CrossRef] [Green Version]
- Myti, D.; Gunjak, M.; Casado, F.; Khaghani Raziabad, S.; Nardiello, C.; Vadasz, I.; Herold, S.; Pryhuber, G.; Seeger, W.; Morty, R.E. Elevated FiO2 increases SARS-CoV-2 co-receptor expression in respiratory tract epithelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 319, L670–L674. [Google Scholar] [CrossRef]
- Berman, J.M.; Awayda, R.G.; Awayda, M.S. Interacting domains in the epithelial sodium channel that mediate proteolytic activation. Channels 2015, 9, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.; Ali, G.; Nie, H.G.; Chang, Y.; Bhattarai, D.; Su, X.; Zhao, X.; Matthay, M.A.; Ji, H.L. Plasmin improves blood-gas barrier function in oedematous lungs by cleaving epithelial sodium channels. Br. J. Pharmacol. 2020, 177, 3091–3106. [Google Scholar] [CrossRef]
- Vallet, V.; Chraibi, A.; Gaeggeler, H.P.; Horisberger, J.D.; Rossier, B.C. An epithelial serine protease activates the amiloride-sensitive sodium channel. Nature 1997, 389, 607–610. [Google Scholar] [CrossRef]
- Chen, D.; Li, X.; Song, Q.; Hu, C.; Su, F.; Dai, J.; Ye, Y.; Huang, J.; Zhang, X. Assessment of Hypokalemia and Clinical Characteristics in Patients With Coronavirus Disease 2019 in Wenzhou, China. JAMA Netw. Open 2020, 3, e2011122. [Google Scholar] [CrossRef]
- Lippi, G.; South, A.M.; Henry, B.M. Electrolyte imbalances in patients with severe coronavirus disease 2019 (COVID-19). Ann. Clin. Biochem. 2020, 57, 262–265. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, T.L.; Candeia-Medeiros, N.; Cavalcante-Araujo, P.M.; Melo, I.S.; Favaro-Pipi, E.; Fatima, L.A.; Rocha, A.A.; Goulart, L.R.; Machado, U.F.; Campos, R.R.; et al. SGLT1 activity in lung alveolar cells of diabetic rats modulates airway surface liquid glucose concentration and bacterial proliferation. Sci. Rep. 2016, 6, 21752. [Google Scholar] [CrossRef] [PubMed]
- Palermo, N.E.; Sadhu, A.R.; McDonnell, M.E. Diabetic Ketoacidosis in COVID-19: Unique concerns and considerations. J. Clin. Endocrinol. Metab. 2020, 105, dgaa360. [Google Scholar] [CrossRef]
- Kang, J.U.; Koo, S.H.; Kwon, K.C.; Park, J.W.; Kim, J.M. Gain at chromosomal region 5p15.33, containing TERT, is the most frequent genetic event in early stages of non-small cell lung cancer. Cancer Genet. Cytogenet. 2008, 182, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yan, X.; Fan, Q.; Liu, H.; Liu, X.; Liu, Z.; Zhang, Z. D-dimer levels on admission to predict in-hospital mortality in patients with Covid-19. J. Thromb. Haemost. 2020, 18, 1324–1329. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wu, D.; Chen, H.; Yan, W.; Yang, D.; Chen, G.; Ma, K.; Xu, D.; Yu, H.; Wang, H.; et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: Retrospective study. BMJ 2020, 368, m1091. [Google Scholar] [CrossRef] [Green Version]
- Richardson, S.; Hirsch, J.S.; Narasimhan, M.; Crawford, J.M.; McGinn, T.; Davidson, K.W.; the Northwell, C.-R.C.; Barnaby, D.P.; Becker, L.B.; Chelico, J.D.; et al. Presenting Characteristics, Comorbidities, and Outcomes Among 5700 Patients Hospitalized With COVID-19 in the New York City Area. JAMA 2020, 323, 2052–2059. [Google Scholar] [CrossRef] [PubMed]
- Gavriilaki, E.; Brodsky, R.A. Severe COVID-19 infection and thrombotic microangiopathy: Success does not come easily. Br. J. Haematol. 2020, 189, e227–e230. [Google Scholar] [CrossRef]
- Nahum, J.; Morichau-Beauchant, T.; Daviaud, F.; Echegut, P.; Fichet, J.; Maillet, J.M.; Thierry, S. Venous Thrombosis Among Critically Ill Patients With Coronavirus Disease 2019 (COVID-19). JAMA Netw. Open 2020, 3, e2010478. [Google Scholar] [CrossRef]
- Long, B.; Brady, W.J.; Koyfman, A.; Gottlieb, M. Cardiovascular complications in COVID-19. Am. J. Emerg. Med. 2020, 38, 1504–1507. [Google Scholar] [CrossRef]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Docherty, A.B.; Harrison, E.M.; Green, C.A.; Hardwick, H.E.; Pius, R.; Norman, L.; Holden, K.A.; Read, J.M.; Dondelinger, F.; Carson, G.; et al. Features of 20 133 UK patients in hospital with covid-19 using the ISARIC WHO Clinical Characterisation Protocol: Prospective observational cohort study. BMJ 2020, 369, m1985. [Google Scholar] [CrossRef]
- Ding, Y.; Wang, H.; Shen, H.; Li, Z.; Geng, J.; Han, H.; Cai, J.; Li, X.; Kang, W.; Weng, D.; et al. The clinical pathology of severe acute respiratory syndrome (SARS): A report from China. J. Pathol. 2003, 200, 282–289. [Google Scholar] [CrossRef]
- Chen, J.; Jiang, Q.; Xia, X.; Liu, K.; Yu, Z.; Tao, W.; Gong, W.; Han, J.J. Individual variation of the SARS-CoV-2 receptor ACE2 gene expression and regulation. Aging Cell 2020, 19, e13168. [Google Scholar] [CrossRef]
- Mogi, M. Effect of renin-angiotensin system on senescence. Geriatr. Gerontol. Int. 2020, 20, 520–525. [Google Scholar] [CrossRef]
- Ellinghaus, K. The ABO blood group locus and a chromosome 3 gene cluster associate with SARS-CoV-2 respiratory failure in an Italian-Spanish genome-wide association analysis. medRxiv 2020. [Google Scholar] [CrossRef]
- Bigiani, A. Does ENaC Work as Sodium Taste Receptor in Humans? Nutrients 2020, 12, 1195. [Google Scholar] [CrossRef]
- Shigemura, N.; Ohkuri, T.; Sadamitsu, C.; Yasumatsu, K.; Yoshida, R.; Beauchamp, G.K.; Bachmanov, A.A.; Ninomiya, Y. Amiloride-sensitive NaCl taste responses are associated with genetic variation of ENaC alpha-subunit in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R66–R75. [Google Scholar] [CrossRef] [Green Version]
- Eylam, S.; Spector, A.C. Oral amiloride treatment decreases taste sensitivity to sodium salts in C57BL/6J and DBA/2J mice. Chem. Senses 2003, 28, 447–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamura, H.; Ugawa, S.; Ueda, T.; Nagao, M.; Joh, T.; Shimada, S. Epithelial Na+ channel delta subunit is an acid sensor in the human oesophagus. Eur. J. Pharmacol. 2008, 600, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Nomura, K.; Nakanishi, M.; Ishidate, F.; Iwata, K.; Taruno, A. All-Electrical Ca(2+)-Independent Signal Transduction Mediates Attractive Sodium Taste in Taste Buds. Neuron 2020, 106, 816–829. [Google Scholar] [CrossRef]
- Chandrashekar, J.; Kuhn, C.; Oka, Y.; Yarmolinsky, D.A.; Hummler, E.; Ryba, N.J.; Zuker, C.S. The cells and peripheral representation of sodium taste in mice. Nature 2010, 464, 297–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigemura, N.; Iwata, S.; Yasumatsu, K.; Ohkuri, T.; Horio, N.; Sanematsu, K.; Yoshida, R.; Margolskee, R.F.; Ninomiya, Y. Angiotensin II modulates salty and sweet taste sensitivities. J. Neurosci. 2013, 33, 6267–6277. [Google Scholar] [CrossRef] [PubMed]
- Kumarhia, D.; He, L.; McCluskey, L.P. Inflammatory stimuli acutely modulate peripheral taste function. J. Neurophysiol. 2016, 115, 2964–2975. [Google Scholar] [CrossRef] [Green Version]
- Bangel, N.; Dahlhoff, C.; Sobczak, K.; Weber, W.M.; Kusche-Vihrog, K. Upregulated expression of ENaC in human CF nasal epithelium. J. Cyst Fibros. 2008, 7, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Bilinska, K.; Jakubowska, P.; Von Bartheld, C.S.; Butowt, R. Expression of the SARS-CoV-2 Entry Proteins, ACE2 and TMPRSS2, in Cells of the Olfactory Epithelium: Identification of Cell Types and Trends with Age. ACS Chem. Neurosci. 2020, 11, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- Ng, R.; Salem, S.S.; Wu, S.T.; Wu, M.; Lin, H.H.; Shepherd, A.K.; Joiner, W.J.; Wang, J.W.; Su, C.Y. Amplification of Drosophila Olfactory Responses by a DEG/ENaC Channel. Neuron 2019, 104, 947–950. [Google Scholar] [CrossRef]
- Chodroff, L.; Bendele, M.; Valenzuela, V.; Henry, M.; Ruparel, S. EXPRESS: BDNF Signaling Contributes to Oral Cancer Pain in a Preclinical Orthotopic Rodent Model. Mol. Pain 2016, 12. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Zhong, L.; Deng, J.; Peng, J.; Dan, H.; Zeng, X.; Li, T.; Chen, Q. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral Sci. 2020, 12, 8. [Google Scholar] [CrossRef]
- Yamamura, H.; Ugawa, S.; Ueda, T.; Nagao, M.; Shimada, S. Protons activate the delta-subunit of the epithelial Na+ channel in humans. J. Biol. Chem. 2004, 279, 12529–12534. [Google Scholar] [CrossRef] [Green Version]
- Yamamura, H.; Ugawa, S.; Ueda, T.; Nagao, M.; Shimada, S. Epithelial Na+ channel delta subunit mediates acid-induced ATP release in the human skin. Biochem. Biophys. Res. Commun. 2008, 373, 155–158. [Google Scholar] [CrossRef]
- Ji, H.L.; Benos, D.J. Degenerin sites mediate proton activation of deltabetagamma-epithelial sodium channel. J. Biol. Chem. 2004, 279, 26939–26947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orr, B.O.; Gorczyca, D.; Younger, M.A.; Jan, L.Y.; Jan, Y.N.; Davis, G.W. Composition and Control of a Deg/ENaC Channel during Presynaptic Homeostatic Plasticity. Cell Rep. 2017, 20, 1855–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasker, J.G.; Prager-Khoutorsky, M.; Teruyama, R.; Lemos, J.R.; Amstrong, W.E. Advances in the neurophysiology of magnocellular neuroendocrine cells. J. Neuroendocrinol. 2020, 32, e12826. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Artunc, F.; Vallon, V. The physiological impact of the serum and glucocorticoid-inducible kinase SGK1. Curr. Opin. Nephrol. Hypertens. 2009, 18, 439–448. [Google Scholar] [CrossRef]
- Wesch, D.; Miranda, P.; Afonso-Oramas, D.; Althaus, M.; Castro-Hernandez, J.; Dominguez, J.; Morty, R.E.; Clauss, W.; Gonzalez-Hernandez, T.; Alvarez de la Rosa, D.; et al. The neuronal-specific SGK1.1 kinase regulates {delta}-epithelial Na+ channel independently of PY motifs and couples it to phospholipase C signaling. Am. J. Physiol. Cell Physiol. 2010, 299, C779–C790. [Google Scholar] [CrossRef]
- Mills, N.J.; Sharma, K.; Haque, M.; Moore, M.; Teruyama, R. Aldosterone Mediated Regulation of Epithelial Sodium Channel (ENaC) Subunits in the Rat Hypothalamus. Neuroscience 2018, 390, 278–292. [Google Scholar] [CrossRef]
- Yanpallewar, S.; Wang, T.; Koh, D.C.; Quarta, E.; Fulgenzi, G.; Tessarollo, L. Nedd4-2 haploinsufficiency causes hyperactivity and increased sensitivity to inflammatory stimuli. Sci. Rep. 2016, 6, 32957. [Google Scholar] [CrossRef]
- Garcia-Caballero, A.; Gandini, M.A.; Huang, S.; Chen, L.; Souza, I.A.; Dang, Y.L.; Stutts, M.J.; Zamponi, G.W. Cav3.2 calcium channel interactions with the epithelial sodium channel ENaC. Mol. Brain 2019, 12, 12. [Google Scholar] [CrossRef]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/search/ENaC (accessed on 17 August 2020).
- Lam, D.K.; Dang, D.; Flynn, A.N.; Hardt, M.; Schmidt, B.L. TMPRSS2, a novel membrane-anchored mediator in cancer pain. Pain 2015, 156, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Kehoe, P.G.; Wong, S.; Al Mulhim, N.; Palmer, L.E.; Miners, J.S. Angiotensin-converting enzyme 2 is reduced in Alzheimer’s disease in association with increasing amyloid-beta and tau pathology. Alzheimers Res. Ther. 2016, 8, 50. [Google Scholar] [CrossRef] [Green Version]
- Krause, E.G.; de Kloet, A.D.; Scott, K.A.; Flak, J.N.; Jones, K.; Smeltzer, M.D.; Ulrich-Lai, Y.M.; Woods, S.C.; Wilson, S.P.; Reagan, L.P.; et al. Blood-borne angiotensin II acts in the brain to influence behavioral and endocrine responses to psychogenic stress. J. Neurosci. 2011, 31, 15009–15015. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; de Kloet, A.D.; Pati, D.; Hiller, H.; Smith, J.A.; Pioquinto, D.J.; Ludin, J.A.; Oh, S.P.; Katovich, M.J.; Frazier, C.J.; et al. Increasing brain angiotensin converting enzyme 2 activity decreases anxiety-like behavior in male mice by activating central Mas receptors. Neuropharmacology 2016, 105, 114–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.A.; de Kloet, A.D.; Smeltzer, M.D.; Cahill, K.M.; Hiller, H.; Bruce, E.B.; Pioquinto, D.J.; Ludin, J.A.; Katovich, M.J.; Raizada, M.K.; et al. Coupling corticotropin-releasing-hormone and angiotensin converting enzyme 2 dampens stress responsiveness in male mice. Neuropharmacology 2018, 133, 85–93. [Google Scholar] [CrossRef]
- de Kloet, A.D.; Cahill, K.M.; Scott, K.A.; Krause, E.G. Overexpression of angiotensin converting enzyme 2 reduces anxiety-like behavior in female mice. Physiol. Behav. 2020, 224, 113002. [Google Scholar] [CrossRef]
- Gu, J.; Gong, E.; Zhang, B.; Zheng, J.; Gao, Z.; Zhong, Y.; Zou, W.; Zhan, J.; Wang, S.; Xie, Z.; et al. Multiple organ infection and the pathogenesis of SARS. J. Exp. Med. 2005, 202, 415–424. [Google Scholar] [CrossRef]
- Song, L. Neuroinvasive potential of SARS-CoV-2 revealed in a human brain organoid model. BioRxiv 2000. [Google Scholar] [CrossRef]
- Song, E.; Zhang, C.; Israelow, B.; Lu-Culligan, A.; Prado, A.V.; Skriabine, S.; Lu, P.; Weizman, O.-E.; Liu, F.; Dai, Y.; et al. Neuroinvasion of SARS-CoV-2 in human and mouse brain. BioRxiv 2020. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020. [Google Scholar] [CrossRef]
- Fotuhi, M.; Mian, A.; Meysami, S.; Raji, C.A. Neurobiology of COVID-19. J. Alzheimers Dis. 2020, 76, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, M.C.; Ascione, T.; Pagliano, P. Neurologic aspects of covid-19: A concise review. Infez. Med. 2020, 28, 42–45. [Google Scholar]
- Sheraton, M.; Deo, N.; Kashyap, R.; Surani, S. A Review of Neurological Complications of COVID-19. Cureus 2020, 12, e8192. [Google Scholar] [CrossRef]
- Helms, J.; Kremer, S.; Merdji, H.; Clere-Jehl, R.; Schenck, M.; Kummerlen, C.; Collange, O.; Boulay, C.; Fafi-Kremer, S.; Ohana, M.; et al. Neurologic Features in Severe SARS-CoV-2 Infection. N. Engl. J. Med. 2020, 382, 2268–2270. [Google Scholar] [CrossRef]
- Lechien, J.R.; Chiesa-Estomba, C.M.; De Siati, D.R.; Horoi, M.; Le Bon, S.D.; Rodriguez, A.; Dequanter, D.; Blecic, S.; El Afia, F.; Distinguin, L.; et al. Olfactory and gustatory dysfunctions as a clinical presentation of mild-to-moderate forms of the coronavirus disease (COVID-19): A multicenter European study. Eur. Arch. Otorhinolaryngol. 2020, 277, 2251–2261. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Sriramula, S.; Xia, H.; Moreno-Walton, L.; Culicchia, F.; Domenig, O.; Poglitsch, M.; Lazartigues, E. Clinical Relevance and Role of Neuronal AT1 Receptors in ADAM17-Mediated ACE2 Shedding in Neurogenic Hypertension. Circ. Res. 2017, 121, 43–55. [Google Scholar] [CrossRef]
- Giacomelli, A.; Pezzati, L.; Conti, F.; Bernacchia, D.; Siano, M.; Oreni, L.; Rusconi, S.; Gervasoni, C.; Ridolfo, A.L.; Rizzardini, G.; et al. Self-reported olfactory and taste disorders in SARS-CoV-2 patients: A cross-sectional study. Clin. Infect. Dis. 2020, 71, 889–890. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Deng, Y.; Dai, Z.; Meng, Z. COVID-19 and anosmia: A review based on up-to-date knowledge. Am. J. Otolaryngol. 2020, 41, 102581. [Google Scholar] [CrossRef] [PubMed]
- Epstein, D.; Andrawis, W.; Lipsky, A.M.; Ziad, H.A.; Matan, M. Anxiety and Suicidality in a Hospitalized Patient with COVID-19 Infection. Eur. J. Case Rep. Intern. Med. 2020, 7, 001651. [Google Scholar] [CrossRef] [PubMed]
- Bouhaddou, M.; Memon, D.; Meyer, B.; White, K.M.; Rezelj, V.V.; Marrero, M.C.; Polacco, B.J.; Melnyk, J.E.; Ulferts, S.; Kaake, R.M.; et al. The Global Phosphorylation Landscape of SARS-CoV-2 Infection. Cell 2020, 182, 685–712. [Google Scholar] [CrossRef]
- Altmayer, S.; Zanon, M.; Pacini, G.S.; Watte, G.; Barros, M.C.; Mohammed, T.L.; Verma, N.; Marchiori, E.; Hochhegger, B. Comparison of the computed tomography findings in COVID-19 and other viral pneumonia in immunocompetent adults: A systematic review and meta-analysis. Eur. Radiol. 2020, 27, 1–12. [Google Scholar]
- Lu, Y.; Liu, D.X.; Tam, J.P. Lipid rafts are involved in SARS-CoV entry into Vero E6 cells. Biochem. Biophys. Res. Commun. 2008, 369, 344–349. [Google Scholar] [CrossRef]
- Tellier, E.; Canault, M.; Rebsomen, L.; Bonardo, B.; Juhan-Vague, I.; Nalbone, G.; Peiretti, F. The shedding activity of ADAM17 is sequestered in lipid rafts. Exp. Cell Res. 2006, 312, 3969–3980. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Zimina, E.P.; Franzke, C.W.; Kutsch, S.; Siebolds, U.; Gordon, M.K.; Bruckner-Tuderman, L.; Koch, M. Shedding of collagen XXIII is mediated by furin and depends on the plasma membrane microenvironment. J. Biol. Chem. 2007, 282, 27424–27435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tellier, E.; Canault, M.; Poggi, M.; Bonardo, B.; Nicolay, A.; Alessi, M.C.; Nalbone, G.; Peiretti, F. HDLs activate ADAM17-dependent shedding. J. Cell. Physiol. 2008, 214, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Bifulco, M.; Gazzerro, P. Statin therapy in COVID-19 infection: Much more than a single pathway. Eur. Heart J. Cardiovasc. Pharmacother. 2020. [Google Scholar] [CrossRef]
- Subir, R.; Jagat, J.M.; Kalyan, K.G. Pros and cons for use of statins in people with coronavirus disease-19 (COVID-19). Diabetes Metab. Syndr. 2020, 14, 1225–1229. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.J.; Qin, J.J.; Cheng, X.; Shen, L.; Zhao, Y.C.; Yuan, Y.; Lei, F.; Chen, M.M.; Yang, H.; Bai, L.; et al. In-Hospital Use of Statins Is Associated with a Reduced Risk of Mortality among Individuals with COVID-19. Cell Metab. 2020, 32, 176–187. [Google Scholar] [CrossRef]
- Rodriguez-Nava, G.; Trelles-Garcia, D.P.; Yanez-Bello, M.A.; Chung, C.W.; Trelles-Garcia, V.P.; Friedman, H.J. Atorvastatin associated with decreased hazard for death in COVID-19 patients admitted to an ICU: A retrospective cohort study. Crit. Care 2020, 24, 429. [Google Scholar] [CrossRef]
- Yamaya, M.; Shimotai, Y.; Hatachi, Y.; Lusamba Kalonji, N.; Tando, Y.; Kitajima, Y.; Matsuo, K.; Kubo, H.; Nagatomi, R.; Hongo, S.; et al. The serine protease inhibitor camostat inhibits influenza virus replication and cytokine production in primary cultures of human tracheal epithelial cells. Pulm. Pharmacol. Ther. 2015, 33, 66–74. [Google Scholar] [CrossRef]
- Solun, B.; Shoenfeld, Y. Inhibition of metalloproteinases in therapy for severe lung injury due to COVID-19. Med. Drug Discov. 2020, 7, 100052. [Google Scholar] [CrossRef]
- Aggarwal, N.K.; Subramanian, A. Antifibrinolytics and cardiac surgery: The past, the present, and the future. Ann. Card Anaesth. 2020, 23, 193–199. [Google Scholar]
- Kim, T.S.; Heinlein, C.; Hackman, R.C.; Nelson, P.S. Phenotypic analysis of mice lacking the Tmprss2-encoded protease. Mol. Cell. Biol. 2006, 26, 965–975. [Google Scholar] [CrossRef] [Green Version]
- Wilson, L.; McKinlay, C.; Gage, P.; Ewart, G. SARS coronavirus E protein forms cation-selective ion channels. Virology 2004, 330, 322–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Morales, A.J.; Cardona-Ospina, J.A.; Gutierrez-Ocampo, E.; Villamizar-Pena, R.; Holguin-Rivera, Y.; Escalera-Antezana, J.P.; Alvarado-Arnez, L.E.; Bonilla-Aldana, D.K.; Franco-Paredes, C.; Henao-Martinez, A.F.; et al. Clinical, laboratory and imaging features of COVID-19: A systematic review and meta-analysis. Travel. Med. Infect. Dis. 2020, 34, 101623. [Google Scholar] [CrossRef] [PubMed]
- Klok, F.A.; Kruip, M.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020, 191, 145–147. [Google Scholar] [CrossRef]
- Wei, W.E.; Li, Z.; Chiew, C.J.; Yong, S.E.; Toh, M.P.; Lee, V.J. Presymptomatic Transmission of SARS-CoV-2 - Singapore, January 23-March 16 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 411–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of Covid-19 - Preliminary Report. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Lei, C.; Qian, K.; Li, T.; Zhang, S.; Fu, W.; Ding, M.; Hu, S. Neutralization of SARS-CoV-2 spike pseudotyped virus by recombinant ACE2-Ig. Nat. Commun. 2020, 11, 2070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libby, P.; Luscher, T. COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 2020, 41, 3038–3044. [Google Scholar] [CrossRef] [PubMed]
- Lowenstein, C.J.; Solomon, S.D. Severe COVID-19 is a Microvascular Disease. Circulation 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Scala, I.; Riccio, M.P.; Marino, M.; Bravaccio, C.; Parenti, G.; Strisciuglio, P. Large Neutral Amino Acids (LNAAs) Supplementation Improves Neuropsychological Performances in Adult Patients with Phenylketonuria. Nutrients 2020, 12, 1092. [Google Scholar] [CrossRef] [Green Version]
- Saeedi Saravi, S.S.; Beer, J.H. Apelin-potential therapy for COVID-19? J. Mol. Cell. Cardiol. 2020, 145, 84–87. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muhanna, D.; Arnipalli, S.R.; Kumar, S.B.; Ziouzenkova, O. Osmotic Adaptation by Na+-Dependent Transporters and ACE2: Correlation with Hemostatic Crisis in COVID-19. Biomedicines 2020, 8, 460. https://doi.org/10.3390/biomedicines8110460
Muhanna D, Arnipalli SR, Kumar SB, Ziouzenkova O. Osmotic Adaptation by Na+-Dependent Transporters and ACE2: Correlation with Hemostatic Crisis in COVID-19. Biomedicines. 2020; 8(11):460. https://doi.org/10.3390/biomedicines8110460
Chicago/Turabian StyleMuhanna, Danah, Shanvanth R. Arnipalli, Shashi B. Kumar, and Ouliana Ziouzenkova. 2020. "Osmotic Adaptation by Na+-Dependent Transporters and ACE2: Correlation with Hemostatic Crisis in COVID-19" Biomedicines 8, no. 11: 460. https://doi.org/10.3390/biomedicines8110460
APA StyleMuhanna, D., Arnipalli, S. R., Kumar, S. B., & Ziouzenkova, O. (2020). Osmotic Adaptation by Na+-Dependent Transporters and ACE2: Correlation with Hemostatic Crisis in COVID-19. Biomedicines, 8(11), 460. https://doi.org/10.3390/biomedicines8110460