Release of Anti-Inflammatory Palmitoleic Acid and Its Positional Isomers by Mouse Peritoneal Macrophages

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Gas Chromatography/Mass Spectrometry (GC/MS) Analyses
2.4. Liquid Chromatography/Mass Spectrometry (LC/MS) Analyses of Phospholipids
2.5. FAHFA Analysis by LC/MS
2.6. iPLA2-VIA Antisense Inhibition Studies
2.7. Data Analysis
3. Results
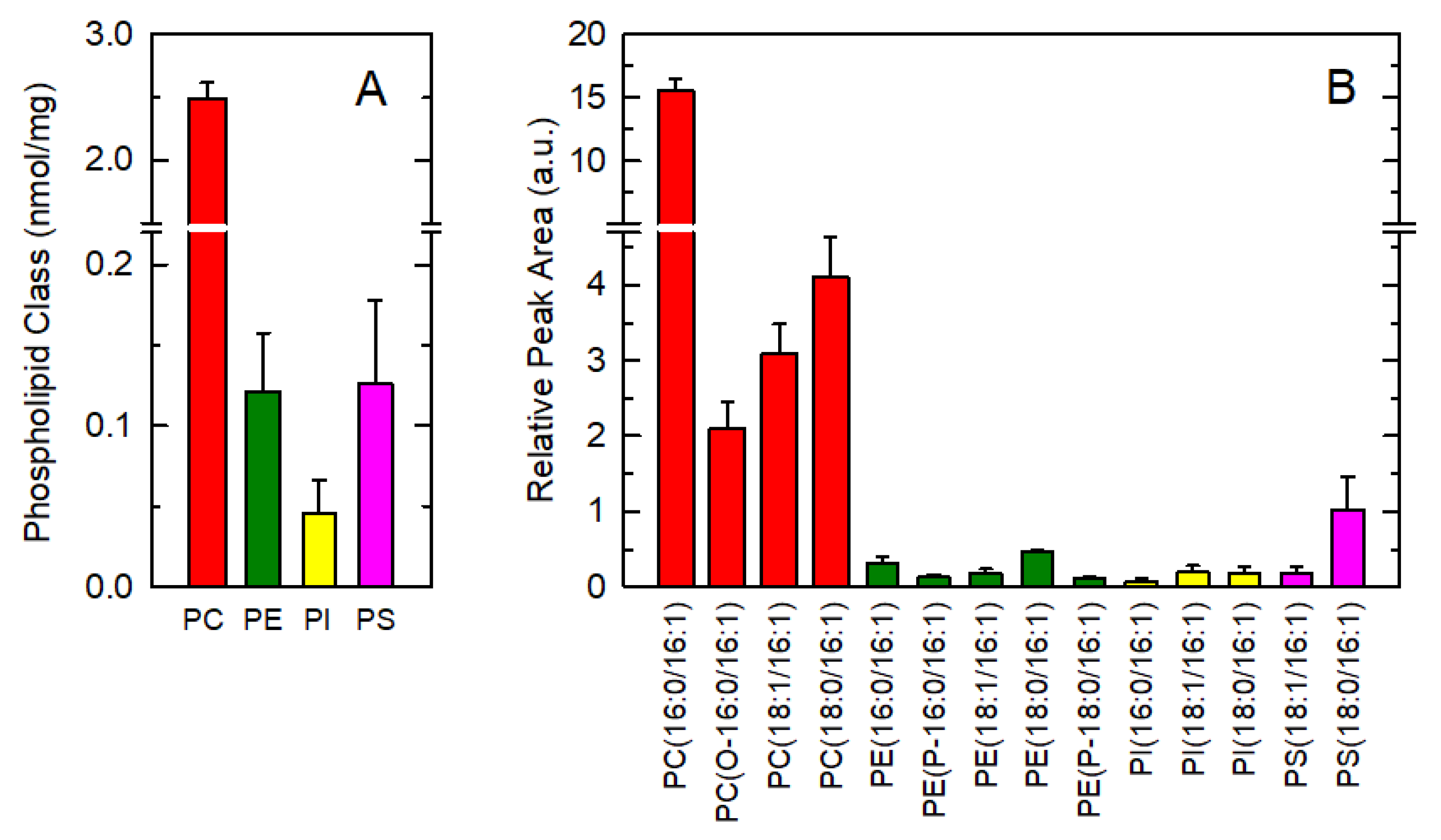
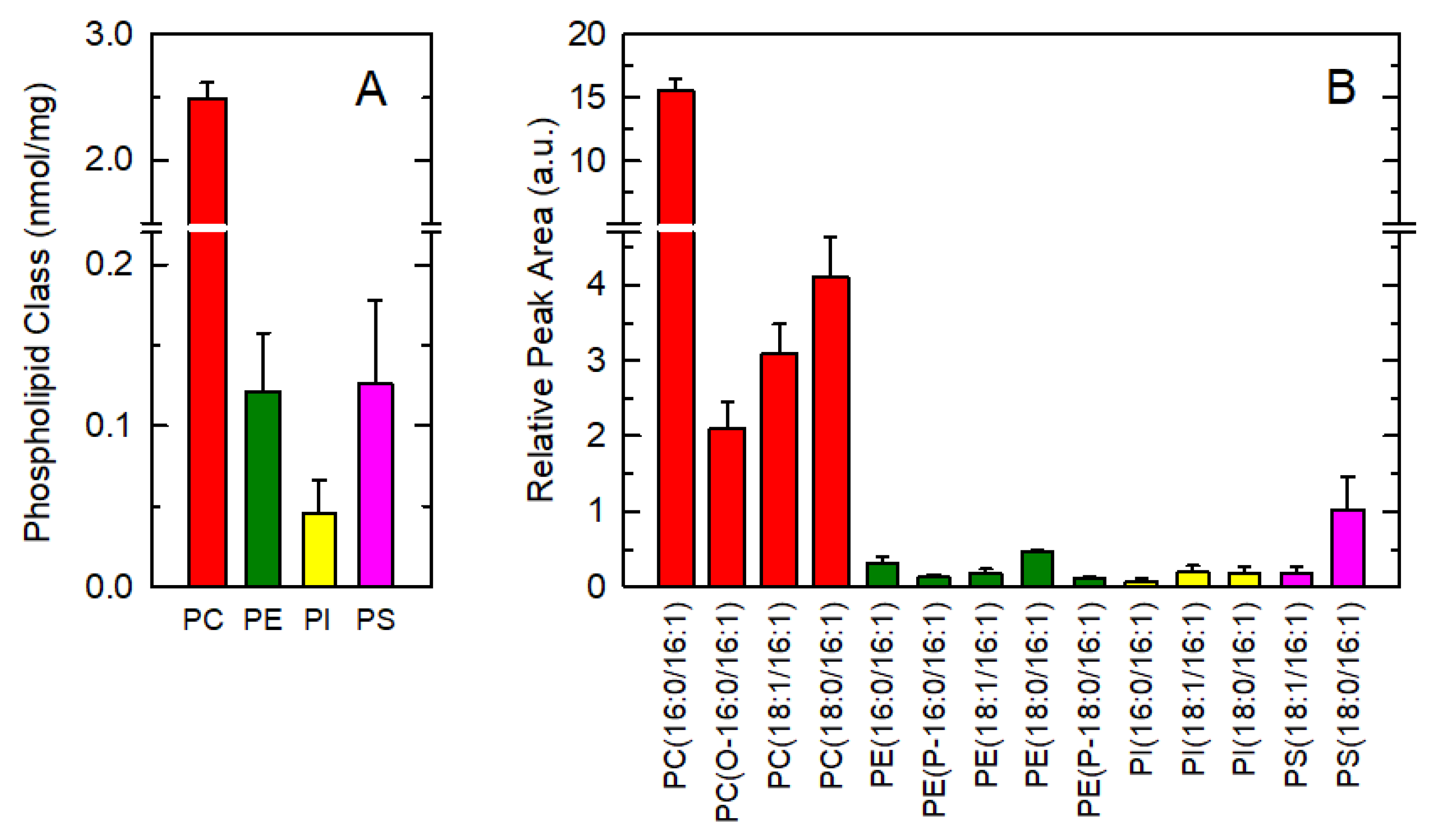
3.1. Endogenous Distribution of 16:1 Fatty Acids among Macrophage Phospholipids
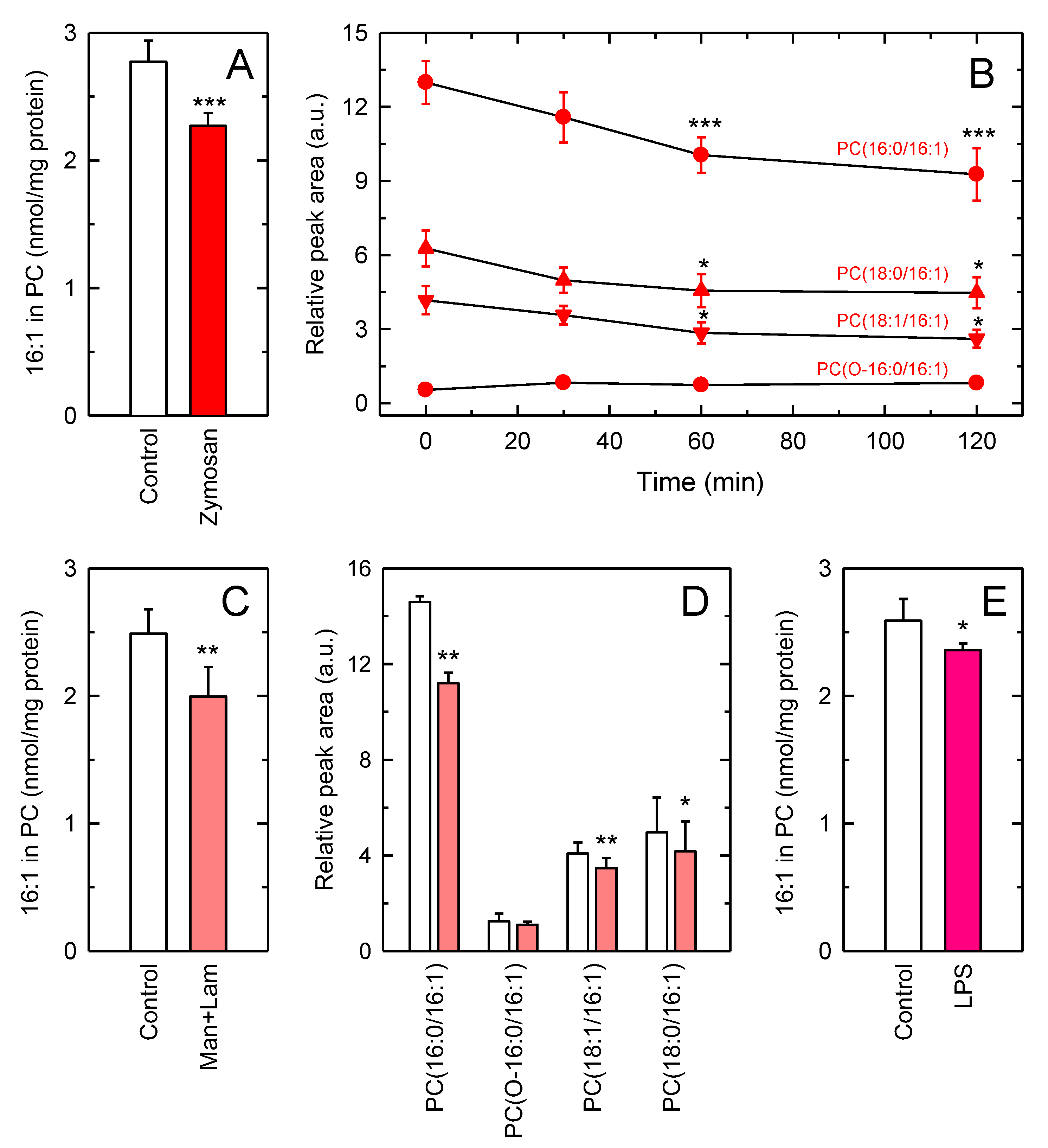
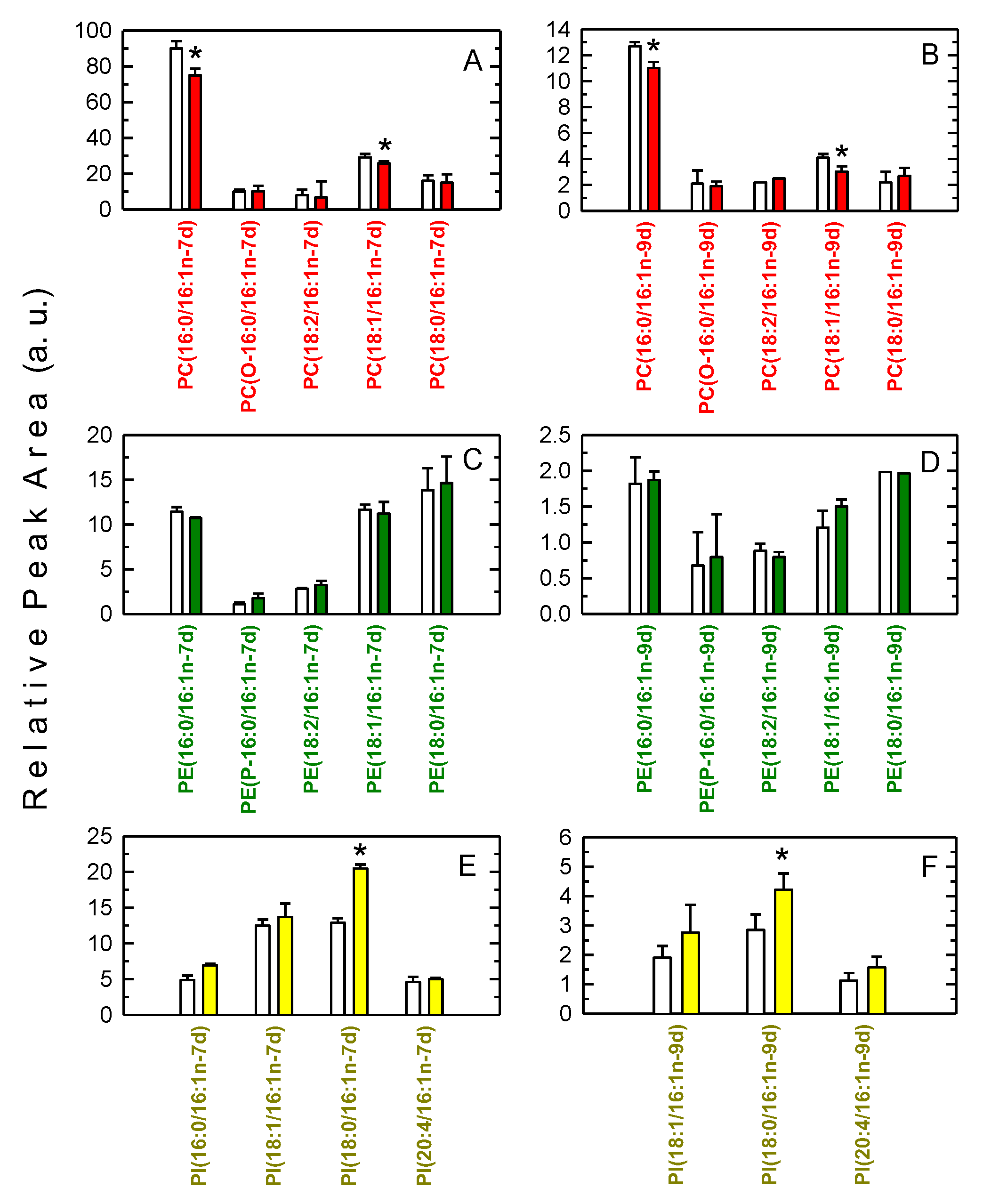
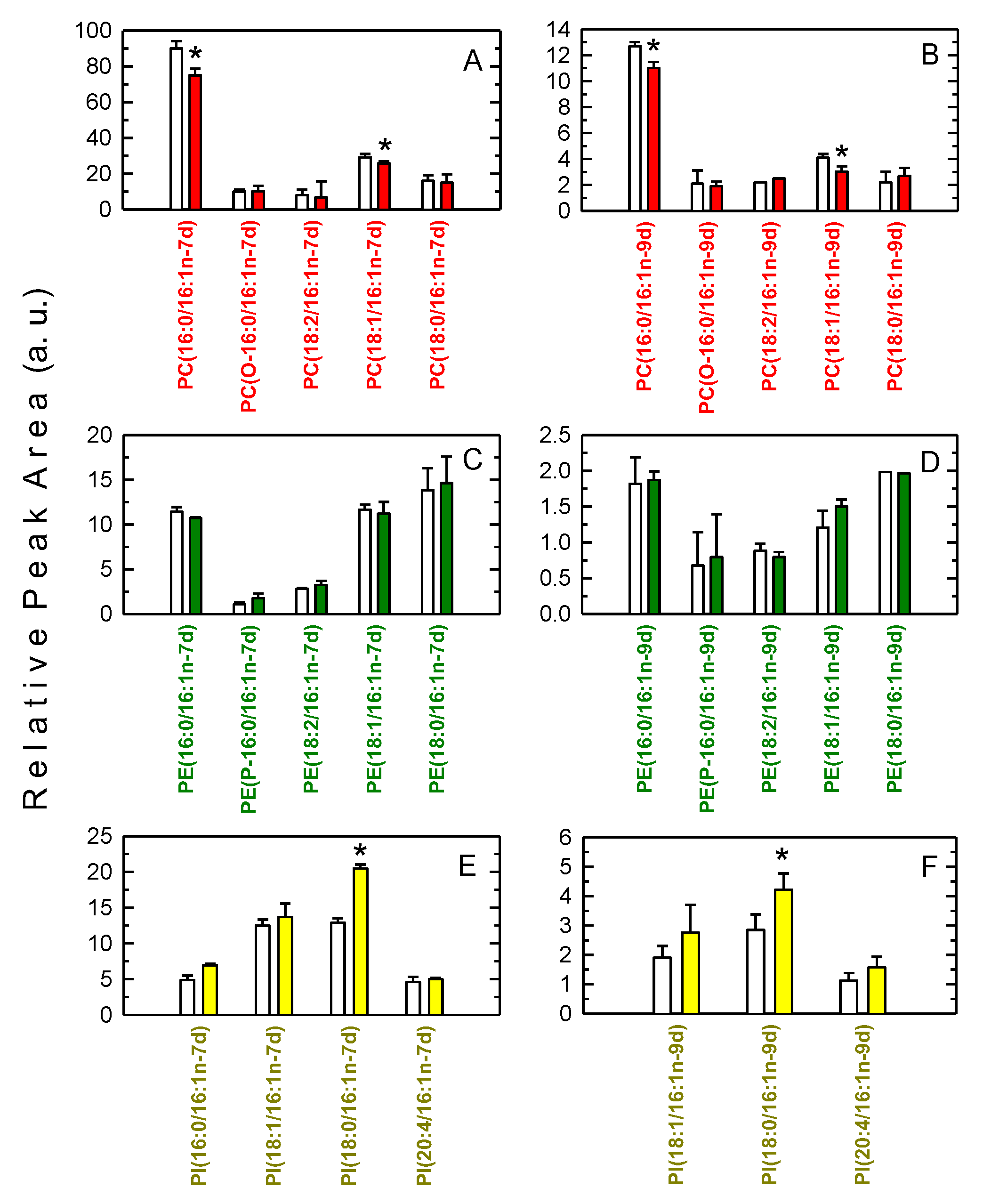
3.2. Decreased Levels of 16:1-Containing Phospholipid Species in Stimulated Macrophages
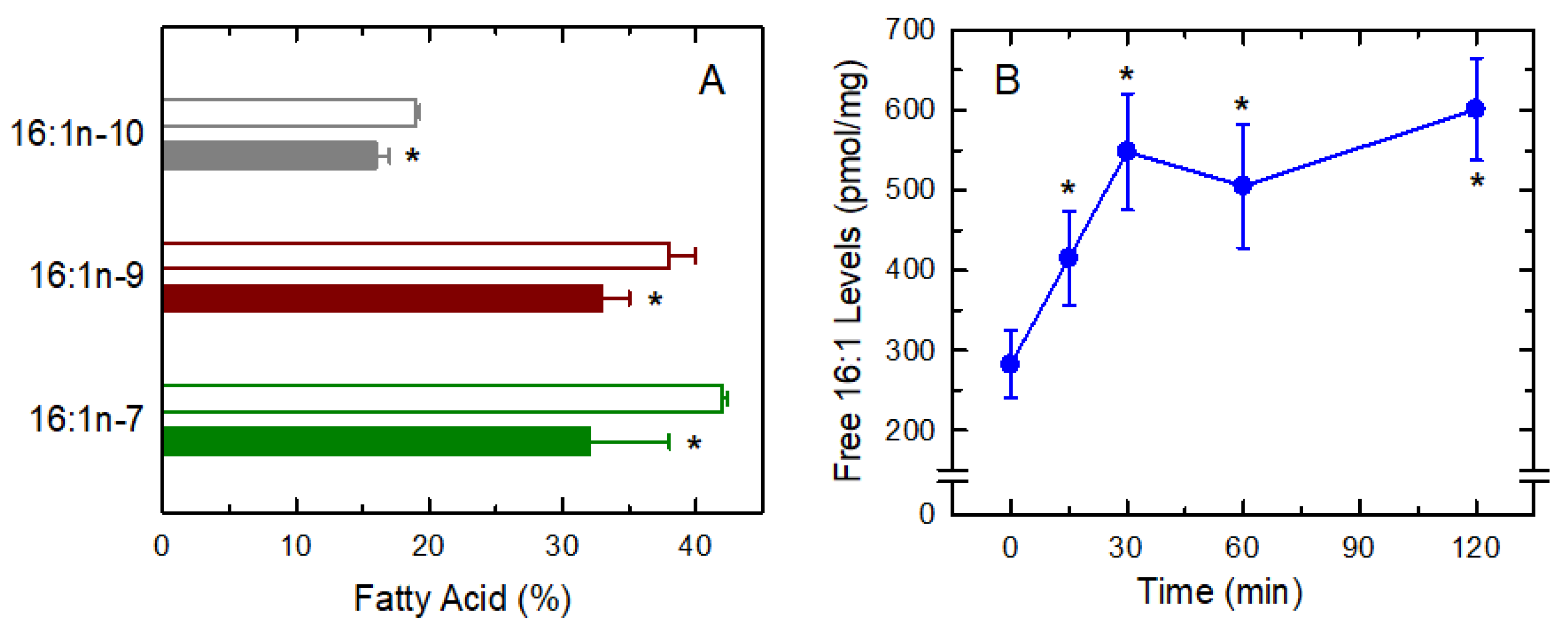
3.3. Fatty Acid Isomers in the Response
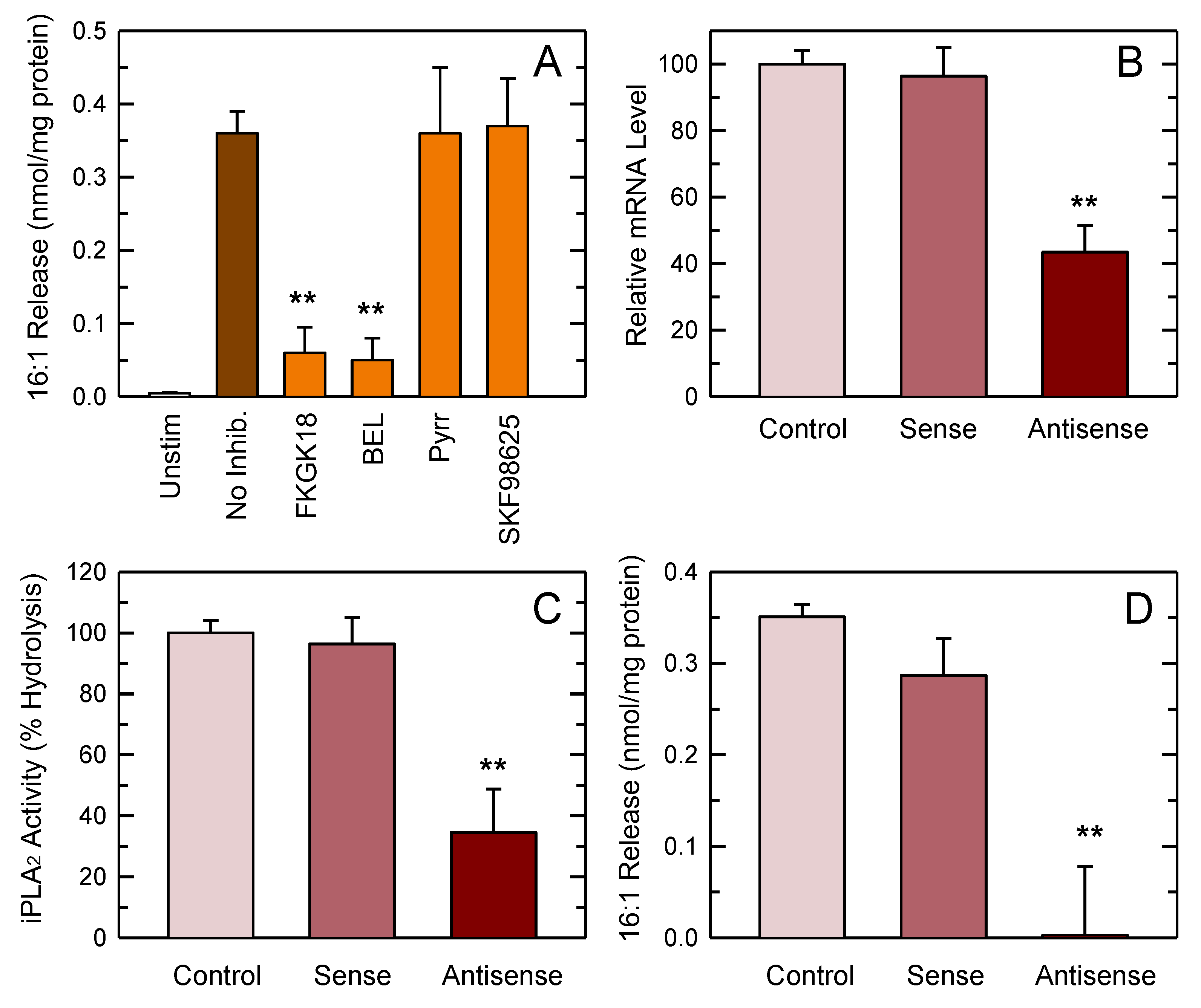
3.4. PLA2 Involvement in 16:1 Release
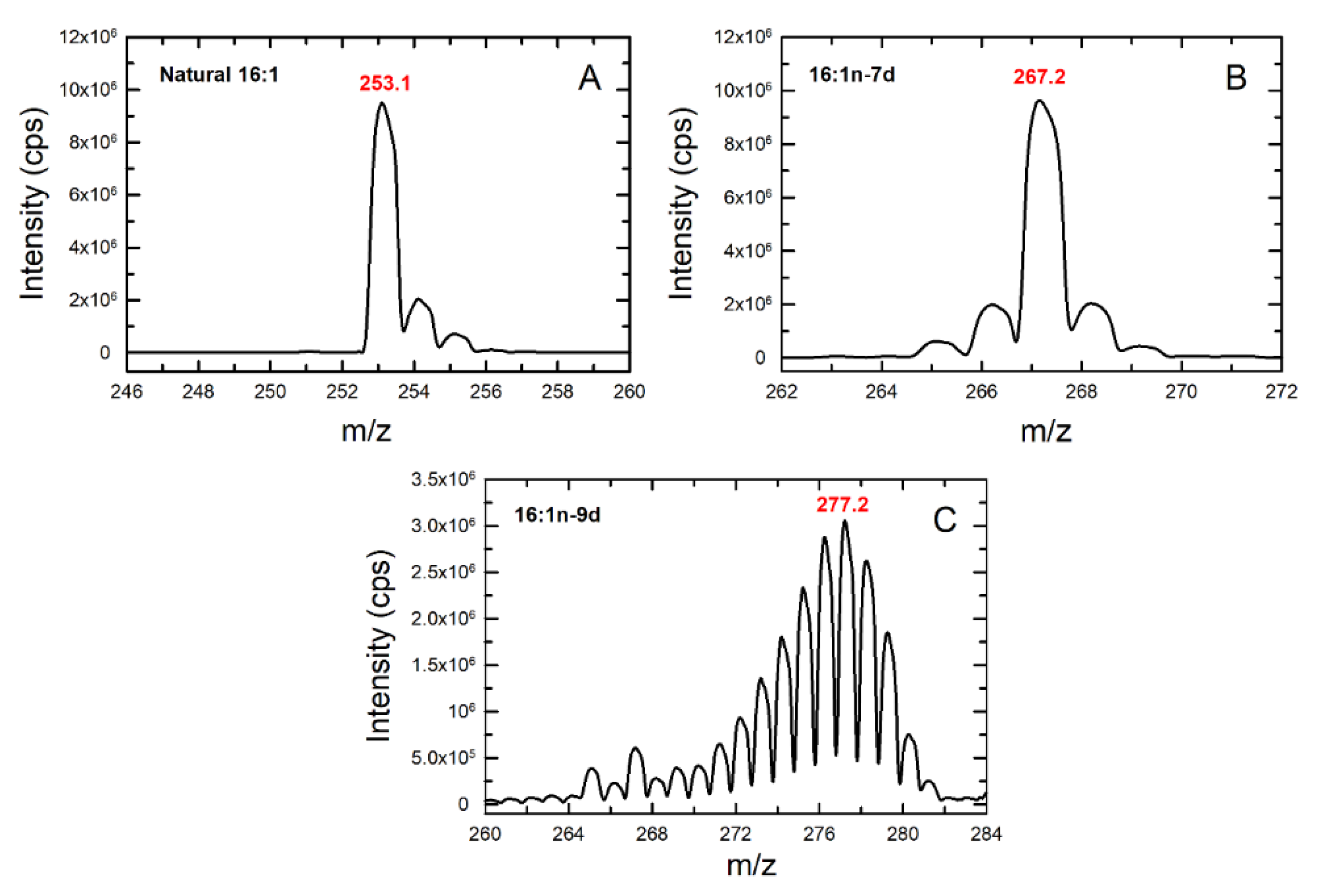
3.5. Studies with Deuterated Fatty Acids
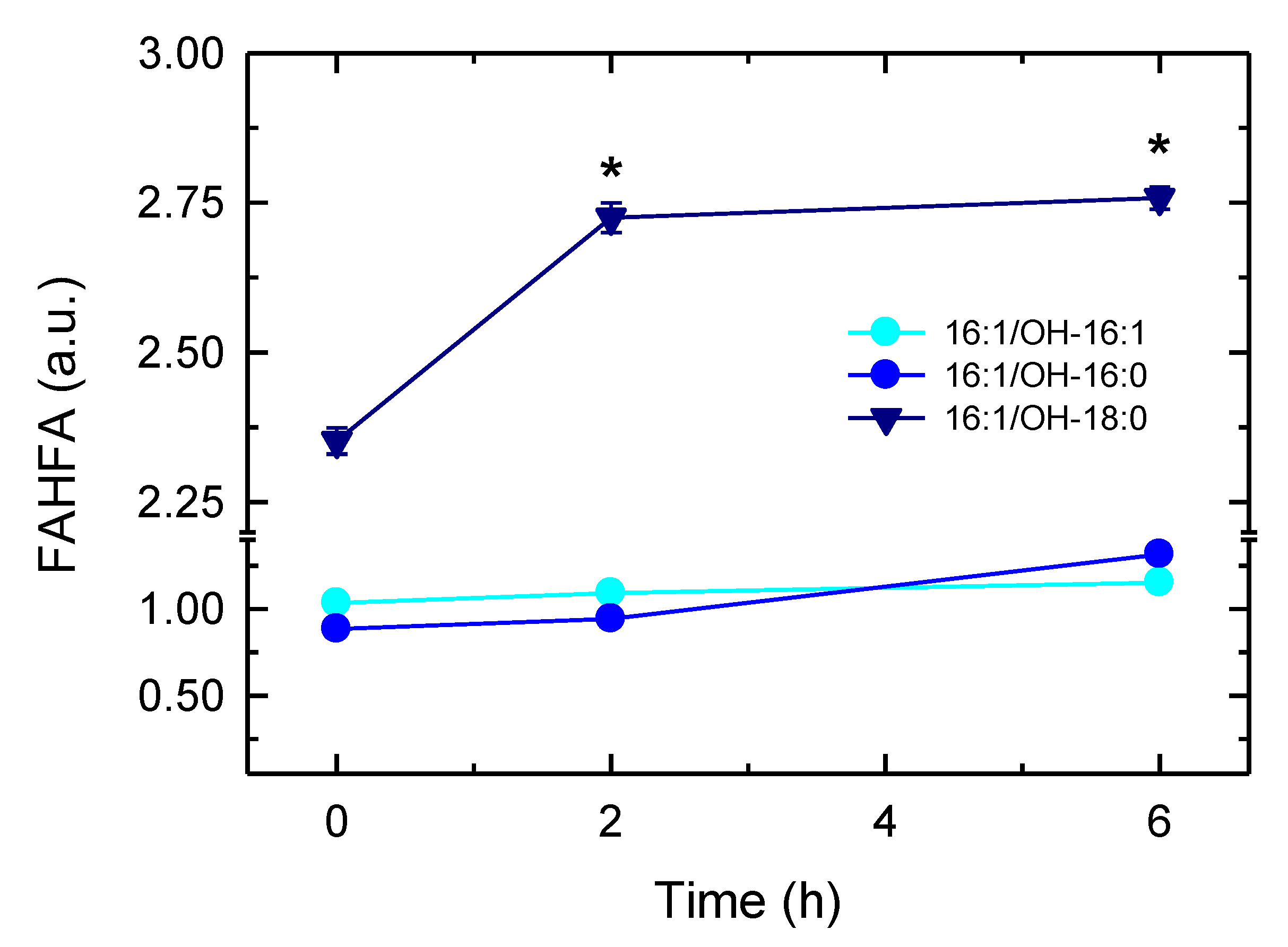
3.6. Synthesis of 16:1-Containing FAHFA
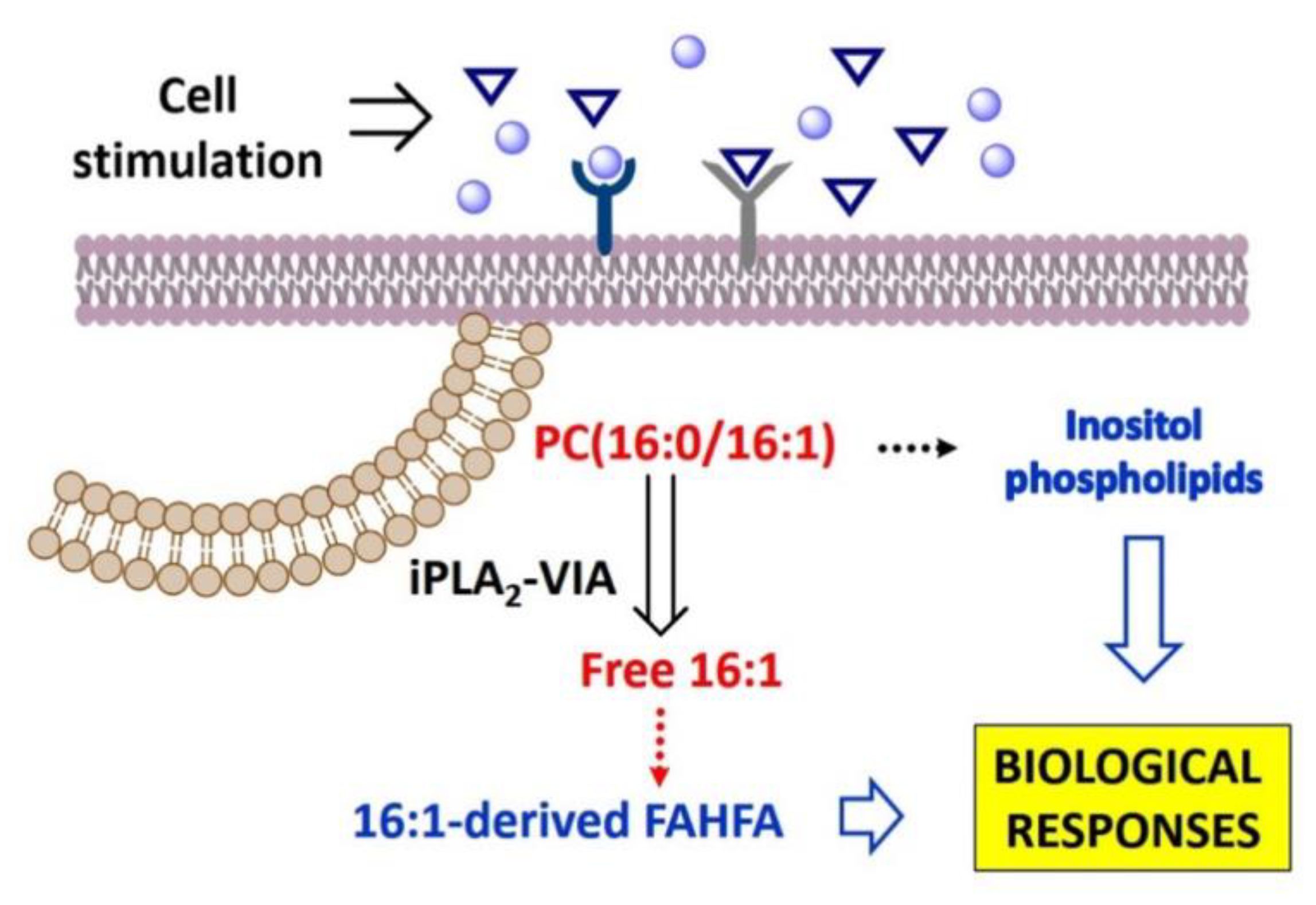
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| GC/MS | Gas chromatography coupled to mass spectrometry |
| LC/MS | Liquid chromatography coupled to mass spectrometry |
| FAHFA | Branched fatty acid esters of hydroxy fatty acids |
| PC | Choline-containing glycerophospholipid |
| PE | Ethanolamine-containing glycerophospholipid |
| PI | Phosphatidylinositol |
| PS | Phosphatidylserine |
| PLA2 | Phospholipase A2 |
| cPLA2α | Cytosolic group IVA phospholipase A2 |
| iPLA2-VIA | Calcium-independent group VIA phospholipase A2 |
Appendix A
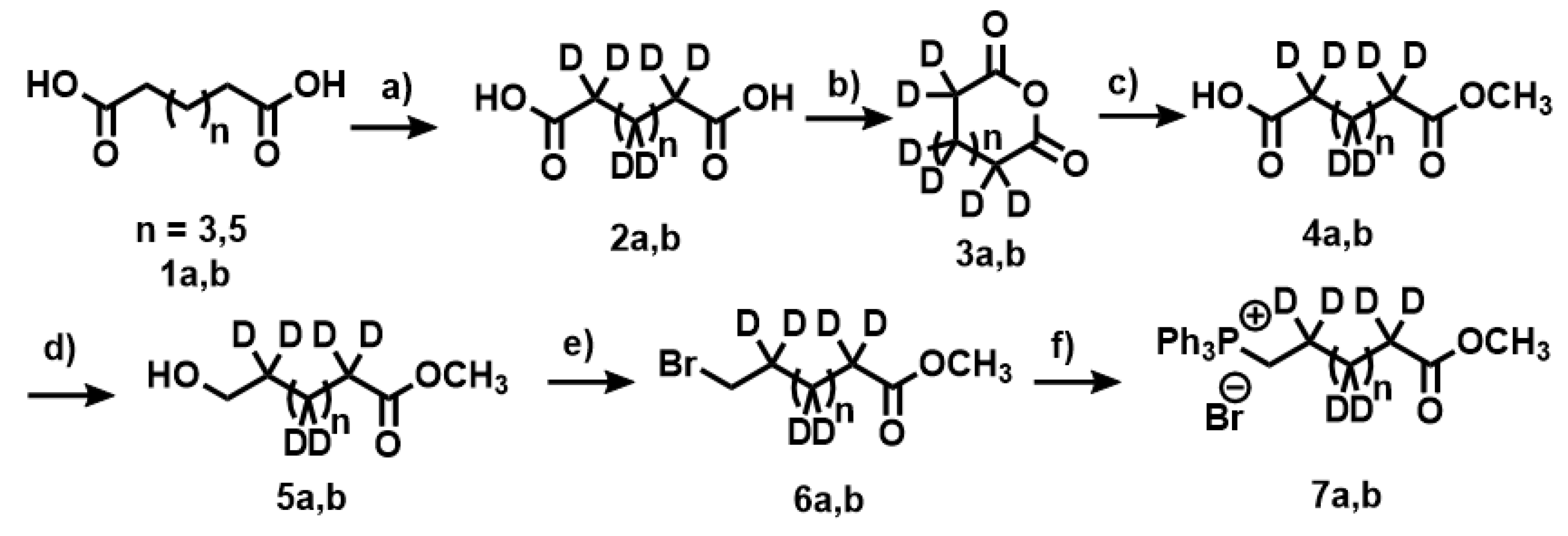
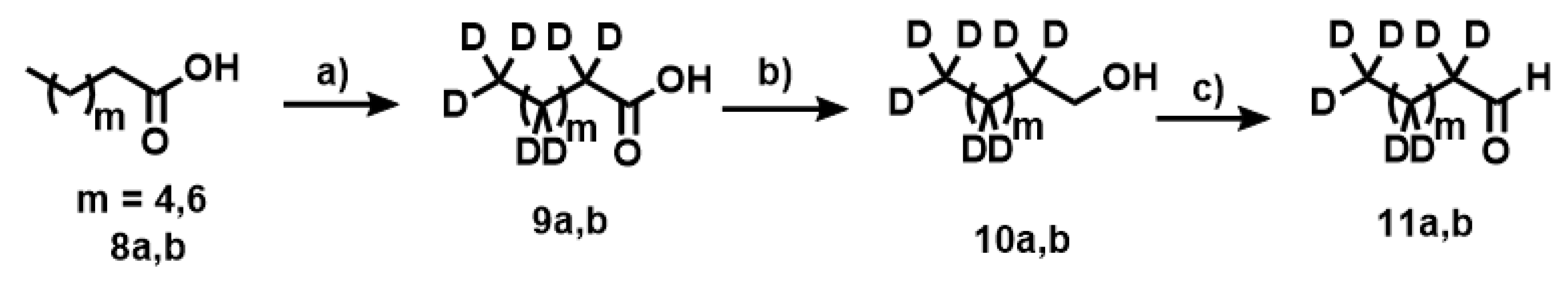
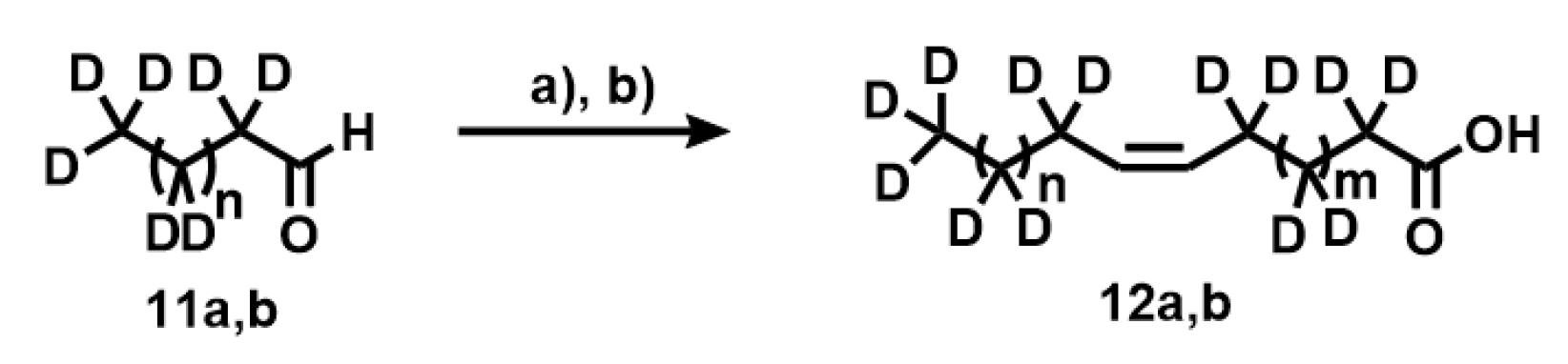
Appendix A.1. Synthesis of Deuterated Fatty Acids



Appendix A.2. Deuteration of Pimelic Acid (General Procedure)
Appendix A.3. Deuteration of Nonanoic Acid and Heptanoic Acid
Appendix A.4. Synthesis of Anhydride 3a,b
Appendix A.5. Synthesis of Compounds 4a,b
Appendix A.6. Synthesis of Compounds 5a,b
Appendix A.7. Synthesis of Compounds 6a,b
Appendix A.8. Synthesis of Compounds 7a,b (Wittig Salt)
Appendix A.9. Synthesis of Compounds 10a,b
Appendix A.10. Synthesis of Compounds 11a,b
Appendix A.11. Synthesis of Compounds 12a,b
References
- De Souza, C.O.; Vannice, G.K.; Rosa Neto, J.C.; Calder, P.C. Is palmitoleic acid a plausible nonpharmacological strategy to prevent or control chronic metabolic and inflammatory disorders? Mol. Nutr. Food Res. 2018, 62, 1700504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Saavedra, D.; Stanford, K.I. The regulation of lipokines by environmental factors. Nutrients 2019, 11, 2422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Fabiani, E. The true story of palmitoleic acid: Between myth and reality. Eur. J. Lipid Sci. Technol. 2011, 113, 809–811. [Google Scholar] [CrossRef]
- Hodson, L.; Karpe, F. Is there something special about palmitoleate? Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.L.; Pillon, N.J.; Sivaloganathan, D.M.; Costford, S.R.; Liu, Z.; Théret, M.; Chazaud, B.; Klip, A. Palmitoleate reverses high fat-induced proinflammatory macrophage polarization via AMP-activated protein kinase. J. Biol. Chem. 2015, 290, 16979–16988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, H.; Gerhold, K.; Mayers, J.R.; Wiest, M.M.; Watkins, S.M.; Hotamisligil, G.S. Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell 2008, 134, 933–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guijas, C.; Pérez-Chacón, G.; Astudillo, A.M.; Rubio, J.M.; Gil-de-Gómez, L.; Balboa, M.A.; Balsinde, J. Simultaneous activation of p38 and JNK by arachidonic acid stimulates the cytosolic phospholipase A2-dependent synthesis of lipid droplets in human monocytes. J. Lipid Res. 2012, 53, 2343–2354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guijas, C.; Meana, C.; Astudillo, A.M.; Balboa, M.A.; Balsinde, J. Foamy monocytes are enriched in cis-7-hexadecenoic fatty acid (16:1n-9), a possible biomarker for early detection of cardiovascular disease. Cell Chem. Biol. 2016, 23, 689–699. [Google Scholar] [CrossRef] [Green Version]
- Astudillo, A.M.; Meana, C.; Guijas, C.; Pereira, L.; Lebrero, R.; Balboa, M.A.; Balsinde, J. Occurrence and biological activity of palmitoleic acid isomers in phagocytic cells. J. Lipid Res. 2018, 59, 237–249. [Google Scholar] [CrossRef] [Green Version]
- Mouchlis, V.D.; Dennis, E.A. Phospholipase A2 catalysis and lipid mediator lipidomics. Biochim. Biophys. Acta 2019, 1864, 766–771. [Google Scholar] [CrossRef]
- Murakami, M.; Sato, H.; Taketomi, Y. Updating phospholipase A2 biology. Biomolecules 2020, 10, 1457. [Google Scholar] [CrossRef] [PubMed]
- Astudillo, A.M.; Balboa, M.A.; Balsinde, J. Selectivity of phospholipid hydrolysis by phospholipase A2 enzymes in activated cells leading to polyunsaturated fatty acid mobilization. Biochim. Biophys. Acta 2019, 1864, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Dennis, E.A.; Cao, J.; Hsu, Y.H.; Magrioti, V.; Kokotos, G. Phospholipase A2 enzymes: Physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem. Rev. 2011, 111, 6130–6185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, M. Novel functions of phospholipase A2s: Overview. Biochim. Biophys. Acta 2019, 1864, 763–765. [Google Scholar] [CrossRef] [PubMed]
- Guijas, C.; Rodríguez, J.P.; Rubio, J.M.; Balboa, M.A.; Balsinde, J. Phospholipase A2 regulation of lipid droplet formation. Biochim. Biophys. Acta 2014, 1841, 1661–1671. [Google Scholar] [CrossRef] [Green Version]
- Leslie, C.C. Cytosolic phospholipase A₂: Physiological function and role in disease. J. Lipid Res. 2015, 56, 1386–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramanadham, S.; Ali, T.; Ashley, J.W.; Bone, R.N.; Hancock, W.D.; Lei, X. Calcium-independent phospholipases A2 and their roles in biological processes and diseases. J. Lipid Res. 2015, 56, 1643–1668. [Google Scholar] [CrossRef] [Green Version]
- Turk, J.; White, T.D.; Nelson, A.J.; Lei, X.; Ramanadham, S. iPLA2β and its role in male fertility, neurological disorders, metabolic disorders, and inflammation. Biochim. Biophys. Acta 2019, 1864, 846–860. [Google Scholar] [CrossRef] [PubMed]
- Gil-de-Gómez, L.; Astudillo, A.M.; Guijas, C.; Magrioti, V.; Kokotos, G.; Balboa, M.A.; Balsinde, J. Cytosolic group IVA and calcium-independent group VIA phospholipase A2s act on distinct phospholipid pools in zymosan-stimulated mouse peritoneal macrophages. J. Immunol. 2014, 192, 752–762. [Google Scholar] [CrossRef] [Green Version]
- Monge, P.; Garrido, A.; Rubio, J.M.; Magrioti, V.; Kokotos, G.; Balboa, M.A.; Balsinde, J. The contribution of cytosolic group IVA and calcium-independent group VIA phospholipase A2s to adrenic acid mobilization in murine macrophages. Biomolecules 2020, 10, 542. [Google Scholar] [CrossRef] [Green Version]
- Yore, M.M.; Syed, I.; Moraes-Vieira, P.M.; Zhang, T.; Herman, M.A.; Homan, E.A.; Patel, R.T.; Lee, J.; Chen, S.; Peroni, O.D.; et al. Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell 2014, 159, 318–332. [Google Scholar] [CrossRef] [Green Version]
- Chilton, F.H.; Fonteh, A.N.; Sung, C.M.; Hickey, D.M.; Torphy, T.J.; Mayer, R.J.; Marshall, L.A.; Heravi, J.D.; Winkler, J.D. Inhibitors of CoA-independent transacylase block the movement of arachidonate into 1-ether-linked phospholipids of human neutrophils. Biochemistry 1995, 34, 5403–5410. [Google Scholar] [CrossRef]
- Pérez, R.; Matabosch, X.; Llebaria, A.; Balboa, M.A.; Balsinde, J. Blockade of arachidonic acid incorporation into phospholipids induces apoptosis in U937 promonocytic cells. J. Lipid Res. 2006, 47, 484–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balsinde, J.; Fernández, B.; Diez, E. Regulation of arachidonic acid release in mouse peritoneal macrophages. The role of extracellular calcium and protein kinase C. J. Immunol. 1990, 144, 4298–4304. [Google Scholar]
- Pindado, J.; Balsinde, J.; Balboa, M.A. TLR3-dependent induction of nitric oxide synthase in RAW 264.7 macrophage-like cells via a cytosolic phospholipase A2/cyclooxygenase-2 pathway. J. Immunol. 2007, 179, 4821–4828. [Google Scholar] [CrossRef] [Green Version]
- Ruipérez, V.; Astudillo, M.A.; Balboa, M.A.; Balsinde, J. Coordinate regulation of TLR-mediated arachidonic acid mobilization in macrophages by group IVA and group V phospholipase A2s. J. Immunol. 2009, 182, 3877–3883. [Google Scholar]
- Balsinde, J.; Fernández, B.; Solís-Herruzo, J.A.; Diez, E. Pathways for arachidonic acid mobilization in zymosan-stimulated mouse peritoneal macrophages. Biochim. Biophys. Acta 1992, 1136, 75–82. [Google Scholar] [CrossRef]
- Balboa, M.A.; Pérez, R.; Balsinde, J. Amplification mechanisms of inflammation: Paracrine stimulation of arachidonic acid mobilization by secreted phospholipase A2 is regulated by cytosolic phospholipase A2-derived hydroperoxyeicosatetraenoic acid. J. Immunol. 2003, 171, 989–994. [Google Scholar] [CrossRef] [Green Version]
- Balboa, M.A.; Pérez, R.; Balsinde, J. Calcium-independent phospholipase A2 mediates proliferation of human promonocytic U937 cells. FEBS J. 2008, 275, 1915–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balsinde, J.; Balboa, M.A.; Insel, P.A.; Dennis, E.A. Differential regulation of phospholipase D and phospholipase A2 by protein kinase C in P388D1 macrophages. Biochem. J. 1997, 321, 805–809. [Google Scholar] [CrossRef] [Green Version]
- Balboa, M.A.; Balsinde, J.; Dennis, E.A. Involvement of phosphatidate phosphohydrolase in arachidonic acid mobilization in human amnionic WISH cells. J. Biol. Chem. 1998, 273, 7684–7690. [Google Scholar] [CrossRef] [Green Version]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [Green Version]
- Diez, E.; Balsinde, J.; Aracil, M.; Schüller, A. Ethanol induces release of arachidonic acid but not synthesis of eicosanoids in mouse peritoneal macrophages. Biochim. Biophys. Acta 1987, 921, 82–89. [Google Scholar] [CrossRef]
- Fine, J.B.; Sprecher, H. Unidimensional thin-layer chromatography of phospholipids on boric acid-impregnated plates. J. Lipid Res. 1982, 23, 660–663. [Google Scholar]
- Astudillo, A.M.; Pérez-Chacón, G.; Balgoma, D.; Gil-de-Gómez, L.; Ruipérez, V.; Guijas, C.; Balboa, M.A.; Balsinde, J. Influence of cellular arachidonic acid levels on phospholipid remodeling and CoA-independent transacylase activity in human monocytes and U937 cells. Biochim. Biophys. Acta 2011, 1811, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Valdearcos, M.; Esquinas, E.; Meana, C.; Gil-de-Gómez, L.; Guijas, C.; Balsinde, J.; Balboa, M.A. Subcellular localization and role of lipin-1 in human macrophages. J. Immunol. 2011, 186, 6004–6013. [Google Scholar] [CrossRef] [Green Version]
- Sansone, A.; Melchiorre, M.; Chatgilialoglu, C.; Ferreri, C. Hexadecenoic fatty acid isomers: A chemical biology approach for human plasma biomarker development. Chem. Res. Toxicol. 2013, 26, 1703–1709. [Google Scholar] [CrossRef]
- Axelsen, P.H.; Murphy, R.C. Quantitative analysis of phospholipids containing arachidonate and docosahexaenoate chains in microdissected regions of mouse brain. J. Lipid Res. 2010, 51, 660–671. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Chen, S.; Syed, I.; Stáhlman, M.; Kolar, M.J.; Homan, E.A.; Chu, Q.; Smith, U.; Borén, J.; Kahn, B.B.; et al. A LC-MS–based workflow for measurement of branched fatty acid esters of hydroxy fatty acids. Nat. Protoc. 2016, 11, 747–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, J.P.; Guijas, C.; Astudillo, A.M.; Rubio, J.M.; Balboa, M.A.; Balsinde, J. Sequestration of 9-hydroxystearic acid in FAHFA (fatty acid esters of hydroxy fatty acids) as a protective mechanism for colon carcinoma cells to avoid apoptotic cell death. Cancers 2019, 11, 524. [Google Scholar] [CrossRef] [Green Version]
- Balsinde, J.; Balboa, M.A.; Dennis, E.A. Antisense inhibition of group VI Ca2+-independent phospholipase A2 blocks phospholipid fatty acid remodeling in murine P388D1 macrophages. J. Biol. Chem. 1997, 272, 29317–29321. [Google Scholar] [CrossRef] [Green Version]
- Balsinde, J.; Balboa, M.A.; Dennis, E.A. Identification of a third pathway for arachidonic acid mobilization and prostaglandin production in activated P388D1 macrophage-like cells. J. Biol. Chem. 2000, 275, 22544–22549. [Google Scholar] [CrossRef] [Green Version]
- Balboa, M.A.; Sáez, Y.; Balsinde, J. Calcium-independent phospholipase A2 is required for lysozyme secretion in U937 promonocytes. J. Immunol. 2003, 170, 5276–5280. [Google Scholar] [CrossRef] [Green Version]
- Pérez, R.; Melero, R.; Balboa, M.A.; Balsinde, J. Role of group VIA calcium-independent phospholipase A2 in arachidonic acid release, phospholipid fatty acid incorporation, and apoptosis in U937 cells responding to hydrogen peroxide. J. Biol. Chem. 2004, 279, 40385–40391. [Google Scholar] [CrossRef] [Green Version]
- Carnevale, K.A.; Cathcart, M.K. Calcium-independent phospholipase A2 is required for human monocyte chemotaxis to monocyte chemoattractant protein 1. J. Immunol. 2001, 167, 3414–3421. [Google Scholar] [CrossRef] [Green Version]
- Mishra, R.S.; Carnevale, K.A.; Cathcart, M.K. iPLA2β: Front and center in human monocyte chemotaxis to MCP-1. J. Exp. Med. 2008, 205, 347–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahy, E.; Subramaniam, S.; Brown, H.A.; Glass, C.K.; Merrill, A.H., Jr.; Murphy, R.C.; Raetz, C.R.; Russell, D.W.; Seyama, Y.; Shaw, W.; et al. A comprehensive classification system for lipids. J. Lipid Res. 2005, 46, 839–861. [Google Scholar] [CrossRef] [Green Version]
- Suram, S.; Gangelhoff, T.A.; Taylor, P.R.; Rosas, M.; Brown, G.D.; Bonventre, J.V.; Akira, S.; Uematsu, S.; Williams, D.L.; Murphy, R.C.; et al. Pathways regulating cytosolic phospholipase A2 activation and eicosanoid production in macrophages by Candida albicans. J. Biol. Chem. 2010, 285, 30676–30685. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Chacón, G.; Astudillo, A.M.; Ruipérez, V.; Balboa, M.A.; Balsinde, J. Signaling role for lysophosphatidylcholine acyltransferase 3 in receptor-regulated arachidonic acid reacylation reactions in human monocytes. J. Immunol. 2010, 184, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Balgoma, D.; Astudillo, A.M.; Pérez-Chacón, G.; Montero, O.; Balboa, M.A.; Balsinde, J. Markers of monocyte activation revealed by lipidomic profiling of arachidonic acid-containing phospholipids. J. Immunol. 2010, 184, 3857–3865. [Google Scholar] [CrossRef] [Green Version]
- Rubio, J.M.; Rodríguez, J.P.; Gil-de-Gómez, L.; Guijas, C.; Balboa, M.A.; Balsinde, J. Group V secreted phospholipase A2 is up-regulated by interleukin-4 in human macrophages and mediates phagocytosis via hydrolysis of ethanolamine phospholipids. J. Immunol. 2015, 194, 3327–3339. [Google Scholar] [CrossRef] [Green Version]
- Nikolaou, A.; Kokotou, M.G.; Vasilakaki, S.; Kokotos, G. Small-molecule inhibitors as potential therapeutics and as tools to understand the role of phospholipases A2. Biochim. Biophys. Acta 2019, 1864, 941–956. [Google Scholar] [CrossRef]
- Yamashita, A.; Hayashi, Y.; Matsumoto, N.; Nemoto-Sasaki, Y.; Koizumi, T.; Inagaki, Y.; Oka, S.; Tanikawa, T.; Sugiura, T. Coenzyme-A-independent transacylation system; possible involvement of phospholipase A2 in transacylation. Biology 2017, 6, 23. [Google Scholar] [CrossRef] [Green Version]
- Ferreri, C.; Masi, A.; Sansone, A.; Giacometti, G.; Larocca, A.V.; Menounou, G.; Scanferlato, R.; Tortorella, S.; Rota, D.; Conti, M.; et al. Fatty acids in membranes as homeostatic, metabolic and nutritional biomarkers: Recent advancements in analytics and diagnostics. Diagnostics 2017, 7, 1. [Google Scholar] [CrossRef]
- Montenegro-Burke, J.R.; Sutton, J.A.; Rogers, L.M.; Milne, G.L.; McLean, J.A.; Aronoff, D.M. Lipid profiling of polarized human monocyte-derived macrophages. Prostaglandins Other Lipid Mediat. 2016, 127, 1–8. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, Y.; Wang, F.; Wang, Z.; Lu, Y.; Xu, Y.; Wang, K.; Shen, H.; Yang, P.; Li, S.; et al. Quantitative profiling of glycerophospholipids during mouse and human macrophage differentiation using targeted mass spectrometry. Sci. Rep. 2017, 7, 412. [Google Scholar] [CrossRef] [Green Version]
- Snider, S.A.; Margison, K.D.; Ghorbani, P.; LeBlond, N.D.; O’Dwyer, C.; Nunes, J.R.C.; Nguyen, T.; Xu, H.; Bennett, S.A.L.; Fullerton, M.D. Choline transport links macrophage phospholipid metabolism and inflammation. J. Biol. Chem. 2018, 293, 11600–11611. [Google Scholar] [CrossRef] [Green Version]
- Rouzer, C.A.; Ivanova, P.T.; Byrne, M.O.; Milne, S.B.; Marnett, L.J.; Brown, H.A. Lipid profiling reveals arachidonate deficiency in RAW264.7 cells: Structural and functional implications. Biochemistry 2006, 45, 14795–14808. [Google Scholar] [CrossRef] [Green Version]
- Murakami, M.; Masuda, S.; Ueda-Semmyo, K.; Yoda, E.; Kuwata, H.; Takanezawa, Y.; Aoki, J.; Arai, H.; Sumimoto, H.; Ishikawa, Y.; et al. Group VIB Ca2+-independent phospholipase A2γ promotes cellular membrane hydrolysis and prostaglandin production in a manner distinct from other intracellular phospholipases A2. J. Biol. Chem. 2005, 280, 14028–14041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouchlis, V.D.; Chen, Y.; McCammon, J.A.; Dennis, E.A. Membrane allostery and unique hydrophobic sites promote enzyme substrate specificity. J. Am. Chem. Soc. 2018, 140, 3285–3291. [Google Scholar] [CrossRef] [Green Version]
- Koeberle, A.; Shindou, H.; Harayama, T.; Shimizu, T. Palmitoleate is a mitogen, formed upon stimulation with growth factors, and converted to palmitoleoyl-phosphatidylinositol. J. Biol. Chem. 2012, 287, 27244–27254. [Google Scholar] [CrossRef] [Green Version]
- Brejchova, K.; Balas, L.; Paluchova, V.; Brezinova, M.; Durand, T.; Kuda, O. Understanding FAHFAs: From structure to metabolic regulation. Prog. Lipid Res. 2020, 79, 101053. [Google Scholar] [CrossRef]
- Darwish, T.A.; Luks, E.; Moraes, G.; Yepuri, N.R.; Holden, P.J.; James, M. Synthesis of deuterated [D32] oleic acid and its phospholipid derivative [D64] dioleoyl-sn-glycero-3-phosphocholine. J. Label. Compd. Radiopharm. 2013, 56, 520–529. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Astudillo, A.M.; Meana, C.; Bermúdez, M.A.; Pérez-Encabo, A.; Balboa, M.A.; Balsinde, J. Release of Anti-Inflammatory Palmitoleic Acid and Its Positional Isomers by Mouse Peritoneal Macrophages. Biomedicines 2020, 8, 480. https://doi.org/10.3390/biomedicines8110480
Astudillo AM, Meana C, Bermúdez MA, Pérez-Encabo A, Balboa MA, Balsinde J. Release of Anti-Inflammatory Palmitoleic Acid and Its Positional Isomers by Mouse Peritoneal Macrophages. Biomedicines. 2020; 8(11):480. https://doi.org/10.3390/biomedicines8110480
Chicago/Turabian StyleAstudillo, Alma M., Clara Meana, Miguel A. Bermúdez, Alfonso Pérez-Encabo, María A. Balboa, and Jesús Balsinde. 2020. "Release of Anti-Inflammatory Palmitoleic Acid and Its Positional Isomers by Mouse Peritoneal Macrophages" Biomedicines 8, no. 11: 480. https://doi.org/10.3390/biomedicines8110480
APA StyleAstudillo, A. M., Meana, C., Bermúdez, M. A., Pérez-Encabo, A., Balboa, M. A., & Balsinde, J. (2020). Release of Anti-Inflammatory Palmitoleic Acid and Its Positional Isomers by Mouse Peritoneal Macrophages. Biomedicines, 8(11), 480. https://doi.org/10.3390/biomedicines8110480