Whole-Transcriptome Analysis in Peripheral Blood Mononuclear Cells from Patients with Lipid-Specific Oligoclonal IgM Band Characterization Reveals Two Circular RNAs and Two Linear RNAs as Biomarkers of Highly Active Disease

 , , and
, , and
Abstract
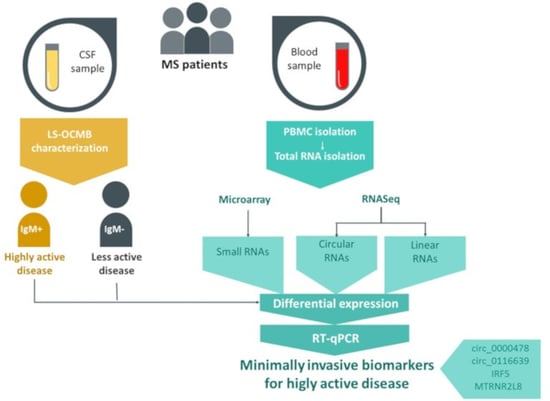
1. Introduction
2. Experimental Section
2.1. Patients, Sample Collection and RNA Isolation
2.2. Microarray Analysis
2.3. RNAseq
2.4. CircRNA Detection and Quantification in RNA-Seq Data
2.5. Linear RNA Detection and Quantification in RNA-Seq Data
2.6. cDNA Synthesis and Quantitative PCR
2.7. Gene Overrepresentation Test
2.8. ROC Curve Analysis
3. Results
3.1. microRNA Expression Profile
3.2. circRNA Expression Profile
3.3. Linear Transcripts Expression Profile
3.4. Evaluation of circRNA and Linear RNAs as Biomarkers of a Highly Active Disease
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Filippi, M.; Bar-Or, A.; Piehl, F.; Preziosa, P.; Solari, A.; Vukusic, S.; Rocca, M.A. Multiple sclerosis. Nat. Rev. Dis. Prim. 2018, 4, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Comabella, M.; Montalban, X. Body fluid biomarkers in multiple sclerosis. Lancet Neurol. 2014, 13, 113–126. [Google Scholar] [CrossRef]
- Paul, A.; Comabella, M.; Gandhi, R. Biomarkers in Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Villar, L.M.; Sadaba, M.C.; Roldan, E.; Masjuan, J.; Gonzalez-Porque, P.; Villarrubia, N.; Espino, M.; Garcia-Trujillo, J.A.; Bootello, A.; Alvarez-Cermeno, J.C. Intrathecal synthesis of oligoclonal IgM against myelin lipids predicts an aggressive disease course in MS. J. Clin. Investig. 2005, 115, 187–194. [Google Scholar] [CrossRef]
- Polivka, J.J.P., Jr.; Krakorova, K.; Peterka, M.; Topolcan, O. Current status of biomarker research in neurology. EPMA J. 2016, 1–13. [Google Scholar] [CrossRef][Green Version]
- Kemppinen, A.K.; Kaprio, J.; Palotie, A.; Saarela, J. Systematic review of genome-wide expression studies in multiple sclerosis. BMJ Open 2011, 1, e000053. [Google Scholar] [CrossRef]
- Nickles, D.; Chen, H.P.; Li, M.M.; Khankhanian, P.; Madireddy, L.; Caillier, S.J.; Santaniello, A.; Cree, B.A.; Pelletier, D.; Hauser, S.L.; et al. Blood RNA profiling in a large cohort of multiple sclerosis patients and healthy controls. Hum. Mol. Genet. 2013, 22, 4194–4205. [Google Scholar] [CrossRef] [PubMed]
- Irizar, H.; Muñoz-Culla, M.; Sáenz-Cuesta, M.; Osorio-Querejeta, I.; Sepúlveda, L.; Castillo-Triviño, T.; Prada, A.; Lopez de Munain, A.; Olascoaga, J.; Otaegui, D. Identification of ncRNAs as potential therapeutic targets in multiple sclerosis through differential ncRNA—mRNA network analysis. BMC Genom. 2015, 16, 250. [Google Scholar] [CrossRef] [PubMed]
- Dolati, S.; Marofi, F.; Babaloo, Z.; Aghebati-Maleki, L.; Roshangar, L.; Ahmadi, M.; Rikhtegar, R.; Yousefi, M. Dysregulated Network of miRNAs Involved in the Pathogenesis of Multiple Sclerosis. Biomed. Pharmacother. 2018, 104, 280–290. [Google Scholar] [CrossRef]
- Du, C.; Liu, C.; Kang, J.; Zhao, G.; Ye, Z.; Huang, S.; Li, Z.; Wu, Z.; Pei, G.; Du, C.; et al. MicroRNA miR-326 regulates T H -17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat. Immunol. 2009, 1259, 1252–1259. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Culla, M.; Irizar, H.; Sáenz-Cuesta, M.; Castillo-Triviño, T.; Osorio-Querejeta, I.; Sepúlveda, L.; López De Munain, A.; Olascoaga, J.; Otaegui, D. SncRNA (microRNA &snoRNA) opposite expression pattern found in multiple sclerosis relapse and remission is sex dependent. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- De Felice, B.; Mondola, P.; Sasso, A.; Orefice, G.; Bresciamorra, V.; Vacca, G.; Biffali, E.; Borra, M.; Pannone, R. Small non-coding RNA signature in multiple sclerosis patients after treatment with interferon-β. BMC Med. Genom. 2014, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef]
- Floris, G.; Zhang, L.; Follesa, P.; Sun, T. Regulatory Role of Circular RNAs and Neurological Disorders. Mol. Neurobiol. 2017, 54, 5156–5165. [Google Scholar] [CrossRef]
- Xia, X.; Tang, X.; Wang, S. Roles of CircRNAs in Autoimmune Diseases. Front. Immunol. 2019, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Paraboschi, E.M.; Cardamone, G.; Soldà, G.; Duga, S.; Asselta, R. Interpreting Non-coding Genetic Variation in Multiple Sclerosis Genome-Wide Associated Regions. Front. Genet. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Cardamone, G.; Paraboschi, E.M.; Rimoldi, V.; Duga, S.; Soldà, G.; Asselta, R. The Characterization of GSDMB Splicing and Backsplicing Profiles Identifies Novel Isoforms and a Circular RNA That Are Dysregulated in Multiple Sclerosis. Int. J. Mol. Sci. 2017, 18, 576. [Google Scholar] [CrossRef]
- Iparraguirre, L.; Muñoz-Culla, M.; Prada-Luengo, I.; Castillo-Triviño, T.; Olascoaga, J.; Otaegui, D. Circular RNA profiling reveals that circular RNAs from ANXA2 can be used as new biomarkers for multiple sclerosis. Hum. Mol. Genet. 2017, 26. [Google Scholar] [CrossRef]
- Kacperska, M.J.; Walenczak, J.; Tomasik, B. Plasmatic microRNA as potential biomarkers of multiple sclerosis: Literature review. Adv. Clin. Exp. Med. 2016, 25, 775–779. [Google Scholar] [CrossRef]
- Stoicea, N.; Du, A.; Lakis, D.C.; Tipton, C.; Arias-Morales, C.E.; Bergese, S.D. The MiRNA Journey from Theory to Practice as a CNS Biomarker. Front. Genet. 2016, 7, 1–8. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, T.; Xiao, J. Circular RNAs: Promising Biomarkers for Human Diseases. EBioMedicine 2018, 34, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Villar, L.M.; González-Porqué, P.; Masjuán, J.; Álvarez-Cermeño, J.; Bootello, A.; Keir, G. A sensitive and reproducible method for the detection of oligoclonal IgM bands. J. Immunol. Methods 2001, 258, 151–155. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, A.D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Zhang, X.; Dong, R.; Zhang, Y.; Zhang, J.; Luo, Z.; Zhang, J.; Chen, L.; Yang, L. Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome Res. 2016, 26, 1277–1287. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, J.; Zhao, F. Circular RNA identification based on multiple seed matching. Brief. Bioinform. 2017, 1–8. [Google Scholar] [CrossRef]
- Hansen, T.B. Improved circRNA Identification by Combining Prediction Algorithms. Front. Cell Dev. Biol. 2018, 6, 1–9. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–528. [Google Scholar] [CrossRef]
- Mi, H.; Thomas, P. PANTHER Pathway: An ontology-based pathway database coupled with data analysis tools. Nat. Protoc. 2019, 14, 703–721. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Ma, W.; Sand, Z.; Finlayson, J.; Wang, T.; Diaz, R.; Willis, W.T.; Mandarino, L.J. Von Willebrand factor A domain-containing protein 8 ( VWA8 ) localizes to the matrix side of the inner mitochondrial membrane. Biochem. Biophys. Res. Commun. 2020, 521, 158–163. [Google Scholar] [CrossRef]
- Moldovan, L.I.; Hansen, T.B.; Venø, M.T.; Okholm, T.L.H.; Andersen, T.L.; Hager, H.; Iversen, L.; Kjems, J.; Johansen, C.; Kristensen, L.S. High-throughput RNA sequencing from paired lesional- and non-lesional skin reveals major alterations in the psoriasis circRNAome. BMC Med. Genom. 2019, 12, 1–17. [Google Scholar] [CrossRef] [PubMed]
- International Multiple Sclerosis Genetics Consortium. The Multiple Sclerosis Genomic Map implicates peripheral immune cells and microglia in susceptibility. Science 2019, 365, 1–10. [Google Scholar] [CrossRef]
- Vandenbroeck, K.; Alloza, I.; Swaminathan, B.; Antiguedad, A.; Otaegui, D.; Olascoaga, J.; Barcina, M.G.; de las Heras, V.; Bartolome, M.; Fernandez-Arquero, M.; et al. Validation of IRF5 as multiple sclerosis risk gene: Putative role in interferon beta therapy and human herpes virus-6 infection. Genes Immun. 2011, 12, 40–45. [Google Scholar] [CrossRef]
- Lindén, M.; Khademi, M.; Bomfim, I.L.; Piehl, F.; Jagodic, M.; Kockum, I.; Olsson, T. Multiple sclerosis risk genotypes correlate with an elevated cerebrospinal fluid level of the suggested prognostic marker CXCL13. Mult. Scler. Jounrla 2012, 19, 863–870. [Google Scholar] [CrossRef]
- Al Mamun, A.; Chauhan, A.; Yu, H.; Xu, Y.; Sharmeen, R.; Liu, F. Interferon regulatory factor 4/5 signaling impacts on microglial activation after ischemic stroke in mice. Eur. J. Neurosci. 2018, 47, 140–149. [Google Scholar] [CrossRef]
- Zhu, D.; Yang, N.; Liu, Y.-Y.; Zheng, J.; Ji, C.; Zuo, P.-P. M2 Macrophage Transplantation Ameliorates Cognitive Dysfunction in Amyloid-β-Treated Rats Through Regulation of Microglial Polarization. J. Alzheimer’s Dis. 2016, 52, 483–495. [Google Scholar] [CrossRef]
- Gil-Varea, E.; Urcelay, E.; Vilariño-Güell, C.; Costa, C.; Midaglia, L.; Matesanz, F.; Rodríguez-antigüedad, A.; Oksenberg, J.; Espino-paisan, L.; Sadovnick, A.D.; et al. Exome sequencing study in patients with multiple sclerosis reveals variants associated with disease course. J. Neuroinflamm. 2018, 15, 1–10. [Google Scholar] [CrossRef]
- Pantazatos, S.P.; Huang, Y.-Y.; Rosoklija, G.B.; Dwork, A.J.; Arango, V.; Mann, J.J. Whole-transcriptome brain expression and exon-usage profiling in major depression and suicide: Evidence for altered glial, endothelial and ATPase activity. Mol. Psychiatry 2017, 22, 760–773. [Google Scholar] [CrossRef]
- Sellebjerg, F.; Christiansen, M.; Garred, P. MBP, anti-MBP and anti-PLP antibodies, and intrathecal complement activation in multiple sclerosis. Mult. Scler. 1998, 4, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Mead, R.J.; Singhrao, S.K.; Neal, J.W.; Lassmann, H.; Morgan, B.P. The Membrane Attack Complex of Complement Causes Severe Demyelination Associated with Acute Axonal Injury. J. Immunol. 2002, 168, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Ingram, G.; Hakobyan, S.; Hirst, C.L.; Harris, C.L.; Loveless, S.; Mitchell, J.P.; Pickersgill, T.P.; Robertson, N.P.; Morgan, B.P. Systemic complement profiling in multiple sclerosis as a biomarker of disease state. Mult. Scler. J. 2012, 18, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar, J.N. Receiver Operating Characteristic Curve in Diagnostic Test Assessment. J. Thorac. Oncol. 2010, 5, 1315–1316. [Google Scholar] [CrossRef] [PubMed]
- Iacobaeus, E.; Ryschkewitsch, C.; Gravell, M.; Khademi, M.; Wallstrom, E.; Olsson, T.; Brundin, L.; Major, E. Analysis of cerebrospinal fluid and cerebrospinal fluid cells from patients with multiple sclerosis for detection of JC virus DNA. Mult. Scler. J. 2009, 15, 28–35. [Google Scholar] [CrossRef]
- Håkansson, I.; Tisell, A.; Cassel, P.; Blennow, K.; Zetterberg, H.; Lundberg, P.; Dahle, C.; Vrethem, M.; Ernerudh, J. Neurofilament levels, disease activity and brain volume during follow-up in multiple sclerosis. J. Neuroinflamm. 2018, 15, 1–10. [Google Scholar] [CrossRef]
- Khademi, M.; Kockum, I.; Andersson, M.L.; Iacobaeus, E.; Brundin, L.; Sellebjerg, F.; Hillert, J.; Piehl, F.; Olsson, T. Cerebrospinal fluid CXCL13 in multiple sclerosis: A suggestive prognostic marker for the disease course. Mult. Scler. J. 2011, 17, 335–343. [Google Scholar] [CrossRef]
- Cantó, E.; Tintore, M.; Villar, L.M.; Costa, C.; Nurtdinov, R.; Deisenhammer, F.; Hegen, H.; Khademi, M.; Olsson, T.; Piehl, F.; et al. Chitinase 3-like 1: Prognostic biomarker in clinically isolated syndromes. Brain 2015, 138, 918–931. [Google Scholar] [CrossRef]
- Quintana, E.; Ortega, F.J.; Robles-Cedeño, R.; Villar, M.L.; Buxó, M.; Mercader, J.M.; Alvarez-Cermeño, J.C.; Pueyo, N.; Perkal, H.; Fernández-Real, J.M.; et al. miRNAs in cerebrospinal fluid identify patients with MS and specifically those with lipid-specific oligoclonal IgM bands. Mult. Scler. J. 2017, 23, 1716–1726. [Google Scholar] [CrossRef]
- Vistbakka, J.; Sumelahti, M.; Lehtimäki, T.; Elovaara, I.; Hagman, S. Evaluation of serum miR-191-5p, miR-24-3p, miR-128-3p and miR-376c-3 in multiple sclerosis patients. Acta Neurol. Scand. 2018, 1–7. [Google Scholar] [CrossRef]
- Gupta, M.; Martens, K.; Metz, L.M.; Jason, A.P.; Koning, D.; Pfeffer, G. Long noncoding RNAs associated with phenotypic severity in multiple sclerosis. Mult. Scler. Relat. Disord. 2019, 36, 101407. [Google Scholar] [CrossRef] [PubMed]
- Iacobaeus, E.; Arrambide, G.; Amato, M.P.; Derfuss, T.; Vukusic, S.; Hemmer, B.; Tintore, M. Aggressive multiple sclerosis (1): Towards a definition of the phenotype. Mult. Scler. J. 2020, 1031–1044. [Google Scholar] [CrossRef] [PubMed]
- Díaz, C.; Zarco, L.A.; Rivera, D.M. Highly active multiple sclerosis: An update. Mult. Scler. Relat. Disord. 2019, 30, 215–224. [Google Scholar] [CrossRef]
- Hindson, C.M.; Chevillet, J.R.; Briggs, H.A.; Gallichotte, E.N.; Ruf, I.K.; Hindson, B.J.; Vessella, R.L.; Tewari, M. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat. Methods 2013, 1–6. [Google Scholar] [CrossRef]
- Taylor, S.C.; Laperriere, G.; Germain, H. Droplet Digital PCR versus qPCR for gene expression analysis with low abundant targets: From variable nonsense to publication quality data. Sci. Rep. 2017, 1–8. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microarrays | RNAseq | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Profiling cohort | LS-OCMB status | Sex | Age | LS-OCMB status | Sex | Age | |||
| P = 5 | F = 5 | 35 (23–58) | P = 4 | F = 4 | 38 (23–58) | ||||
| M = 0 | M = 0 | ||||||||
| N = 6 | F = 6 | 34.5 (27–53) | N = 4 | F = 4 | 35 (27–44) | ||||
| M = 0 | M = 0 | ||||||||
| Validation cohort | miRNA validation * | circRNA validation * | linear RNA validation | ||||||
| LS-OCMB status | Sex | Age | LS-OCMB status | Sex | Age | LS-OCMB status | Sex | Age | |
| P = 42 | F = 24 | 34 (17–58) | P = 42 | F = 24 | 34 (17–58) | P = 30 | F = 18 | 33.4 (17–51) | |
| M = 18 | M = 18 | M = 12 | |||||||
| N = 47 | F = 39 | 37 (20–53) | N = 46 | F = 38 | 37 (20–53) | N = 30 | F = 24 | 36.9 (20–49) | |
| M = 8 | M = 8 | M = 6 | |||||||
| Marker/Combination | Transcripts | Sample Size | AUC (%) | CI (%) |
|---|---|---|---|---|
| circRNA-1 | circ_0000478 | 80 (43 LS-OCMB−; 37 LS-OCMB+) | 65.9 | 53.9–77.9 |
| circRNA-2 | circ_0116639 | 66.4 | 54.2–78.5 | |
| Combi_circ | circ_0000478-circ_0116639 | 68.9 | 57.2–80.5 | |
| linearRNA-1 | IRF5 | 55 (29 LS-OCMB−; 26 LS-OCMB+) | 65.6 | 50.7–80.6 |
| linearRNA-2 | MTRNR2L8 | 67.0 | 52.4–81.7 | |
| Combi_linear | IRF5-MTRNR2L8 | 68.6 | 54.1–83.1 | |
| circRNA-1 | circ_0000478 | 51 (26 LS-OCMB−; 25 LS-OCMB+) | 65.1 | 49.8–80.4 |
| circRNA-2 | circ_0116639 | 66.2 | 50.6–81.7 | |
| linearRNA-1 | IRF5 | 67.1 | 51.8–82.3 | |
| linearRNA-2 | MTRNR2L8 | 69.0 | 54.1–83.9 | |
| CombiALL | circ_0000478-circ_0116639-IRF5-MTRNR2L8 | 69.8 | 55.3–84.4 | |
| combi1 | circ_0000478-circ_0116639 | 66.3 | 51.1–81.6 | |
| combi2 | IRF5-MTRNR2L8 | 70.3 | 55.6–85.0 | |
| combi3 | circ_0000478-IRF5 | 68.3 | 52.8–83.8 | |
| combi4 | circ_0000478-MTRNR2L8 | 68.6 | 53.7–83.6 | |
| combi5 | circ_0116639-IRF5 | 69.8 | 55.0–84.7 | |
| combi6 | circ_0116639-MTRNR2L8 | 67.8 | 52.8–82.9 | |
| combi7 | circ_0000478-circ_0116639-IRF5 | 69.8 | 55.0–84.6 | |
| combi8 | circ_0000478-circ_0116639-MTRNR2L8 | 68.2 | 53.3–83.0 | |
| combi9 | circ_0000478-IRF5-MTRNR2L8 | 68.8 | 53.7–83.8 | |
| combi10 | circ_0116639-IRF5-MTRNR2L8 | 68.8 | 53.8–83.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iparraguirre, L.; Olaverri, D.; Blasco, T.; Sepúlveda, L.; Castillo-Triviño, T.; Espiño, M.; Costa-Frossard, L.; Prada, Á.; Villar, L.M.; Otaegui, D.; et al. Whole-Transcriptome Analysis in Peripheral Blood Mononuclear Cells from Patients with Lipid-Specific Oligoclonal IgM Band Characterization Reveals Two Circular RNAs and Two Linear RNAs as Biomarkers of Highly Active Disease. Biomedicines 2020, 8, 540. https://doi.org/10.3390/biomedicines8120540
Iparraguirre L, Olaverri D, Blasco T, Sepúlveda L, Castillo-Triviño T, Espiño M, Costa-Frossard L, Prada Á, Villar LM, Otaegui D, et al. Whole-Transcriptome Analysis in Peripheral Blood Mononuclear Cells from Patients with Lipid-Specific Oligoclonal IgM Band Characterization Reveals Two Circular RNAs and Two Linear RNAs as Biomarkers of Highly Active Disease. Biomedicines. 2020; 8(12):540. https://doi.org/10.3390/biomedicines8120540
Chicago/Turabian StyleIparraguirre, Leire, Danel Olaverri, Telmo Blasco, Lucía Sepúlveda, Tamara Castillo-Triviño, Mercedes Espiño, Lucienne Costa-Frossard, Álvaro Prada, Luisa María Villar, David Otaegui, and et al. 2020. "Whole-Transcriptome Analysis in Peripheral Blood Mononuclear Cells from Patients with Lipid-Specific Oligoclonal IgM Band Characterization Reveals Two Circular RNAs and Two Linear RNAs as Biomarkers of Highly Active Disease" Biomedicines 8, no. 12: 540. https://doi.org/10.3390/biomedicines8120540
APA StyleIparraguirre, L., Olaverri, D., Blasco, T., Sepúlveda, L., Castillo-Triviño, T., Espiño, M., Costa-Frossard, L., Prada, Á., Villar, L. M., Otaegui, D., & Muñoz-Culla, M. (2020). Whole-Transcriptome Analysis in Peripheral Blood Mononuclear Cells from Patients with Lipid-Specific Oligoclonal IgM Band Characterization Reveals Two Circular RNAs and Two Linear RNAs as Biomarkers of Highly Active Disease. Biomedicines, 8(12), 540. https://doi.org/10.3390/biomedicines8120540