Modulating the Heat Sensitivity of Prostate Cancer Cell Lines In Vitro: A New Impact for Focal Therapies

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Pre-Treatment
2.2. Heat Stress Induction
2.3. Measurement of Metabolic Activity
2.4. PI/Annexin V Staining and Flow Cytometry
2.5. RNA Sequencing (RNAseq)
2.6. Sequencing Validation
3. Results
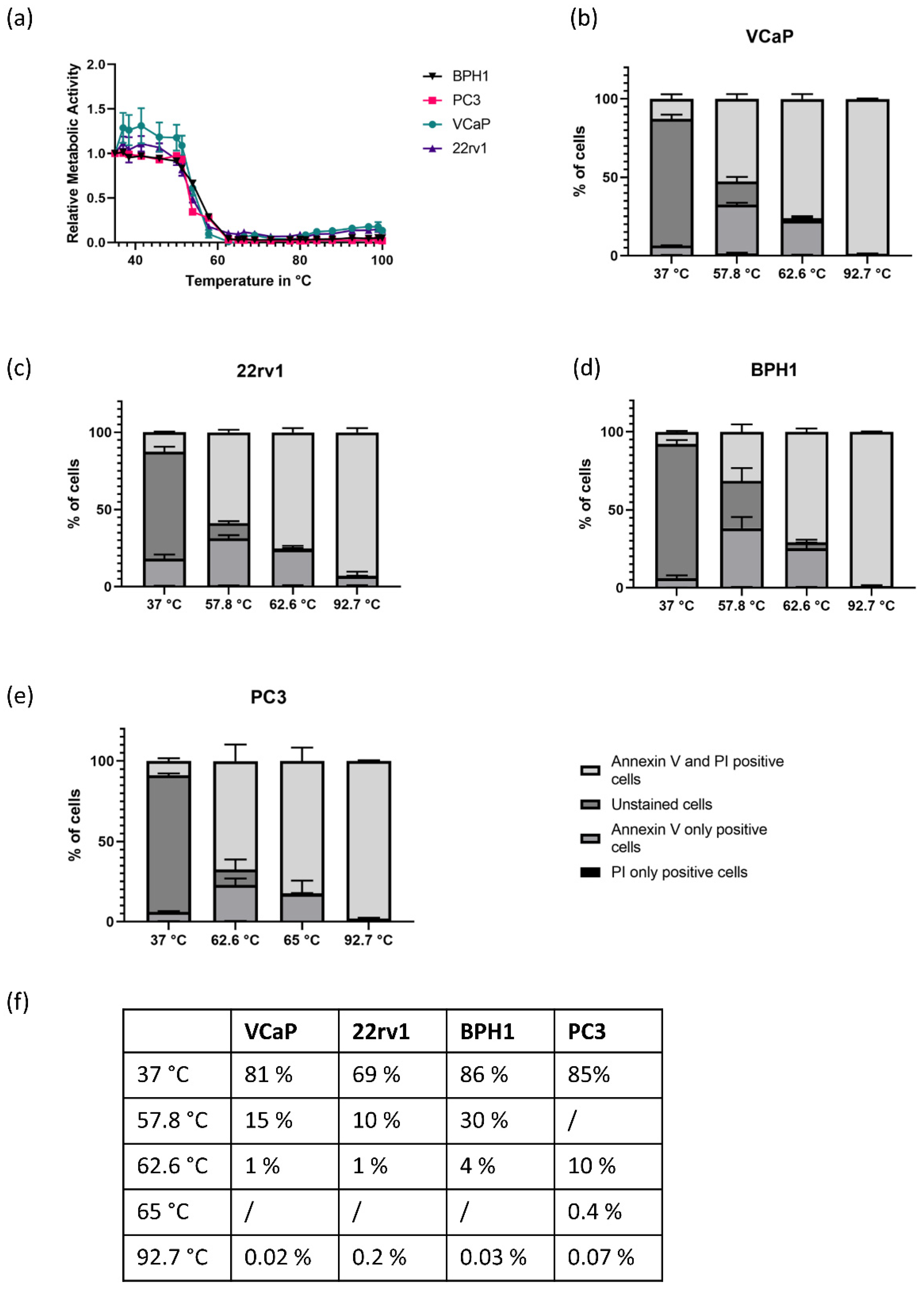
3.1. Heat Response Differs Significantly between AR-Positive and AR-Negative Cell Lines
3.2. Inhibition of the Androgen Axis Using Bicalutamide and Finasteride Does Not Affect Heat Sensitivity
3.3. The VCaPrev Cell Model of Castration Sensitive Prostate Canwcer Shows a Higher Increase in Metabolic Activity Compared to the Other Cell Lines
3.4. Pre-Treatment with Mitoxantrone and Docetaxel Decreases Metabolic Activity but Does Not Increase Heat-Induced Cell Death
3.5. RNA Sequencing of VCaP and VCaPrev Reveals a Highly Significant Enrichment of Genes Normally Repressed by H3K27me3 in VCaPrev Cells
3.6. VCaPrev Cells Show a De-Differentiation towards a Neuroendocrine Phenotype Compared to Wildtype VCaP Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Wilt, T.J.; Jones, K.M.; Barry, M.J.; Andriole, G.L.; Culkin, D.; Wheeler, T.; Aronson, W.J.; Brawer, M.K. Follow-Up of Prostatectomy versus Observation for Early Prostate Cancer. Available online: https://www.nejm.org/doi/10.1056/NEJMoa1615869?url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org&rfr_dat=cr_pub%3Dwww.ncbi.nlm.nih.gov (accessed on 28 November 2019).
- Perera, M.; Krishnananthan, N.; Lindner, U.; Lawrentschuk, N. An update on focal therapy for prostate cancer. Nat. Rev. Urol. 2016, 13, 641–653. [Google Scholar] [CrossRef]
- Liu, W.; Laitinen, S.; Khan, S.; Vihinen, M.; Kowalski, J.; Yu, G.; Chen, L.; Ewing, C.M.; Eisenberger, M.A.; Carducci, M.A.; et al. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat. Med. 2009, 15, 559–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valerio, M.; Cerantola, Y.; Eggener, S.E.; Lepor, H.; Polascik, T.J.; Villers, A.; Emberton, M. New and Established Technology in Focal Ablation of the Prostate: A Systematic Review. Eur. Urol. 2017, 71, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Dewhirst, M.W.; Viglianti, B.L.; Lora-Michiels, M.; Hanson, M.; Hoopes, P.J. Basic principles of thermal dosimetry and thermal thresholds for tissue damage from hyperthermia. Int. J. Hyperth. 2003, 19, 267–294. [Google Scholar] [CrossRef] [PubMed]
- Haar, G.T.; Coussios, C. High intensity focused ultrasound: Physical principles and devices. Int. J. Hyperth. 2007, 23, 89–104. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Wilson Gao, X. Variations of bubble cavitation and temperature elevation during lesion formation by high-intensity focused ultrasound. J. Acoust. Soc. Am. 2013, 134, 1683–1694. [Google Scholar] [CrossRef]
- Hoogenboom, M.; Eikelenboom, D.; den Brok, M.H.; Heerschap, A.; Fütterer, J.J.; Adema, G.J. Mechanical High-Intensity Focused Ultrasound Destruction of Soft Tissue: Working Mechanisms and Physiologic Effects. Ultrasound Med. Biol. 2015, 41, 1500–1517. [Google Scholar] [CrossRef]
- Hill, C.R.; ter Haar, G.R. High intensity focused ultrasound—Potential for cancer treatment. BJR 1995, 68, 1296–1303. [Google Scholar] [CrossRef]
- Chu, K.F.; Dupuy, D.E. Thermal ablation of tumours: Biological mechanisms and advances in therapy. Nat. Rev. Cancer 2014, 14, 199–208. [Google Scholar] [CrossRef]
- Markovsky, E.; Baabur-Cohen, H.; Satchi-Fainaro, R. Anticancer polymeric nanomedicine bearing synergistic drug combination is superior to a mixture of individually-conjugated drugs. J. Control. Release 2014, 187, 145–157. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.; Xu, W.; Xiao, G.; Ding, J.; Chen, X. Tumor microenvironment-labile polymer–doxorubicin conjugate thermogel combined with docetaxel for in situ synergistic chemotherapy of hepatoma. Acta Biomater. 2018, 77, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Kesch, C.; Schmitt, V.; Bidnur, S.; Thi, M.; Beraldi, E.; Moskalev, I.; Yago, V.; Bowden, M.; Adomat, H.; Fazil, L.; et al. A polymeric paste-drug formulation for intratumoral treatment of prostate cancer. Prostate Cancer Prostatic Dis. 2019, 1–9. [Google Scholar] [CrossRef]
- Bremmer, F.; Jarry, H.; Unterkircher, V.; Kaulfuss, S.; Burfeind, P.; Radzun, H.-J.; Ströbel, P.; Thelen, P. Testosterone metabolites inhibit proliferation of castration- and therapy-resistant prostate cancer. Oncotarget 2018, 9, 16951–16961. [Google Scholar] [CrossRef] [Green Version]
- Bremmer, F.; Jarry, H.; Strauß, A.; Behnes, C.L.; Trojan, L.; Thelen, P. Increased expression of CYP17A1 indicates an effective targeting of the androgen receptor axis in castration resistant prostate cancer (CRPC). SpringerPlus 2014, 3, 574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thelen, P.; Heinrich, E.; Bremmer, F.; Trojan, L.; Strauss, A. Testosterone boosts for treatment of castration resistant prostate cancer: An experimental implementation of intermittent androgen deprivation. Prostate 2013, 73, 1699–1709. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [Green Version]
- Goecks, J.; Eberhard, C.; Too, T.; Nekrutenko, A.; Taylor, J. The Galaxy Team Web-based visual analysis for high-throughput genomics. BMC Genom. 2013, 14, 397. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, K.; Kohno, K.; Sato, S.; Uchiumi, T.; Tanimura, H.; Yamada, Y.; Kuwano, M. Enhanced Expression of the DNA Topoisomerase II Gene in Response to Heat Shock Stress in Human Epidermoid Cancer KB Cells. Cancer Res. 1993, 53, 1085–1090. [Google Scholar]
- Hirohashi, Y.; Hidaka, K.; Sato, S.; Kuwano, M.; Kohno, K.; Hisatsugu, T. Biomodulation by Hyperthermia of Topoisomerase II-Targeting Drugs in Human Colorectal Cancer Cells. Jpn. J. Cancer Res. 1995, 86, 1097–1105. [Google Scholar] [CrossRef]
- Kampinga, H.H. Hyperthermia, thermotolerance and topoisomerase II inhibitors. Br. J. Cancer 1995, 72, 333–338. [Google Scholar] [CrossRef] [Green Version]
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular Characterization of Neuroendocrine Prostate Cancer and Identification of New Drug Targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef] [Green Version]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.S.K.; Varambally, S.; et al. Divergent clonal evolution of castration resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [Green Version]
- Tsai, H.K.; Lehrer, J.; Alshalalfa, M.; Erho, N.; Davicioni, E.; Lotan, T.L. Gene expression signatures of neuroendocrine prostate cancer and primary small cell prostatic carcinoma. BMC Cancer 2017, 17, 759. [Google Scholar] [CrossRef] [Green Version]
- Bakht, M.K.; Derecichei, I.; Li, Y.; Ferraiuolo, R.-M.; Dunning, M.; Oh, S.W.; Hussein, A.; Youn, H.; Stringer, K.F.; Jeong, C.W.; et al. Neuroendocrine differentiation of prostate cancer leads to PSMA suppression. Endocr.-Relat. Cancer 2019, 26, 131–146. [Google Scholar] [CrossRef]
- Bonekamp, D.; Wolf, M.B.; Roethke, M.C.; Pahernik, S.; Hadaschik, B.A.; Hatiboglu, G.; Kuru, T.H.; Popeneciu, I.V.; Chin, J.L.; Billia, M.; et al. Twelve-month prostate volume reduction after MRI-guided transurethral ultrasound ablation of the prostate. Eur. Radiol. 2019, 29, 299–308. [Google Scholar] [CrossRef]
- Richter, K.; Haslbeck, M.; Buchner, J. The heat shock response: Life on the verge of death. Mol. Cell 2010, 40, 253–266. [Google Scholar] [CrossRef]
- Morey, L.; Helin, K. Polycomb group protein-mediated repression of transcription. Trends Biochem. Sci. 2010, 35, 323–332. [Google Scholar] [CrossRef]
- Tai, S.; Sun, Y.; Squires, J.M.; Zhang, H.; Oh, W.K.; Liang, C.-Z.; Huang, J. PC3 is a cell line characteristic of prostatic small cell carcinoma. Prostate 2011, 71, 1668–1679. [Google Scholar] [CrossRef] [Green Version]
- Göllner, S.; Oellerich, T.; Agrawal-Singh, S.; Schenk, T.; Klein, H.-U.; Rohde, C.; Pabst, C.; Sauer, T.; Lerdrup, M.; Tavor, S.; et al. Loss of the Histone Methyltransferase EZH2 induces Resistance to Multiple Drugs in Acute Myeloid Leukemia. Nat. Med. 2017, 23, 69–78. [Google Scholar] [CrossRef]
- Huang, Q.; He, S.; Tian, Y.; Gu, Y.; Chen, P.; Li, C.; Huang, J.; Liu, Y.; Yu, H.; Jin, M.; et al. Hsp90 inhibition destabilizes Ezh2 protein in alloreactive T cells and reduces graft-versus-host disease in mice. Blood 2017, 129, 2737–2748. [Google Scholar] [CrossRef] [Green Version]
- Powers, M.V.; Workman, P. Targeting of multiple signalling pathways by heat shock protein 90 molecular chaperone inhibitors. Endocr.-Relat. Cancer 2006, 13, S125–S135. [Google Scholar] [CrossRef]
- Bradley, W.D.; Arora, S.; Busby, J.; Balasubramanian, S.; Gehling, V.S.; Nasveschuk, C.G.; Vaswani, R.G.; Yuan, C.-C.; Hatton, C.; Zhao, F.; et al. EZH2 Inhibitor Efficacy in Non-Hodgkin’s Lymphoma Does Not Require Suppression of H3K27 Monomethylation. Chem. Biol. 2014, 21, 1463–1475. [Google Scholar] [CrossRef] [Green Version]
- Barker, C.R.; Hamlett, J.; Pennington, S.R.; Burrows, F.; Lundgren, K.; Lough, R.; Watson, A.J.M.; Jenkins, J.R. The topoisomerase II–Hsp90 complex: A new chemotherapeutic target? Int. J. Cancer 2006, 118, 2685–2693. [Google Scholar] [CrossRef]
- Barker, C.R.; McNamara, A.V.; Rackstraw, S.A.; Nelson, D.E.; White, M.R.; Watson, A.J.M.; Jenkins, J.R. Inhibition of Hsp90 acts synergistically with topoisomerase II poisons to increase the apoptotic killing of cells due to an increase in topoisomerase II mediated DNA damage. Nucleic Acids Res. 2006, 34, 1148–1157. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hahn, O.; Heining, F.M.; Janzen, J.; Becker, J.C.R.; Bertlich, M.; Thelen, P.; Mansour, J.J.; Duensing, S.; Pahernik, S.; Trojan, L.; et al. Modulating the Heat Sensitivity of Prostate Cancer Cell Lines In Vitro: A New Impact for Focal Therapies. Biomedicines 2020, 8, 585. https://doi.org/10.3390/biomedicines8120585
Hahn O, Heining FM, Janzen J, Becker JCR, Bertlich M, Thelen P, Mansour JJ, Duensing S, Pahernik S, Trojan L, et al. Modulating the Heat Sensitivity of Prostate Cancer Cell Lines In Vitro: A New Impact for Focal Therapies. Biomedicines. 2020; 8(12):585. https://doi.org/10.3390/biomedicines8120585
Chicago/Turabian StyleHahn, Oliver, Franziska M. Heining, Jörn Janzen, Johanna C. R. Becker, Marina Bertlich, Paul Thelen, Josef J. Mansour, Stefan Duensing, Sascha Pahernik, Lutz Trojan, and et al. 2020. "Modulating the Heat Sensitivity of Prostate Cancer Cell Lines In Vitro: A New Impact for Focal Therapies" Biomedicines 8, no. 12: 585. https://doi.org/10.3390/biomedicines8120585
APA StyleHahn, O., Heining, F. M., Janzen, J., Becker, J. C. R., Bertlich, M., Thelen, P., Mansour, J. J., Duensing, S., Pahernik, S., Trojan, L., & Popeneciu, I. V. (2020). Modulating the Heat Sensitivity of Prostate Cancer Cell Lines In Vitro: A New Impact for Focal Therapies. Biomedicines, 8(12), 585. https://doi.org/10.3390/biomedicines8120585