Fatty Acid Binding Protein 5 Mediates Cell Death by Psychosine Exposure through Mitochondrial Macropores Formation in Oligodendrocytes



Abstract
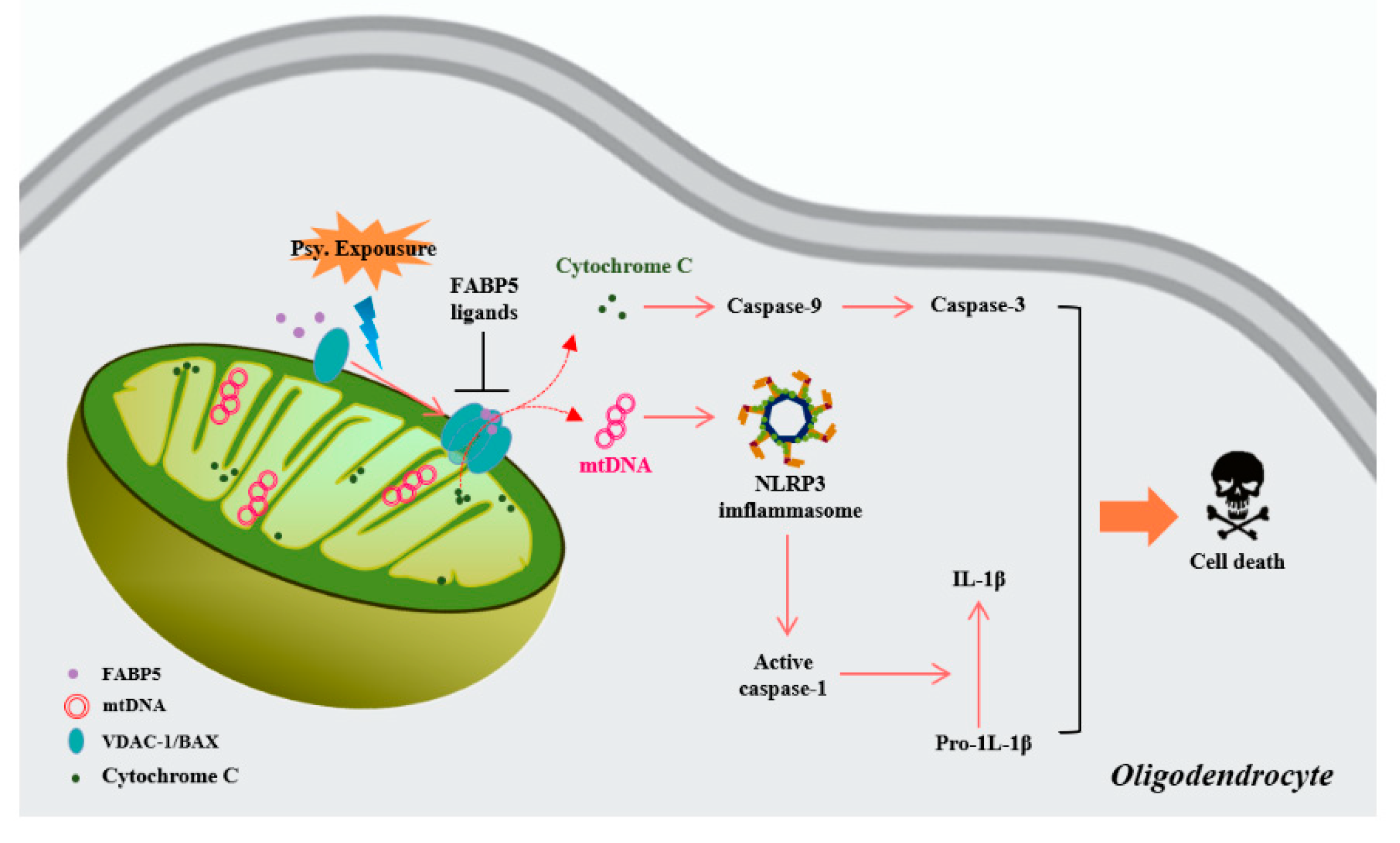
:1. Introduction
2. Experimental Section
2.1. Cell Culture
2.2. Animals and Oligodendrocyte Precursor Cell Culture
2.3. Protein Extraction
2.4. Mitochondria Isolation
2.5. Immunoblotting Analysis
2.6. Immunoprecipitation Analysis
2.7. Sucrose Gradient Centrifugation
2.8. Dot Blot Assay
2.9. Immunofluorescent Staining and Confocal Microscopy
2.10. Cell Death Assay
2.11. FABP5 shRNA Plasmid
2.12. shRNA Delivery
2.13. Analysis of Mitochondrial Membrane Potential (JC-1 Assay)
2.14. FABP5 Ligands
2.15. Statistical Analysis
3. Results
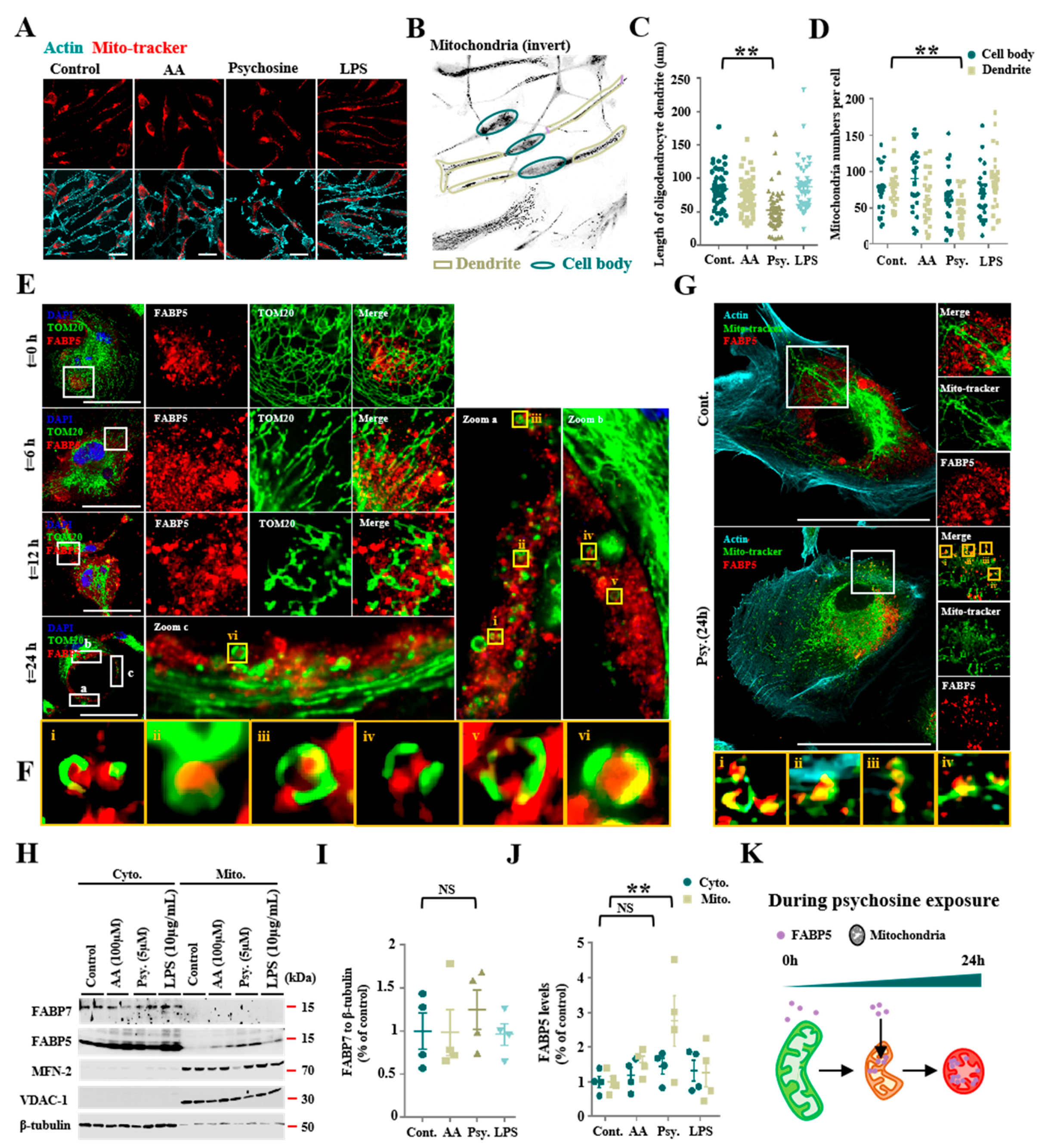
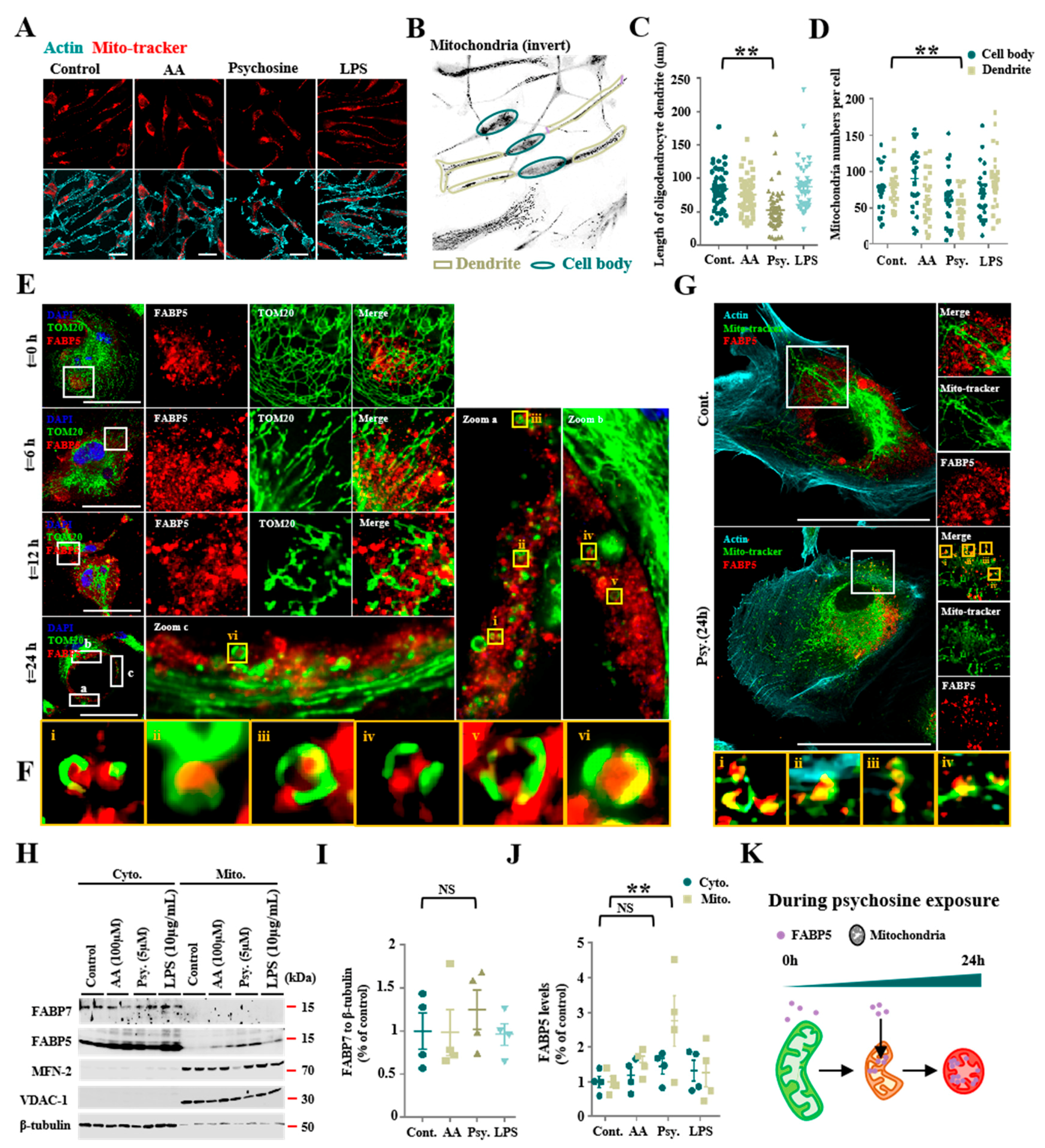
3.1. Psychosine Triggered Mitochondria Loss and Abnormal Accumulations of FABP5 in Mitochondria
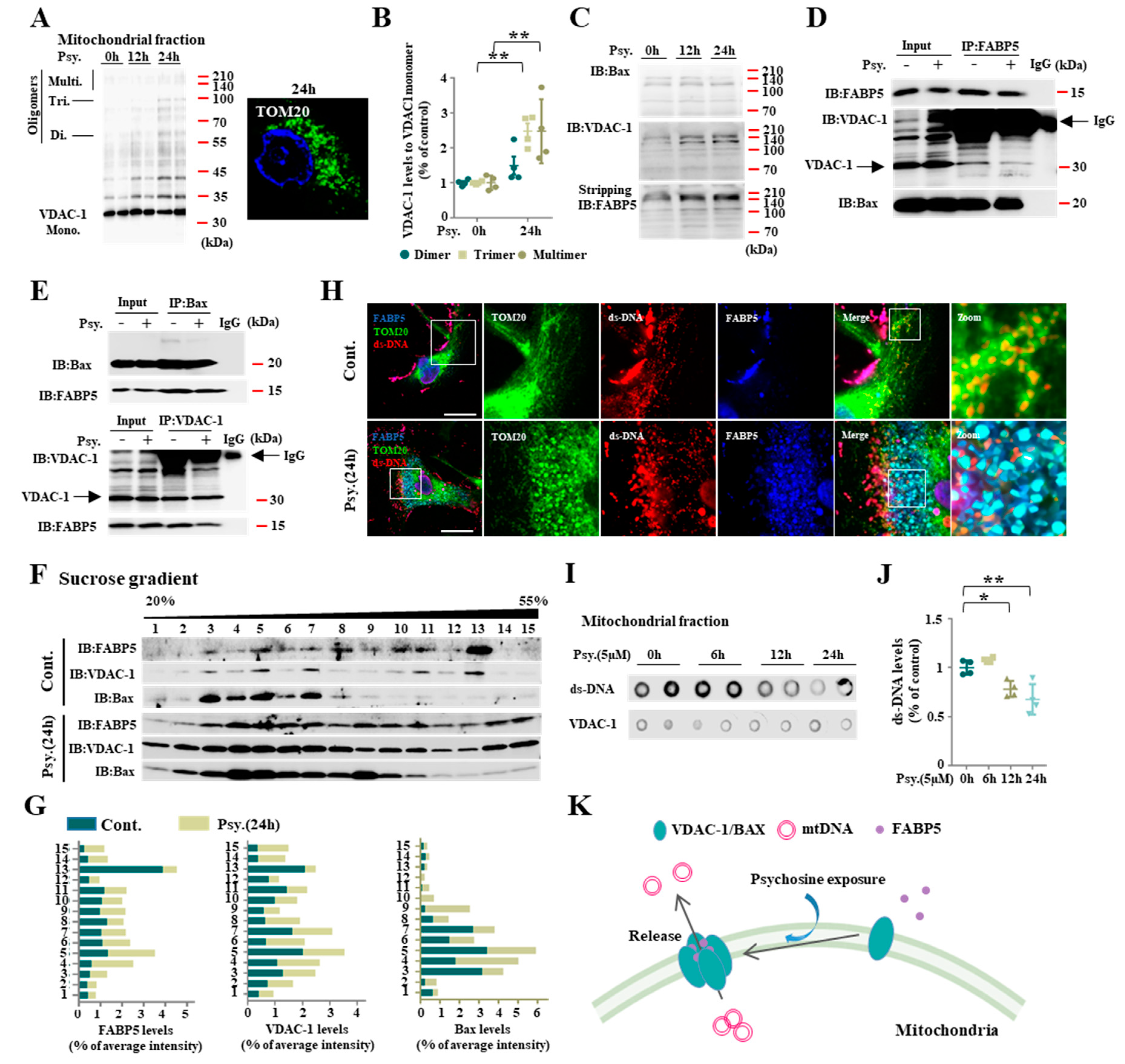
3.2. FABP5 Engaged in Mitochondrial Pores Formation under Psychosine Stress
3.3. FABP5 Is Essential in Psychosine-Induced Mitochondrial Pores Formation
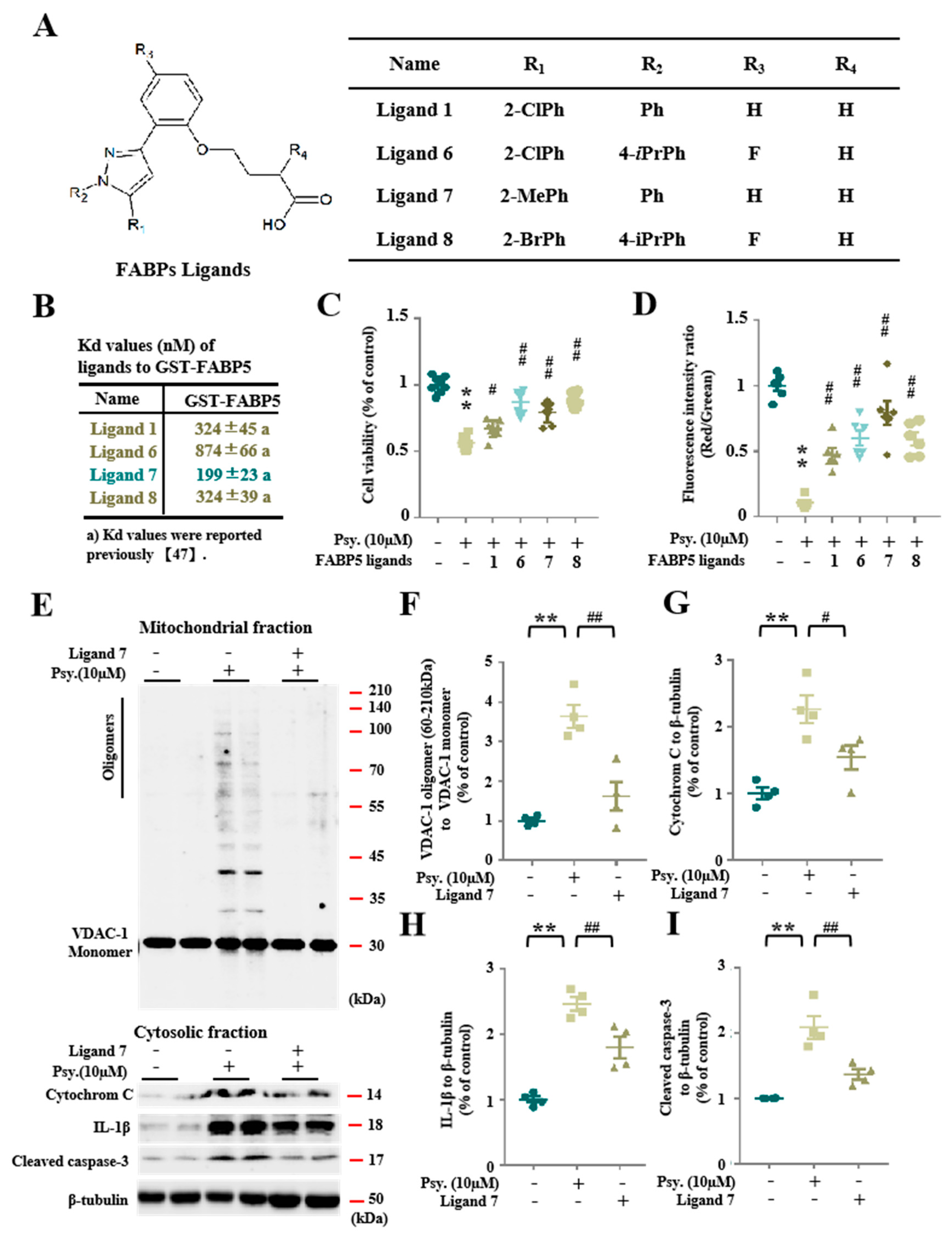
3.4. Pharmacological Inhibition of FABP5 in Psychosine Toxicity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Griffiths, I.; Klugmann, M.; Anderson, T.; Yool, D.; Thomson, C.; Schwab, M.H.; Schneider, A.; Zimmermann, F.; McCulloch, M.; Nadon, N.; et al. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science 1998, 280, 1610–1613. [Google Scholar] [CrossRef] [PubMed]
- Lappe-Siefke, C.; Goebbels, S.; Gravel, M.; Nicksch, E.; Lee, J.; Braun, P.E.; Griffiths, I.R.; Nave, K.A. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat. Enet. 2003, 33, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Gibson, E.; Purger, D.; Mount, C.W.; Goldstein, A.K.; Lin, G.L.; Wood, L.S.; Inema, I.; Miller, S.E.; Bieri, G.; Zuchero, J.B.; et al. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 2014, 344, 1252304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K. Twenty-five years of the ‘‘psychosine hypothesis’’: A personal perspective of its history and present status. Neurochem. Res. 1998, 23, 251–259. [Google Scholar] [CrossRef]
- Tapasi, S.; Padma, P.; Setty, O.H. Effect of psychosine on mitochondrial function. Indian J. Biochem. Biophys. 1998, 35, 161–165. [Google Scholar]
- Haq, E.; Giri, S.; Singh, I.; Singh, A.K. Molecular mechanism of psychosine-induced cell death in human oligodendrocyte cell line. J. Neurochem. 2003, 86, 1428–1440. [Google Scholar] [CrossRef]
- White, A.B.; Givogri, M.I.; Lopez-Rosas, A.; Cao, H.; van Breemen, R.; Thinakaran, G.; Bongarzone, E.R. Psychosine accumulates in membrane microdomains in the brain of krabbe patients, disrupting the raft architecture. J. Neurosci. 2009, 29, 6068–6077. [Google Scholar] [CrossRef] [Green Version]
- D’Auria, L.; Reiter, C.; Ward, E.; Moyano, A.L.; Marshall, M.S.; Nguyen, D.; Scesa, G.; Hauck, Z.; van Breemen, R.; Givogri, M.I.; et al. Psychosine enhances the shedding of membrane microvesicles: Implications in demyelination in Krabbe’s disease. PLoS ONE 2017, 12, e0178103. [Google Scholar] [CrossRef]
- Díaz, Y.L.M.; Caby, S.; Bongarzone, E.R.; Fanani, M.L. Psychosine remodels model lipid membranes at neutral pH. Biochim. Biophys. Acta Biomembr. 2018, 1860, 2515–2526. [Google Scholar] [CrossRef]
- Chmurzyńska, A. The multigene family of fatty acid-binding proteins (FABPs): Function, structure and polymorphism. J. Appl. Genet. 2006, 47, 39–48. [Google Scholar] [CrossRef]
- Smathers, R.L.; Petersen, D.R. The human fatty acid-binding protein family: Evolutionary divergences and function. Hum. Genom. 2011, 5, 170–191. [Google Scholar] [CrossRef] [PubMed]
- Shioda, N.; Yabuki, Y.; Kobayashi, Y.; Onozato, M.; Owada, Y.; Fukunaga, K. FABP3 Protein Promotes α-Synuclein Oligomerization Associated with 1-Methyl-1,2,3,6-tetrahydro- piridine-induced Neurotoxicity. J. Biol. Chem. 2014, 289, 18957–18965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.; Shinoda, Y.; Yamamoto, T.; Miyachi, H.; Fukunaga, K. Development of FABP3 ligands that inhibit arachidonic acid-induced α-synuclein oligomerization. Brain Res. 2019, 1707, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Cheng, A.; Yabuki, Y.; Takahata, I.; Miyachi, H.; Fukunaga, K. Inhibition of MPTP-induced α-synuclein oligomerization by fatty acid-binding protein 3 ligand in MPTP-treated mice. Neuropharmacology 2019, 150, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Rao, E.; Singh, P.; Li, Y.; Zhang, Y.; Chi, Y.I.; Suttles, J.; Li, B. Targeting epidermal fatty acid binding protein for treatment of experimental autoimmune encephalomyelitis. BMC Immunol. 2015, 16, 28. [Google Scholar] [CrossRef] [Green Version]
- Kamizato, K.; Sato, S.; Shil, S.K.; Umaru, B.A.; Kagawa, Y.; Yamamoto, Y.; Ogata, M.; Yasumoto, Y.; Okuyama, Y.; Ishii, N.; et al. The role of fatty acid binding protein 7 in spinal cord astrocytes in a mouse model of experimental autoimmune encephalomyelitis. Neuroscience 2019, 409, 120–129. [Google Scholar] [CrossRef]
- Field, C.S.; Baixauli, F.; Kyle, R.L.; Puleston, D.J.; Cameron, A.M.; Sanin, D.E.; Hippen, K.L.; Loschi, M.; Thangavelu, G.; Corrado, M.; et al. Mitochondrial Integrity Regulated by Lipid Metabolism Is a Cell-Intrinsic Checkpoint for Treg Suppressive Function. Cell Metab. 2020, 31, 422–437. [Google Scholar] [CrossRef] [Green Version]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [Green Version]
- Grünewald, A.; Kumar, K.R.; Sue, C.M. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef]
- Saffari, A.; Kölker, S.; Hoffmann, G.F.; Ebrahimi-Fakhari, D. Linking mitochondrial dysfunction to neurodegeneration in lysosomal storage diseases. J. Inherit. Metab. Dis. 2017, 40, 631–640. [Google Scholar] [CrossRef]
- Briones, R.; Weichbrodt, C.; Paltrinieri, L.; Mey, I.; Villinger, S.; Giller, K.; Lange, A.; Zweckstetter, M.; Griesinger, C.; Becker, S.; et al. Voltage Dependence of Conformational Dynamics and Subconducting States of VDAC-1. Biophys. J. 2016, 111, 1223–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Gupta, R.; Blanco, L.P.; Yang, S.; Shteinfer-Kuzmine, A.; Wang, K.; Zhu, J.; Yoon, H.E.; Wang, X.; Kerkhofs, M.; et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 2019, 366, 1531–1536. [Google Scholar] [CrossRef] [PubMed]
- McArthur, K.; Whitehead, L.W.; Heddleston, J.M.; Li, L.; Padman, B.S.; Oorschot, V.; Geoghegan, N.D.; Chappaz, S.; Davidson, S.; San Chin, H.; et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 2018, 359, eaao6047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carré, M.; André, N.; Carles, G.; Borghi, H.; Brichese, L.; Briand, C.; Braguer, D. Tubulin is an inherent component of mitochondrial membranes that interacts with the voltage-dependent anion channel. J. Biol. Chem. 2002, 277, 33664–33669. [Google Scholar] [CrossRef] [Green Version]
- Martins-Branco, D.; Esteves, A.R.; Santos, D.; Arduino, D.M.; Swerdlow, R.H.; Oliveira, C.R.; Januario, C.; Cardoso, S.M. Ubiquitin proteasome system in Parkinson’s disease: A keeper or a witness? Exp. Neurol. 2012, 238, 89–99. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Shinoda, Y.; Fukunaga, K. KY-226 Protects Blood-brain Barrier Function through the Akt/FoxO1 Signaling Pathway in Brain Ischemia. Neuroscience 2019, 399, 89–102. [Google Scholar] [CrossRef]
- Keinan, N.; Tyomkin, D.; Shoshan-Barmatz, V. Oligomerization of the mitochondrial protein voltage-dependent anion channel is coupled to the induction of apoptosis. Mol. Cell Biol. 2010, 30, 5698–5709. [Google Scholar] [CrossRef] [Green Version]
- Shinoda, Y.; Wang, Y.F.; Yamamoto, T.; Miyachi, H.; Fukunaga, K. Analysis of binding affinity and docking of novel fatty acid-binding protein (FABP) ligands. J. Pharmacol. Sci. 2020, 143, 264–271. [Google Scholar] [CrossRef]
- O’Sullivan, C.; Dev, K.K. Galactosylsphingosine (psychosine)-induced demyelination is attenuated by sphingosine 1-phosphate signalling. J. Cell Sci. 2015, 128, 3878–3887. [Google Scholar] [CrossRef] [Green Version]
- Folts, C.J.; Scott-Hewitt, N.; Pröschel, C.; Mayer-Pröschel, M.; Noble, M. Lysosomal Re-acidification Prevents Lysosphingolipid-Induced Lysosomal Impairment and Cellular Toxicity. PLoS Biol. 2016, 14, e1002583. [Google Scholar] [CrossRef] [Green Version]
- Storch, J.; Corsico, B. The emerging functions and mechanisms of mammalian fatty acid-binding proteins. Annu. Rev. Nutr. 2008, 28, 73–95. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.S.; Quarato, G.; Cloix, C.; Lopez, J.; O’Prey, J.; Pearson, M.; Chapman, J.; Sesaki, H.; Carlin, L.M.; Passos, J.F.; et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 2018, 37, e99238. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, J.; Cervantes, C.; Liu, J.; He, S.; Zhou, H.; Zhang, B.; Cai, H.; Yin, D.; Hu, D.; Li, Z.; et al. DsbA-L prevents obesity-induced inflammation and insulin resistance by suppressing the mtDNA release-activated cGAS-cGAMP-STING pathway. Proc. Natl. Acad. Sci. USA 2017, 114, 12196–12201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Fedurco, M. Redox reactions of heme-containing metalloproteins: Dynamic effects of self-assembled monolayers on thermodynamics and kinetics of cytochrome c electron-transfer reactions. Coordin. Chem. Rev. 2000, 209, 263–331. [Google Scholar] [CrossRef]
- Pelicci, P.G. Electron transfer between cytochrome C and P66SHC generates reactive oxygen species that trigger mitochondrial apoptosis. FEBS J. 2005, 272, 319–320. [Google Scholar]
- Jiang, X.J.; Wang, X.D. Cytochrome C-mediated apoptosis. Annu. Rev. Biochem. 2004, 73, 87–106. [Google Scholar] [CrossRef]
- Nakagawa, I.; Nakata, M.; Kawabata, S.; Hamada, S. Cytochrome c-mediated caspase-9 activation triggers apoptosis in Streptococcus pyogenes-infected epithelial cells. Cell Microbiol. 2001, 3, 395–405. [Google Scholar] [CrossRef] [Green Version]
- Skulachev, V.P. Cytochrome c in the apoptotic and antioxidant cascades. FEBS Lett. 1998, 423, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Madesh, M.; Hajnóczky, G. VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. J. Cell Biol. 2001, 155, 1003–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, S.; Matsuoka, Y.; Shinohara, Y.; Yoneda, Y.; Tsujimoto, Y. Essential role of voltage-dependent anion channel in various forms of apoptosis in mammalian cells. J. Cell Biol. 2001, 152, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.Y.; Shi, C.; Tian, C.; Jiang, H.; Jin, J.; Chen, A.; Almasan, H. Essential role of the voltage-dependent anion channel (VDAC) in mitochondrial permeability transition pore opening and cytochrome c release induced by arsenic trioxide. Oncogene 2004, 23, 1239–1247. [Google Scholar] [CrossRef] [Green Version]
- Abu-Hamad, S.; Zaid, H.; Israelson, A.; Nahon, E.; Shoshan-Barmatz, V. Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: mapping the site of binding. J. Biol. Chem. 2008, 19, 13482–13490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaid, H.; Abu-Hamad, S.; Israelson, A.; Nathan, I.; Shoshan-Barmatz, V. The voltage-dependent anion channel-1 modulates apoptotic cell death. Cell Death Differ. 2005, 12, 751–760. [Google Scholar] [CrossRef]
- White, M.J.; McArthur, K.; Metcalf, D.; Lane, R.M.; Cambier, J.C.; Herold, M.J.; van Delft, M.F.; Bedoui, S.; Lessene, G.; Ritchie, M.E.; et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 2014, 159, 1549–1562. [Google Scholar] [CrossRef] [Green Version]
- Rongvaux, A.; Jackson, R.; Harman, C.C.; Li, T.; West, A.P.; de Zoete, M.R.; Wu, Y.; Yordy, B.; Lakhani, S.A.; Kuan, C.Y.; et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 2014, 159, 1563–1577. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Designation | Source | Identifiers | Dilution Ratio |
|---|---|---|---|
| FABP7 | R&D Systems | AF3166 | 1:200 |
| FABP5 | R&D Systems | AF3077 | 1:200 |
| MFN-2 | abcam | ab56889 | 1:1000 |
| VDAC-1 | CST | 4866 | 1:500 |
| β-tubulin | Sigma-Aldrich | T0198 | 1:4000 |
| BAX | Santa Cruz | SC-493 | 1:500 |
| dsDNA | abcam | ab27156 | 1:1000 |
| Cytochrome C | CST | 4272 | 1:1000 |
| IL-1β | abcam | ab9722 | 1:1000 |
| Cleaved Caspase-3 | CST | 9661 | 1:200 |
| Olig2 | Sigma-Aldrich | MABN50 | 1:500 |
| Anti-mouse IgG (H&L) | SouthernBiotech | 1031-05 | 1:5000 |
| Anti-rabbitIgG (H&L) | SouthernBiotech | 4050-05 | 1:5000 |
| Anti-goatIgG (H&L) | Rockland Immunochemicals | 605-4302 | 1:5000 |
| Alexa 405-labeled ant-mouse IgG | Thermo Fisher Scientific | A-31553 | 1:500 |
| Alexa 488-labeled ant-goat IgG | Thermo Fisher Scientific | A-11055 | 1:500 |
| Alexa 594-labeled ant-mouse IgG | Thermo Fisher Scientific | A-21203 | 1:500 |
| Alexa 594-labeled ant-rabbit IgG | Thermo Fisher Scientific | A-21207 | 1:500 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, A.; Kawahata, I.; Fukunaga, K. Fatty Acid Binding Protein 5 Mediates Cell Death by Psychosine Exposure through Mitochondrial Macropores Formation in Oligodendrocytes. Biomedicines 2020, 8, 635. https://doi.org/10.3390/biomedicines8120635
Cheng A, Kawahata I, Fukunaga K. Fatty Acid Binding Protein 5 Mediates Cell Death by Psychosine Exposure through Mitochondrial Macropores Formation in Oligodendrocytes. Biomedicines. 2020; 8(12):635. https://doi.org/10.3390/biomedicines8120635
Chicago/Turabian StyleCheng, An, Ichiro Kawahata, and Kohji Fukunaga. 2020. "Fatty Acid Binding Protein 5 Mediates Cell Death by Psychosine Exposure through Mitochondrial Macropores Formation in Oligodendrocytes" Biomedicines 8, no. 12: 635. https://doi.org/10.3390/biomedicines8120635