PP1C and PP2A are p70S6K Phosphatases Whose Inhibition Ameliorates HLD12-Associated Inhibition of Oligodendroglial Cell Morphological Differentiation



Abstract
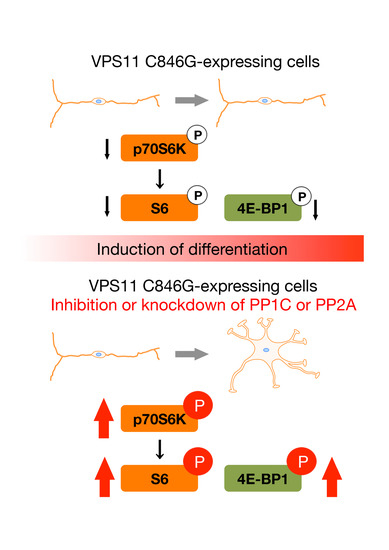
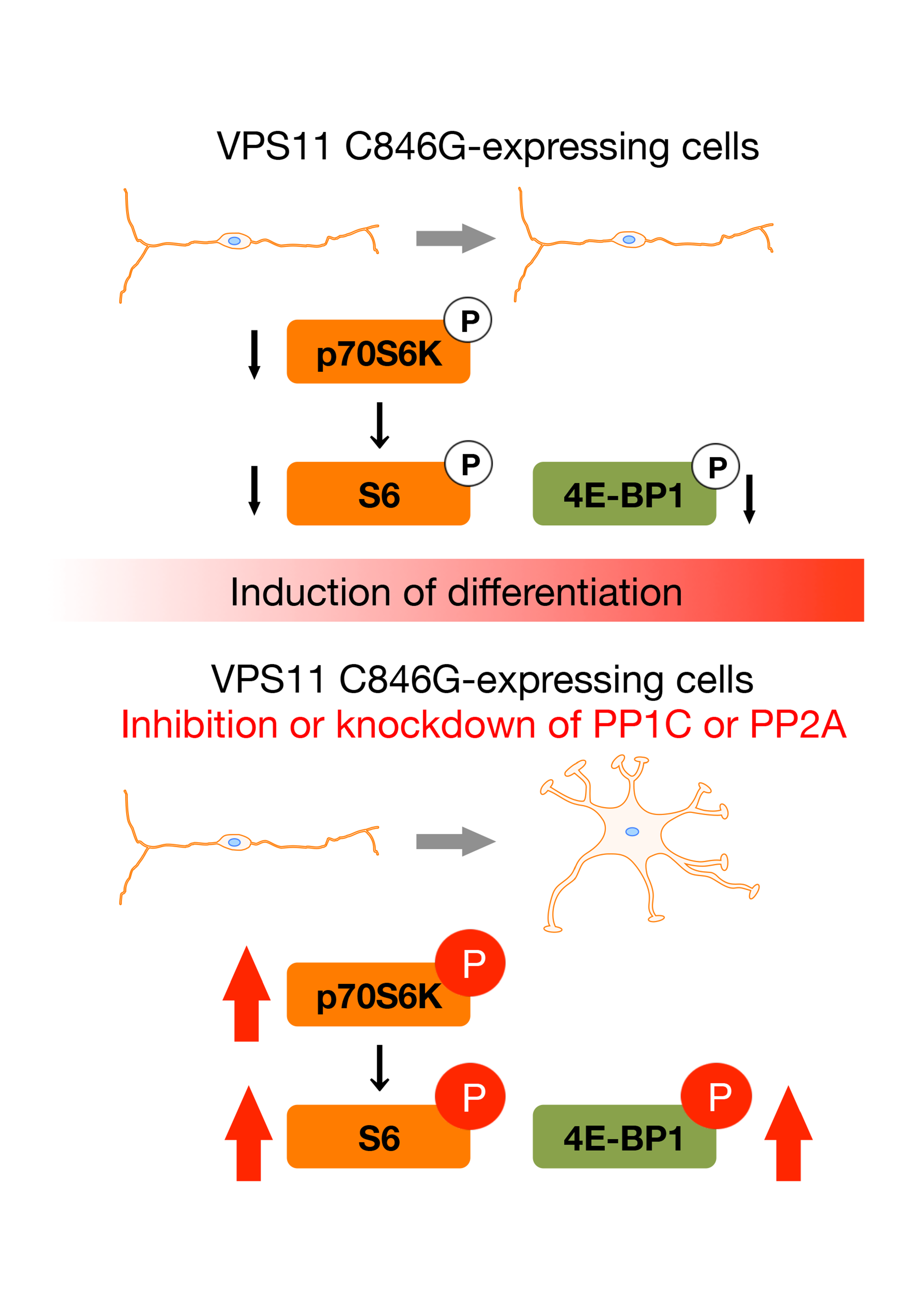
:
1. Introduction
2. Materials and Methods
2.1. Primary Antibodies
2.2. Inhibitors
2.3. Plasmid Constructions
2.4. RT-PCR and PCR Primers
2.5. Cell Culture, Differentiation, Plasmid Transfection, and Stable Clone Isolation
2.6. siRNA Transfection and Target Sequences
2.7. Fluorescence Images
2.8. Polyacrylamide Gel Electrophoresis and Immunoblotting
2.9. Transcriptome Analysis
2.10. Statistical Analysis
2.11. Ethics Statement
3. Results
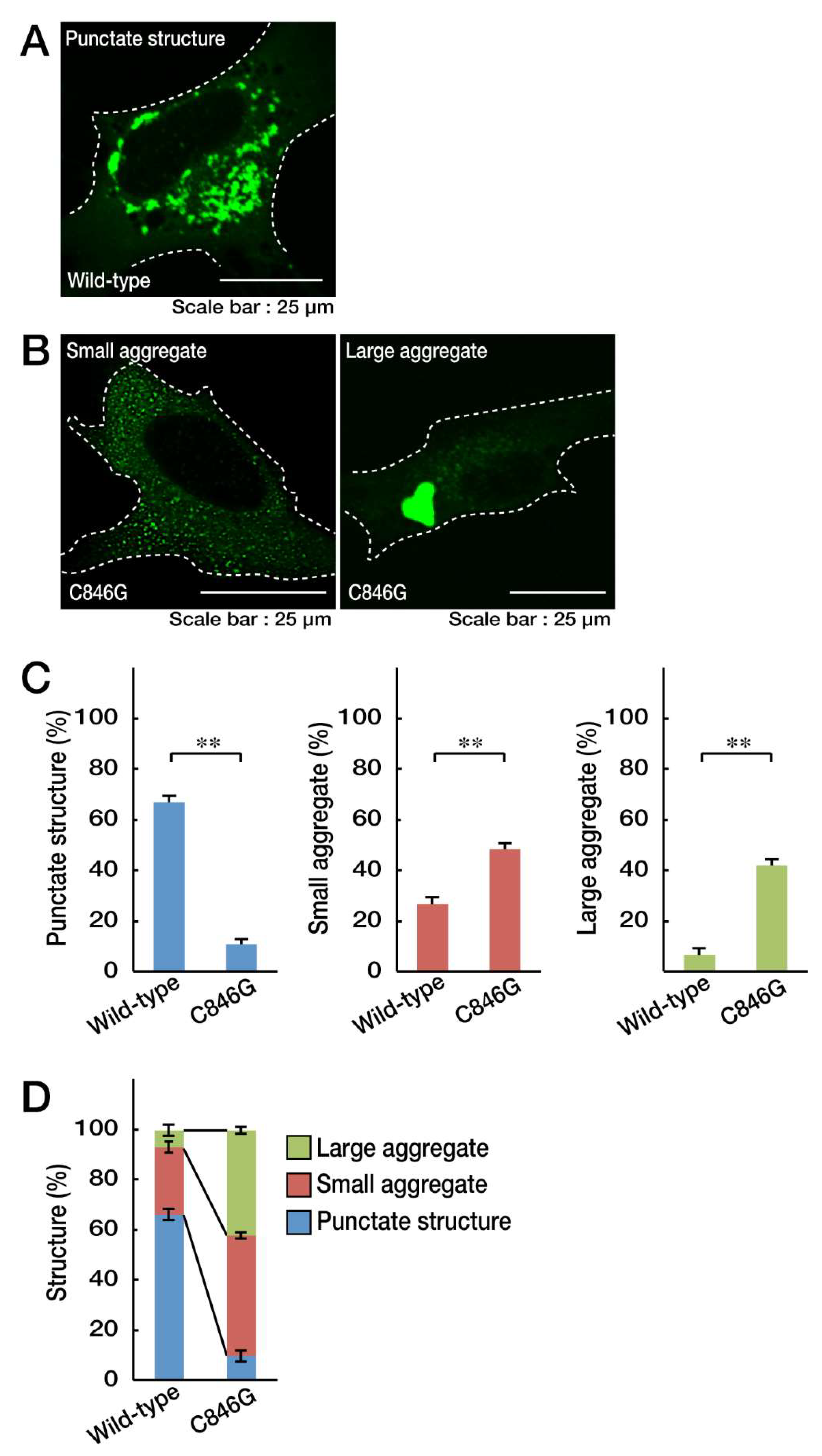
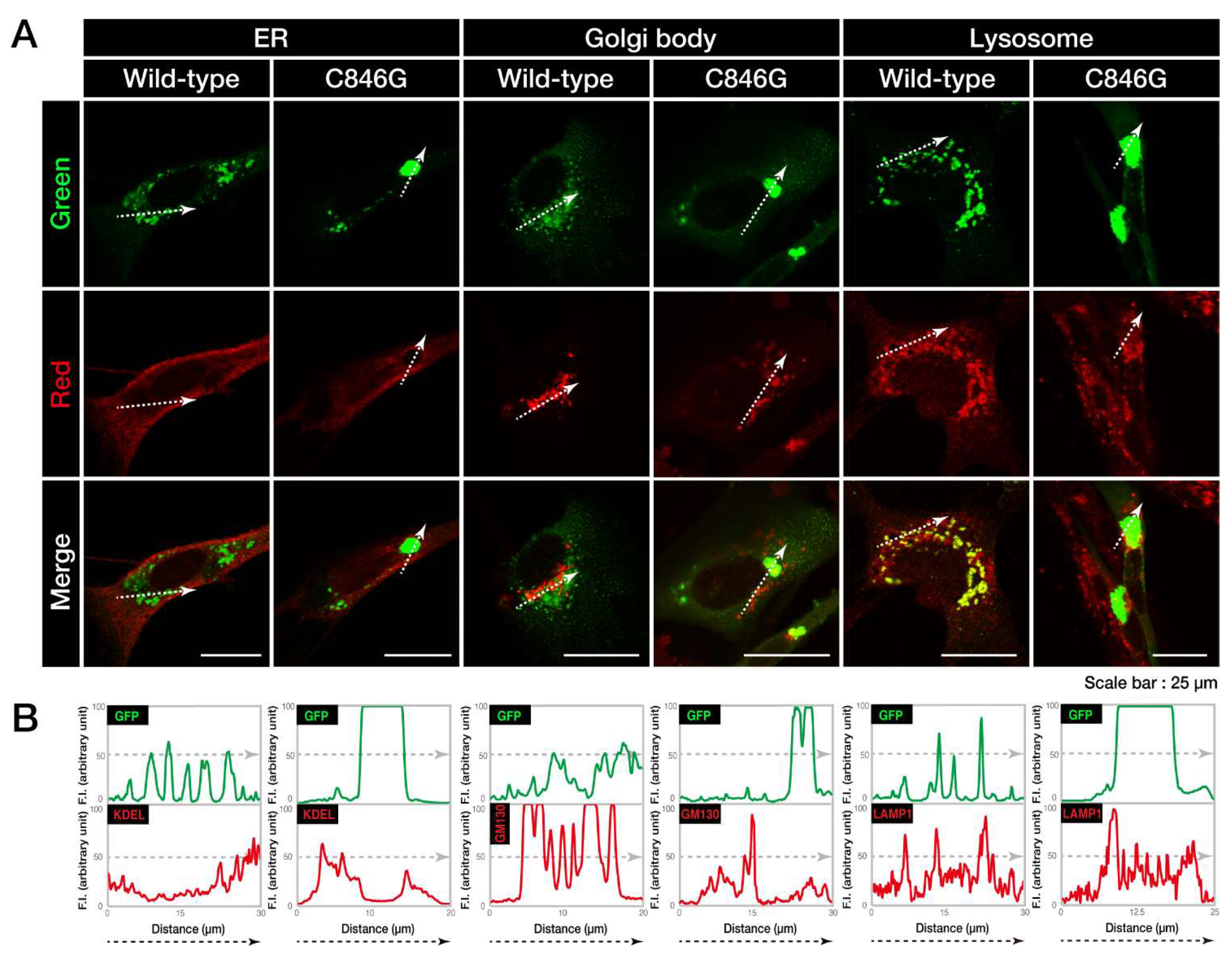
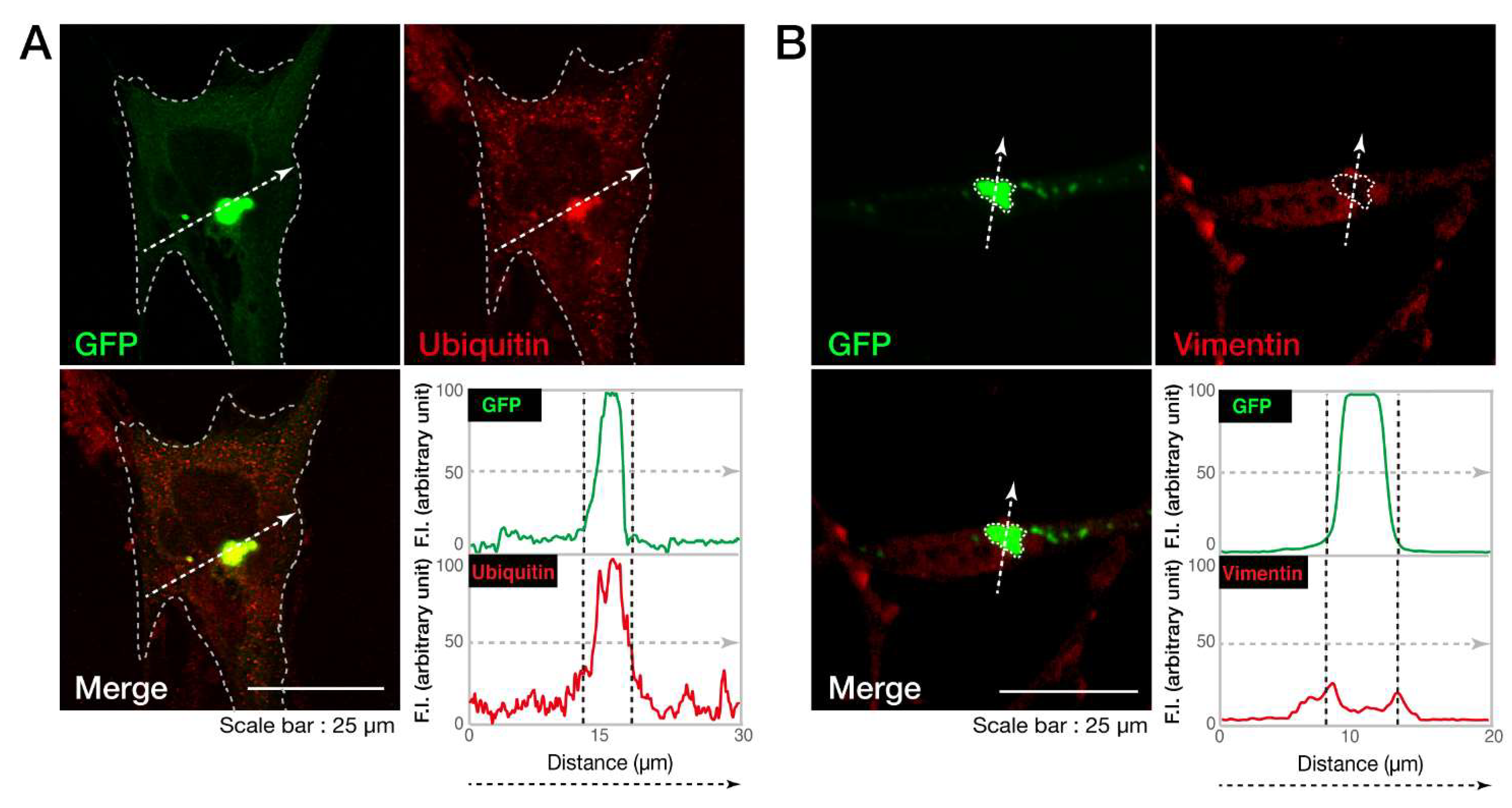
3.1. The C846G Mutation Renders VPS11 Proteins to Form Aggresomes
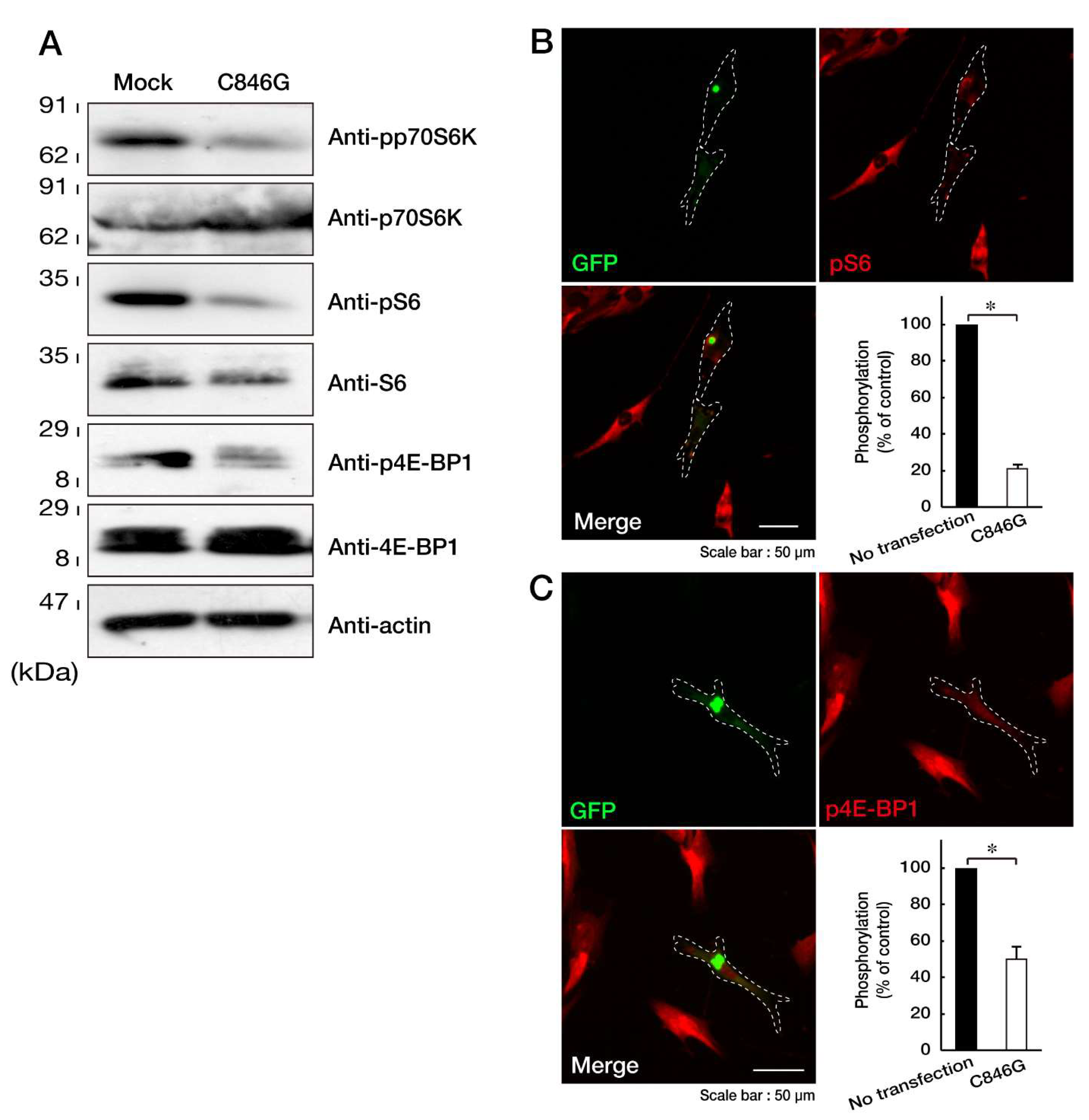
3.2. Expression of the C846G Mutant Proteins in Cells Leads to Downregulation of p70S6K Signaling
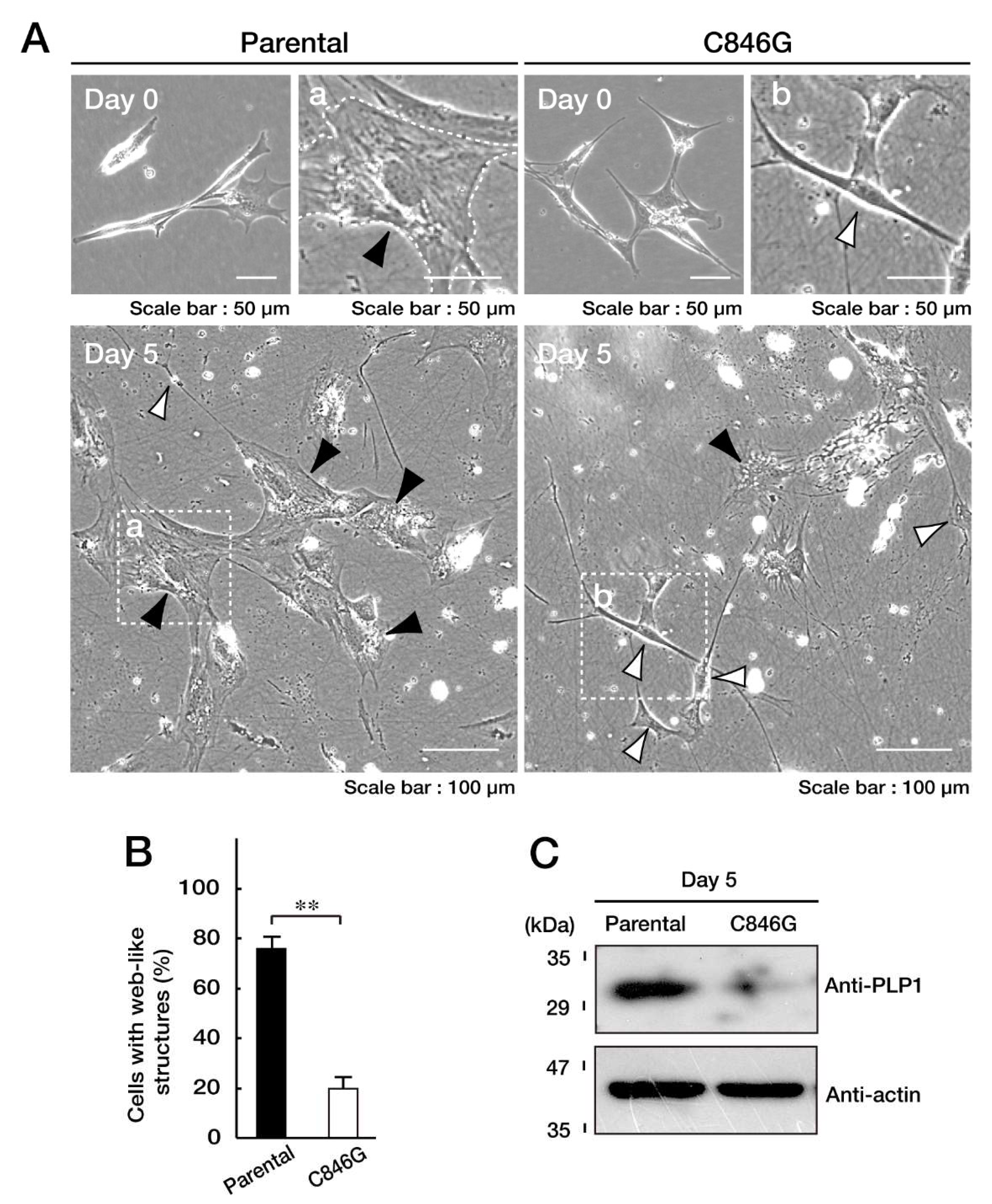
3.3. Cells Stably Expressing the C846G Mutant Constructs Exhibit Undifferentiated Phenotypes
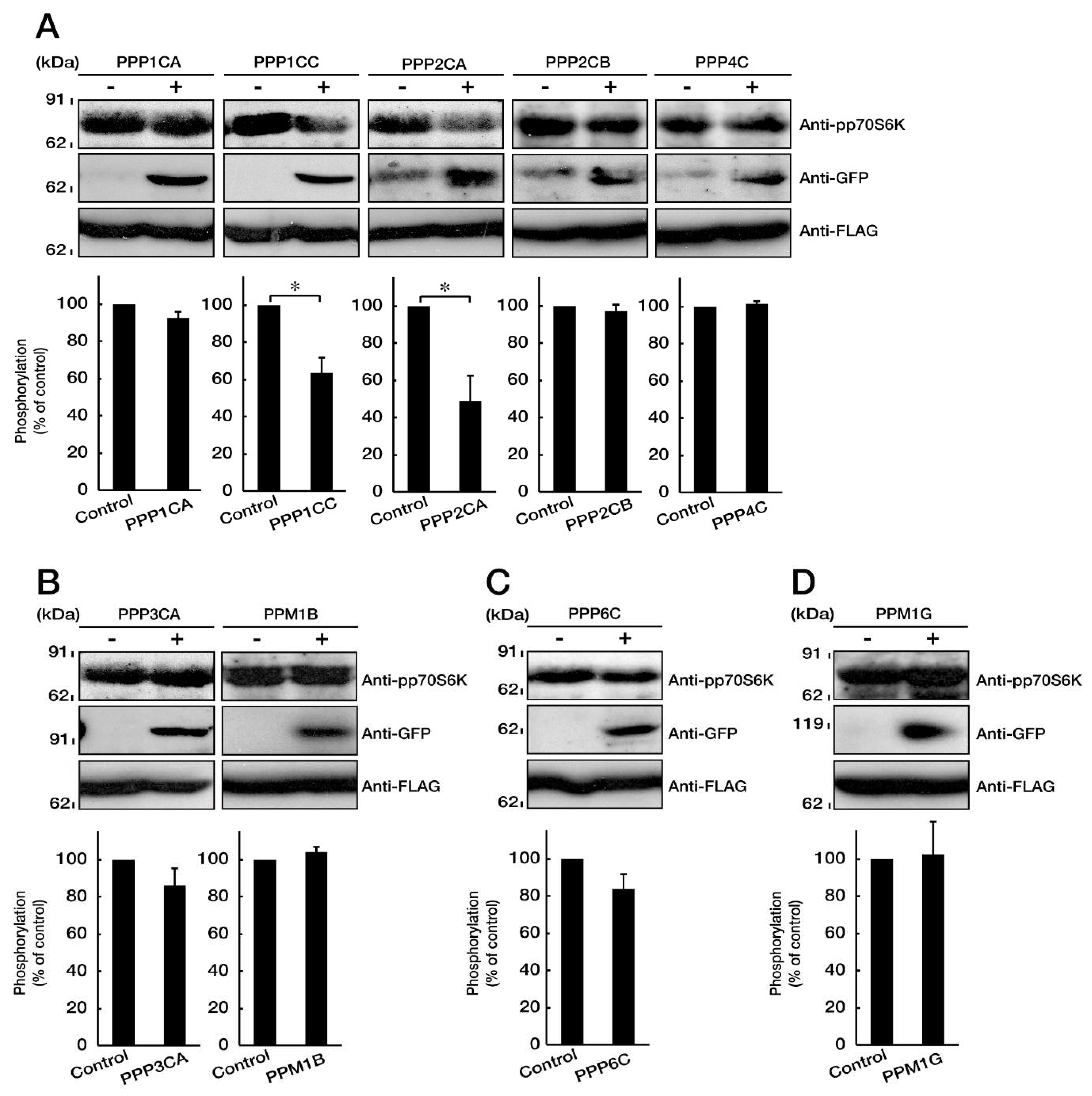
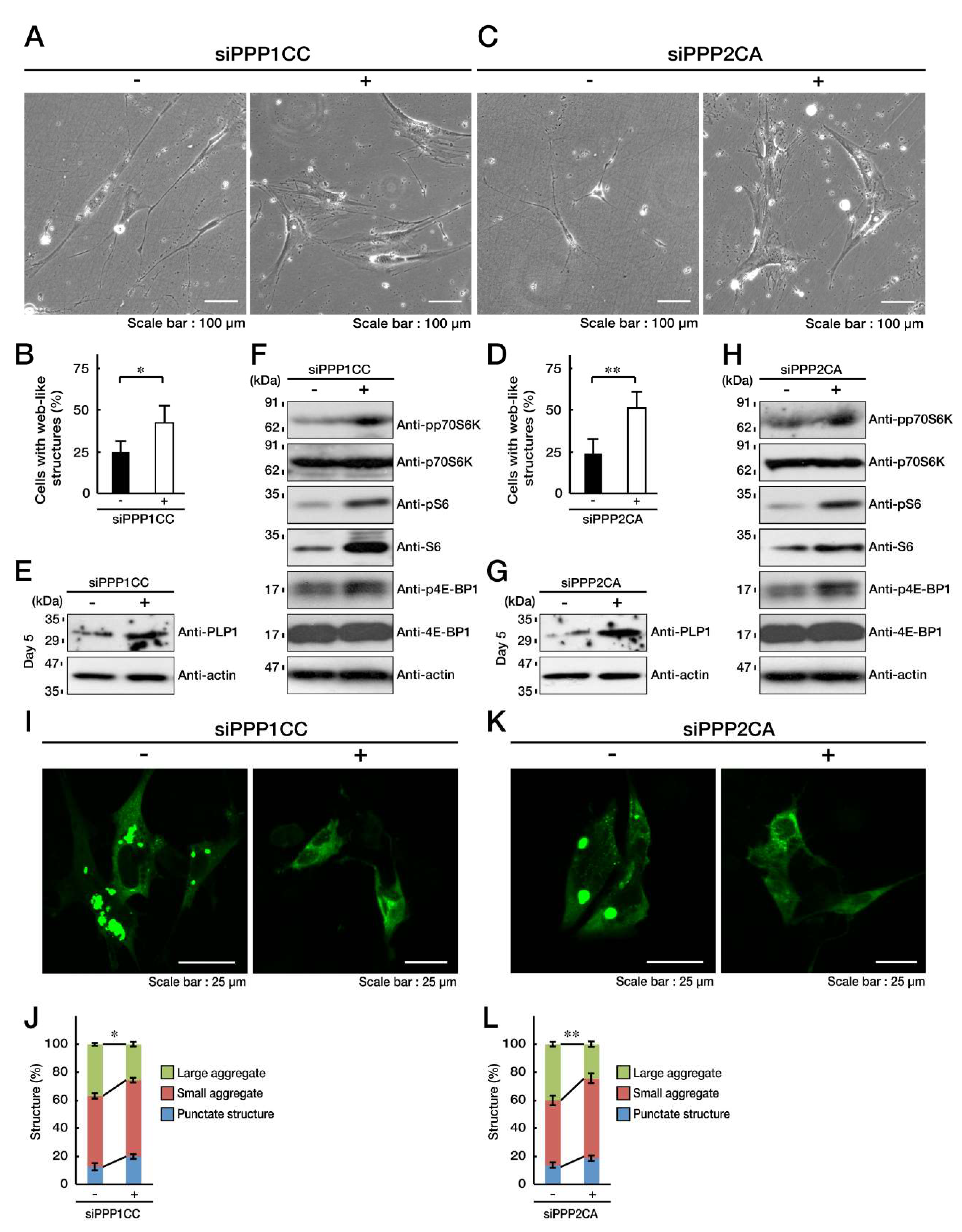
3.4. PP1C and PP2A are p70S6K Phosphatases Whose Inhibition Impairs HLD12 Mutation-Associated Undifferentiated Phenotypes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Simons, M.; Lyons, D.A. Axonal selection and myelin sheath generation in the central nervous system. Curr. Opin. Cell Biol. 2013, 25, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Morton, P.D.; Ishibashi, N.; Jonas, R.A.; Gallo, V. Congenital cardiac anomalies and white matter injury. Trends Neurosci. 2015, 38, 353–563. [Google Scholar] [CrossRef] [Green Version]
- Saab, A.S.; Nave, K.A. Myelin dynamics: Protecting and shaping neuronal functions. Curr. Opin. Neurobiol. 2017, 47, 104–112. [Google Scholar] [CrossRef]
- Abu-Rub, M.; Miller, R.H. Emerging cellular and molecular strategies for enhancing central nervous system (CNS) remyelination. Brain Sci. 2018, 8, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbern, J.; Cambi, F.; Shy, M.; Kamholz, J. The molecular pathogenesis of Pelizaeus-Merzbacher disease. Arch. Neurol. 1999, 56, 1210–1214. [Google Scholar] [CrossRef] [PubMed]
- Dhaunchak, A.S.; Colman, D.R.; Nave, K.A. Misalignment of PLP/DM20 transmembrane domains determines protein misfolding in Pelizaeus-Merzbacher disease. J. Neurosci. 2011, 31, 14961–14971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.; Lin, Y.; Li, J.; Fenstermaker, A.G.; Way, S.W.; Clayton, B.; Jamison, S.; Harding, H.P.; Ron, D.; Popko, B. Oligodendrocyte-specific activation of PERK signaling protects mice against experimental autoimmune encephalomyelitis. J. Neurosci. 2013, 33, 5980–5991. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K. Cellular pathology of Pelizaeus-Merzbacher disease involving chaperones associated with endoplasmic reticulum stress. Front. Mol. Biosci. 2017, 4, 7. [Google Scholar] [CrossRef] [Green Version]
- Biancheri, R.; Rosano, C.; Denegri, L.; Lamantea, E.; Pinto, F.; Lanza, F.; Severino, M.; Filocamo, M. Expanded spectrum of Pelizaeus-Merzbacher-like disease: Literature revision and description of a novel GJC2 mutation in an unusually severe form. Eur. J. Hum. Genet. 2013, 21, 34–39. [Google Scholar] [CrossRef] [Green Version]
- Van der Kant, R.; Jonker, C.T.; Wijdeven, R.H.; Bakker, J.; Janssen, L.; Klumperman, J.; Neefjes, J. Characterization of the mammalian CORVET and HOPS complexes and their modular restructuring for endosome specificity. J. Biol. Chem. 2015, 290, 30280–30290. [Google Scholar] [CrossRef] [Green Version]
- Edvardson, S.; Gerhard, F.; Jalas, C.; Lachmann, J.; Golan, D.; Saada, A.; Shaag, A.; Ungermann, C.; Elpeleg, O. Hypomyelination and developmental delay associated with VPS11 mutation in Ashkenazi-Jewish patients. J. Med. Genet. 2015, 52, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lachance, V.; Schaffner, A.; Li, X.; Fedick, A.; Kaye, L.E.; Liao, J.; Rosenfeld, J.; Yachelevich, N.; Chu, M.L.; et al. A founder mutation in VPS11 causes an autosomal recessive leukoencephalopathy linked to autophagic defects. PLoS Genet. 2016, 12, e1005848. [Google Scholar] [CrossRef] [Green Version]
- Hörtnagel, K.; Krägeloh-Mann, I.; Bornemann, A.; Döcker, M.; Biskup, S.; Mayrhofer, H.; Battke, F.; Bois, G.d.; Harzer, K. The second report of a new hypomyelinating disease due to a defect in the VPS11 gene discloses a massive lysosomal involvement. J. Inherit. Metab. Dis. 2016, 39, 849–857. [Google Scholar] [CrossRef] [Green Version]
- Heard, J.J.; Fong, V.; Bathaie, S.Z.; Tamanoi, F. Recent progress in the study of the Rheb family GTPases. Cell. Signal. 2014, 26, 1950–1957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayanan, S.P.; Flores, A.I.; Wang, F.; Macklin, W.B. Akt signals through the mammalian target of rapamycin pathway to regulate CNS myelination. J. Neurosci. 2009, 29, 6860–6870. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Zhou, L.; Du, X.X.; Ji, Y.; Xu, J.; Tian, J.; Jiang, W.; Zou, Y.; Yu, S.; Gan, L.; et al. Rheb1 is required for mTORC1 and myelination in postnatal brain development. Dev. Cell. 2011, 20, 97–108. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, Y.; Torii, T.; Tanoue, A.; Yamauchi, J. VCAM1 acts in parallel with CD69 and is required for the initiation of oligodendrocyte myelination. Nat. Commun. 2016, 7, 13478. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, Y.; Torii, T.; Tago, K.; Tanoue, A.; Takashima, S.; Yamauchi, J. BIG1/Arfgef1 and Arf1 regulate the initiation of myelination by Schwann cells in mice. Sci. Adv. 2018, 4, eaar4471. [Google Scholar] [CrossRef] [Green Version]
- Sha, Y.; Pandit, L.; Zeng, S.; Eissa, N.T. A critical role for CHIP in the aggresome pathway. Mol. Cell. Biol. 2009, 29, 116–128. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Sala, D.; Oeste, C.L.; Martínez, A.E.; Carrasco, M.J.; Garzón, B.; Cañada, F.J. Vimentin filament organization and stress sensing depend on its single cysteine residue and zinc binding. Nat. Commun. 2015, 6, 7287. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Ikenoue, T.; Chen, X.; Li, L.; Inoki, K.; Guan, K.L. Rheb controls misfolded protein metabolism by inhibiting aggresome formation and autophagy. Proc. Natl. Acad. Sci. USA 2009, 106, 8923–8928. [Google Scholar] [CrossRef] [Green Version]
- Zhu, T.; Zhao, D.; Song, Z.; Yuan, Z.; Li, C.; Wang, Y.; Zhou, X.; Yin, X.; Hassan, M.F.; Yang, L. HDAC6 alleviates prion peptide-mediated neuronal death via modulating PI3K-Akt-mTOR pathway. Neurobiol. Aging. 2016, 37, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Belham, C.; Wu, S.; Avruch, J. Intracellular signaling: PDK1-a kinase at the hub of things. Curr. Biol. 1999, 9, R93–R96. [Google Scholar] [CrossRef] [Green Version]
- Dann, S.G.; Selvaraj, A.; Thomas, G. mTOR Complex1-S6K1 signaling: At the crossroads of obesity, diabetes and cancer. Trends. Mol. Med. 2007, 13, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.H.; Yu, D.; Hu, Z.; Liu, X.; Yang, Y.; Luo, Y.; Zhu, J.; Li, Z. miR-26a and miR-384-5p are required for LTP maintenance and spine enlargement. Nat. Commun. 2015, 6, 6789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, Y.; Yamauchi, J.; Tanoue, A. Cdk5 phosphorylation of WAVE2 regulates oligodendrocyte precursor cell migration through nonreceptor tyrosine kinase Fyn. J. Neurosci. 2008, 28, 8326–8337. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, Y.; Yamauchi, J.; Chan, J.R.; Okada, A.; Tomooka, Y.; Hisanaga, S.; Tanoue, A. Cdk5 regulates differentiation of oligodendrocyte precursor cells through the direct phosphorylation of paxillin. J. Cell Sci. 2007, 120, 4355–4366. [Google Scholar] [CrossRef] [Green Version]
- Sabatini, D.M. Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc. Natl. Acad. Sci. USA 2017, 114, 11818–11825. [Google Scholar] [CrossRef] [Green Version]
- Wainszelbaum, M.J.; Liu, J.; Kong, C.; Srikanth, P.; Samovski, D.; Su, X.; Stahl, P.D. TBC1D3, a hominoid-specific gene, delays IRS-1 degradation and promotes insulin signaling by modulating p70 S6 kinase activity. PLoS ONE 2012, 7, e31225. [Google Scholar] [CrossRef]
- Sheppeck, J.E., 2nd; Gauss, C.M.; Chamberlin, A.R. Inhibition of the Ser-Thr phosphatases PP1 and PP2A by naturally occurring toxins. Bioorg. Med. Chem. 1997, 5, 739–750. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Torii, T.; Eguchi, T.; Nakamura, K.; Tanoue, A.; Yamauchi, J. Hypomyelinating leukodystrophy-associated missense mutant of FAM126A/hyccin/DRCTNNB1A aggregates in the endoplasmic reticulum. J. Clin. Neurosci. 2014, 21, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.C.; Shooter, E.M.; Notterpek, L. Aggresome formation in neuropathy models based on peripheral myelin protein 22 mutations. Neurobiol. Dis. 2002, 10, 109–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortun, J.; Dunn, W.A., Jr.; Joy, S.; Li, J.; Notterpek, L. Emerging role for autophagy in the removal of aggresomes in Schwann cells. J. Neurosci. 2003, 23, 10672–10680. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Names for Genes or Uses | 5’ Primers | 3’ Primers |
|---|---|---|
| the C846G mutagenesis of human VPS11 | 5’-ggctttgagagttactcggaaagtg-3’ | 5’-gtgttggtggaaggagtggc-3’ |
| human PPP1CA coding region (GenBank Acc. No. NM_002708) | 5’-ccgggatccatgtccgacagcgagaagctc-3’ | 5’-ccgggatccctatttcttggctttggcggaattg-3’ |
| human PPP1CC coding region plus 3’-non-coding region (GenBank Acc. No. NM_002710) | 5’-ccgggatccatggcggatttagataaactcaacatcgac-3’ | 5’-ccgggatcctcactcgtatagaacagtattgtttctataatttgaag-3’ |
| human PPP2CA coding region (GenBank Acc. No. NM_002715) | 5’-ccgggatccatggacgagaaggtgttcaccaag-3’ | 5’-ccgggatccttacaggaagtagtctggggtacgac-3’ |
| human PPP2CB coding region (GenBank Acc. No. NM_001009552) | 5’-ccgggatccatggacgacaaggcgttcac-3’ | 5’-ccgggatccttataggaagtagtctggggtgcg-3’ |
| human PPP3CAcoding region (GenBank Acc. No. NM_000944) | 5’-ccgggatccatgtccgagcccaaggcaattg-3’ | 5’-ccgggatcctcactgaatattgctgctattactgccattg-3’ |
| human PPP4C coding region (GenBank Acc. No. NM_001303503) | 5’-ccgggatccatggcggagatcagcgacc-3’ | 5’-ccgggatcctcacaggaagtagtcggccac-3’ |
| human PPP6C coding region (GenBank Acc. No. NM_001123355) | 5’-ccgggatccatggcgccgctagacctg-3’ | 5’-ccgggatcctcaaaggaaatatggcgttgtcgttctg-3’ |
| human PPM1B coding region (GenBank Acc. No. NM_002706) | 5’-ccgagatctatgggtgcatttttggataaacccaaaac-3’ | 5’-ccgagatcttcatattttttcaccactcatctttgtccctg-3’ |
| human PPM1G coding region (GenBank Acc. No. NM_177983) | 5’-ccgggatccatgggtgcctacctctcccag-3’ | 5’-ccgggatccctagtctcgcttggccttcttcttc-3’ |
| mouse PLP1 | 5’-atgggcttgttagagtgttgtgctagatgtctg-3’ | 5’-gaacttggtgcctcggcccatgag-3’ |
| mouse MBP | 5’-atggcatcacagaagagaccctcac-3’ | 5’-cccttgaatcccttgtgagccg-3’ |
| rodent actin | 5’-atggatgacgatatcgctgcgctc-3’ | 5’-ctagaagcatttgcggtgcacgatg-3’ |
| mouse PPP1CC | 5’-atggcggatatcgacaaactcaacatcg-3’ | 5’-ctttgcttgctttgtgatcataccccgtg-3’ |
| mouse PPP2CA | 5’-atggacgagaagttgttcaccaaggag-3’ | 5’-caggaagtagtctggggtacgacgag-3’ |
| human and rodent VPS11 | 5’-tctgaggagttcatccccatctttg-3’ | 5’-gccacacaggaagtggactgag-3’ |
| Names | Target Sequences |
|---|---|
| 78th nucleotide number of mouse PPP1CC | 5’-aagaatgtccagctccaggag-3’ |
| 123th nucleotide number of mouse PPP1CC | 5’-aagtctcgggagatcttcctc-3’ |
| 163th nucleotide number of mouse PPP1CC | 5’-aacttgaagcaccactcaaga-3’ |
| 258th nucleotide number of mouse PPP1CC | 5’-aactatttgtttctcggggac-3’ |
| 87th nucleotide number of mouse PPP2CA | 5’-aagagcctctgcgagaaggct-3’ |
| 109th nucleotide number of mouse PPP2CA | 5’-aagaaatcctgacaaaagaat-3’ |
| 139th nucleotide number of mouse PPP2CA | 5’-aagaggttcgatgtccagtca-3’ |
| 312th nucleotide number of mouse PPP2CA | 5’-aaggttcgttaccgagagcgc-3’ |
| control luciferase | 5’-aagccattctatcctctagag-3’ |
| GenBank Acc. No. | Name | Sample 1 | Sample 2 | Sample 3 | Average | S.D. | Coefficient of Variation |
|---|---|---|---|---|---|---|---|
| NM_031527.1 | PPP1CA | 1436 | 2355 | 2147 | 1979 | 481.743 | 0.243 |
| NM_022498.1 | PPP1CC | 1457 | 1673 | 1356 | 1495 | 161.750 | 0.108 |
| NM_017039.2 | PPP2CA | 770 | 1385 | 994 | 1049 | 311.223 | 0.297 |
| NM_134359.1 | PPP4C | 279 | 745 | 391 | 472 | 242.878 | 0.515 |
| NM_017040.1 | PPP2CB | 287 | 527 | 366 | 393 | 122.327 | 0.311 |
| NM_147209.2 | PPM1G | 254 | 415 | 333 | 334 | 80.172 | 0.24 |
| NM_133589.2 | PPP6C | 251 | 356 | 249 | 285 | 61.148 | 0.215 |
| NM_033096.1 | PPM1B | 178 | 116 | 97 | 130 | 42.433 | 0.325 |
| NM_017041.1 | PPP3CA | 133 | 124 | 73 | 110 | 32.030 | 0.291 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsumoto, N.; Miyamoto, Y.; Hattori, K.; Ito, A.; Harada, H.; Oizumi, H.; Ohbuchi, K.; Mizoguchi, K.; Yamauchi, J. PP1C and PP2A are p70S6K Phosphatases Whose Inhibition Ameliorates HLD12-Associated Inhibition of Oligodendroglial Cell Morphological Differentiation. Biomedicines 2020, 8, 89. https://doi.org/10.3390/biomedicines8040089
Matsumoto N, Miyamoto Y, Hattori K, Ito A, Harada H, Oizumi H, Ohbuchi K, Mizoguchi K, Yamauchi J. PP1C and PP2A are p70S6K Phosphatases Whose Inhibition Ameliorates HLD12-Associated Inhibition of Oligodendroglial Cell Morphological Differentiation. Biomedicines. 2020; 8(4):89. https://doi.org/10.3390/biomedicines8040089
Chicago/Turabian StyleMatsumoto, Naoto, Yuki Miyamoto, Kohei Hattori, Akihiro Ito, Hironori Harada, Hiroaki Oizumi, Katsuya Ohbuchi, Kazushige Mizoguchi, and Junji Yamauchi. 2020. "PP1C and PP2A are p70S6K Phosphatases Whose Inhibition Ameliorates HLD12-Associated Inhibition of Oligodendroglial Cell Morphological Differentiation" Biomedicines 8, no. 4: 89. https://doi.org/10.3390/biomedicines8040089
APA StyleMatsumoto, N., Miyamoto, Y., Hattori, K., Ito, A., Harada, H., Oizumi, H., Ohbuchi, K., Mizoguchi, K., & Yamauchi, J. (2020). PP1C and PP2A are p70S6K Phosphatases Whose Inhibition Ameliorates HLD12-Associated Inhibition of Oligodendroglial Cell Morphological Differentiation. Biomedicines, 8(4), 89. https://doi.org/10.3390/biomedicines8040089