Transcriptome-Wide Analysis of CXCR5 Deficient Retinal Pigment Epithelial (RPE) Cells Reveals Molecular Signatures of RPE Homeostasis




Abstract
1. Introduction
2. Results
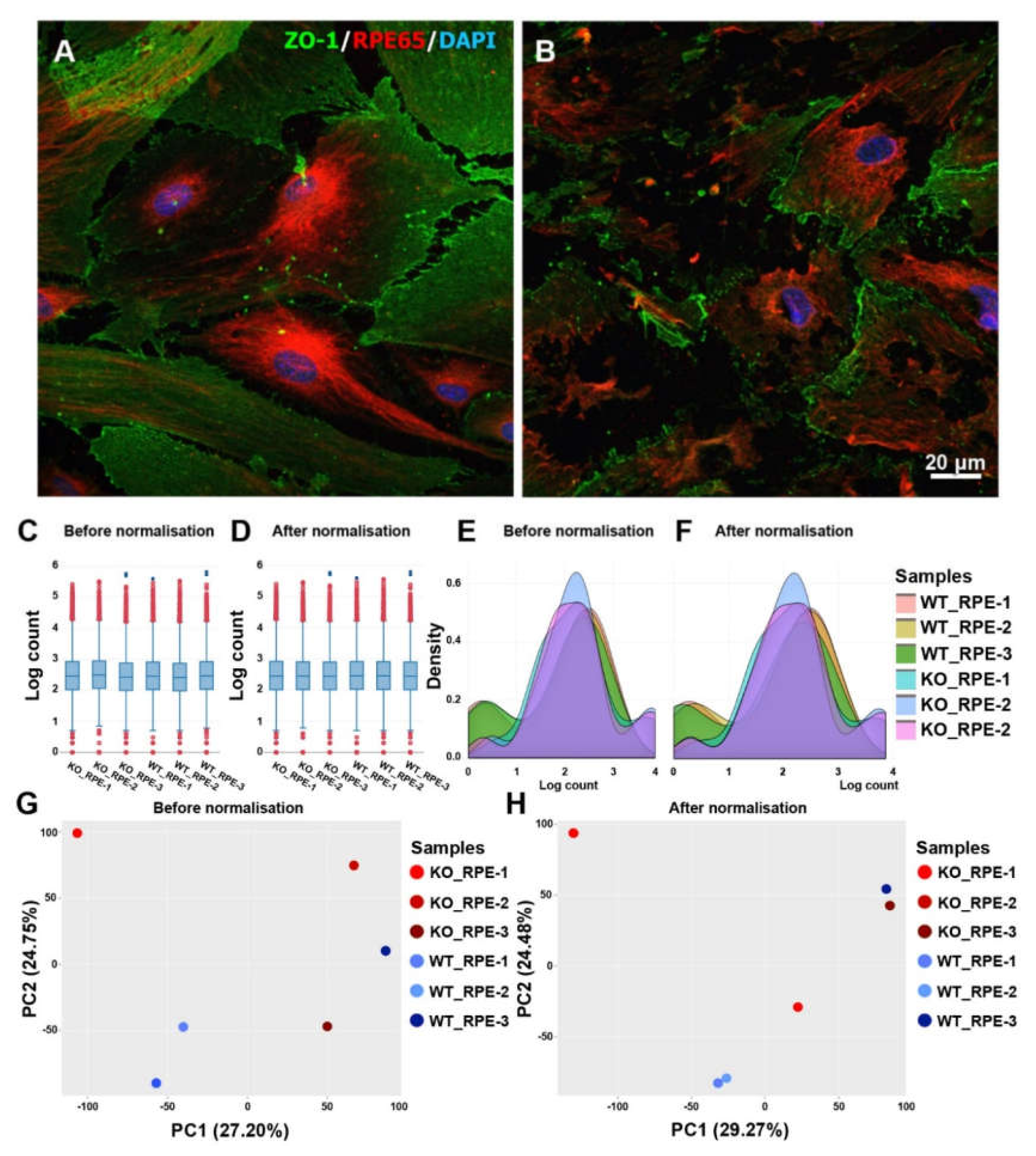
2.1. Abnormalities of the CXCR5-Deficient RPE in Primary Cell Cultures
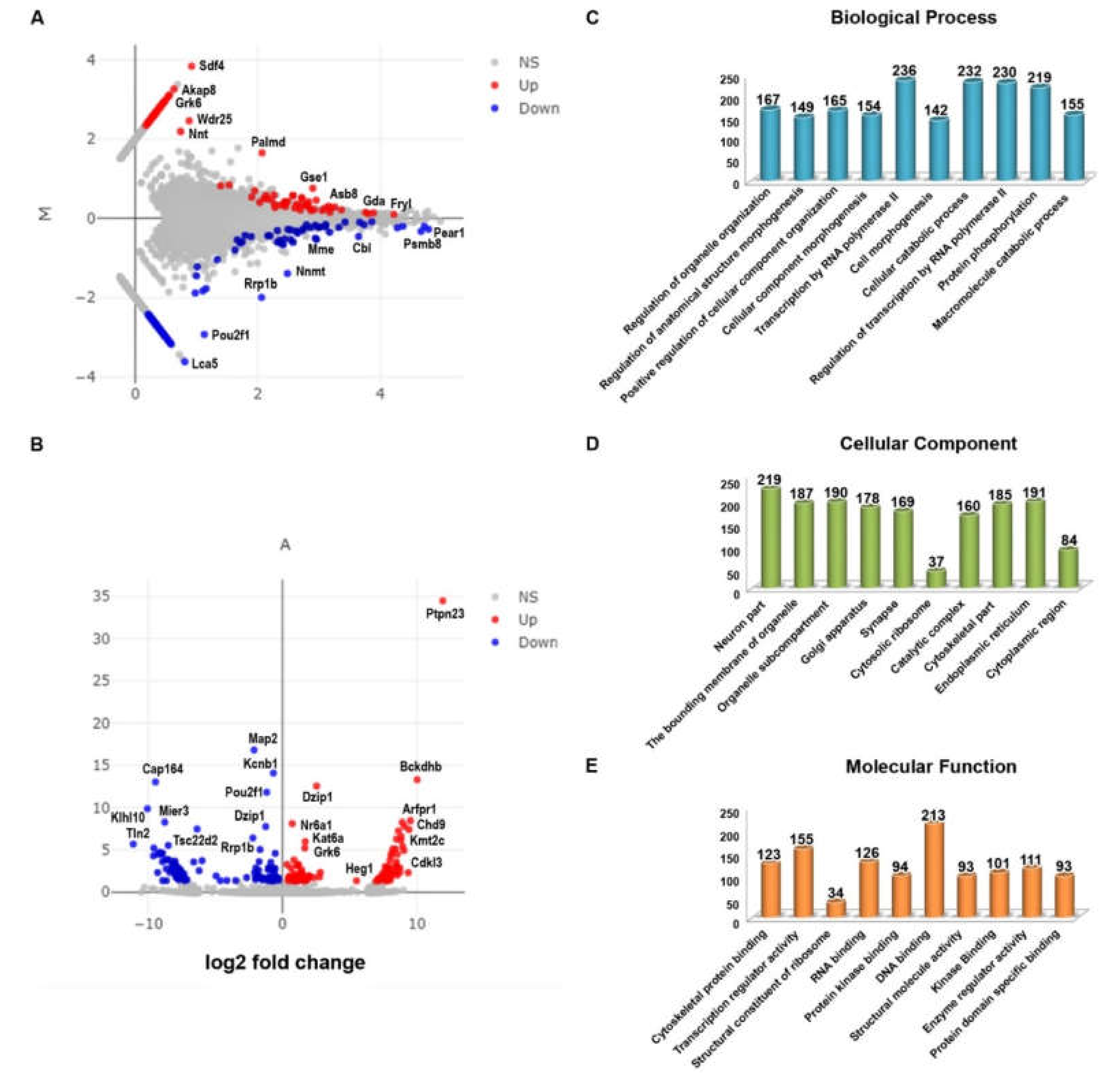
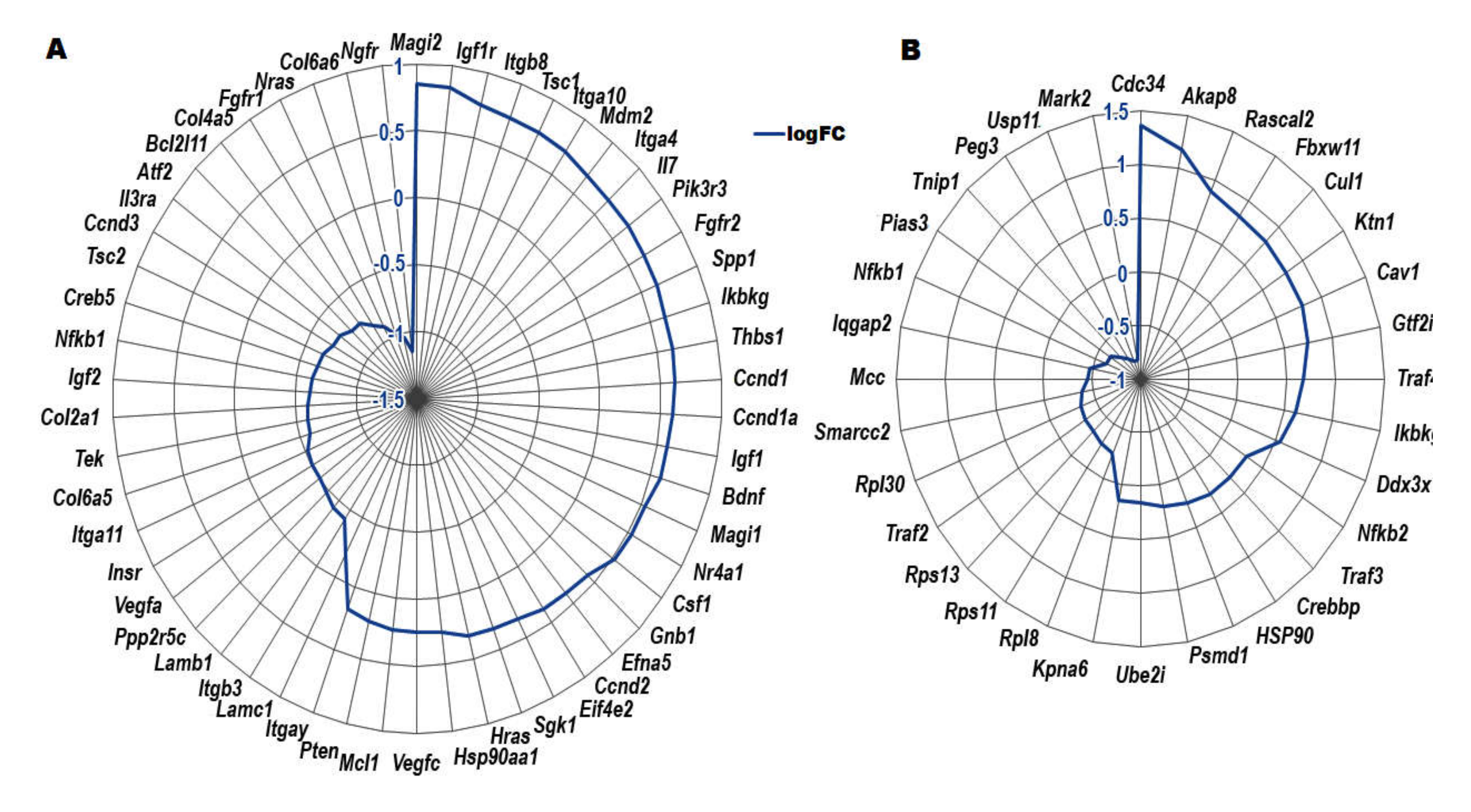
2.2. Differentially Expressed Genes in the CXCR5 KO
2.3. Gene Ontology of the Differentially Expressed Genes
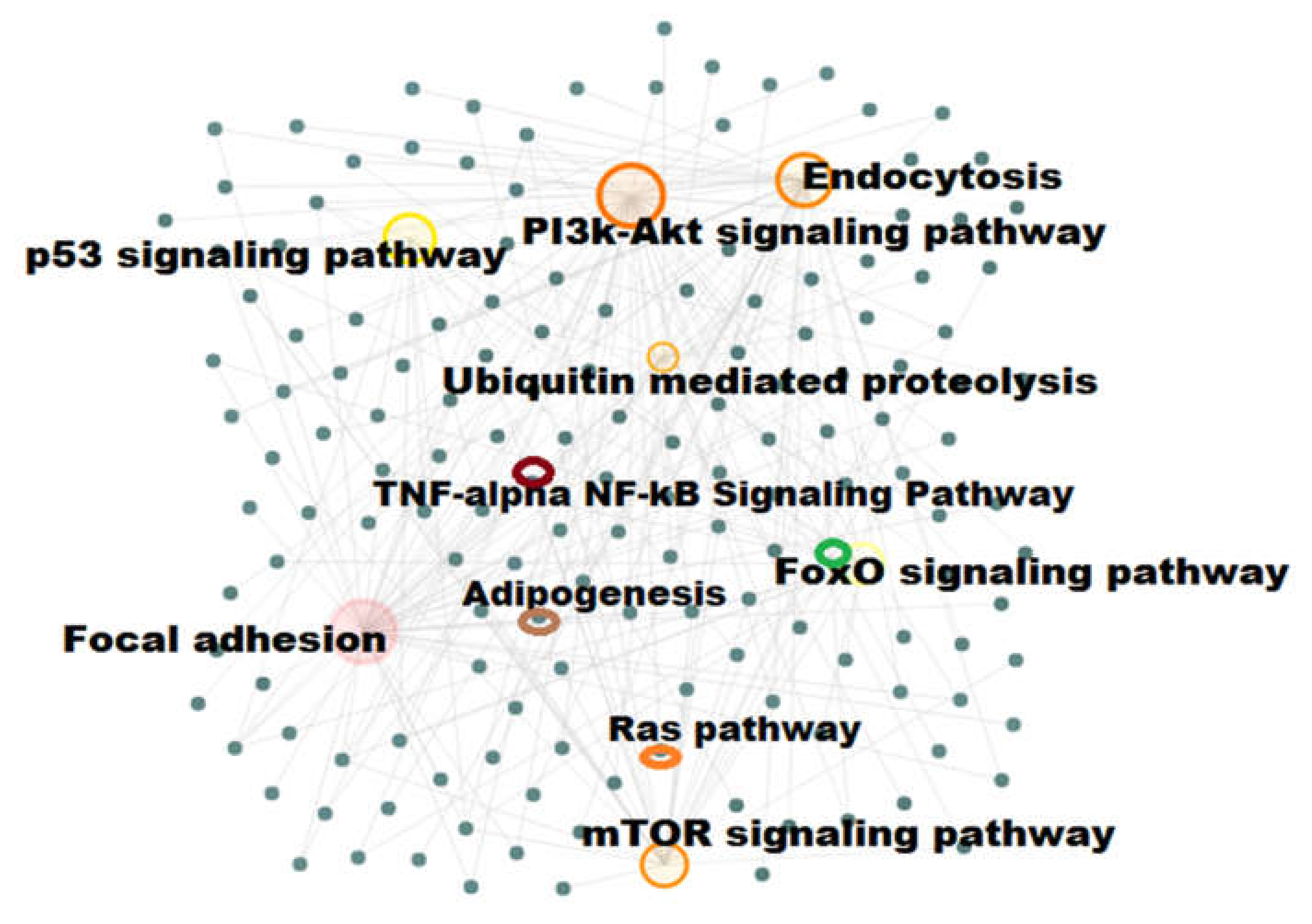
2.4. Pathway Enrichment Analysis
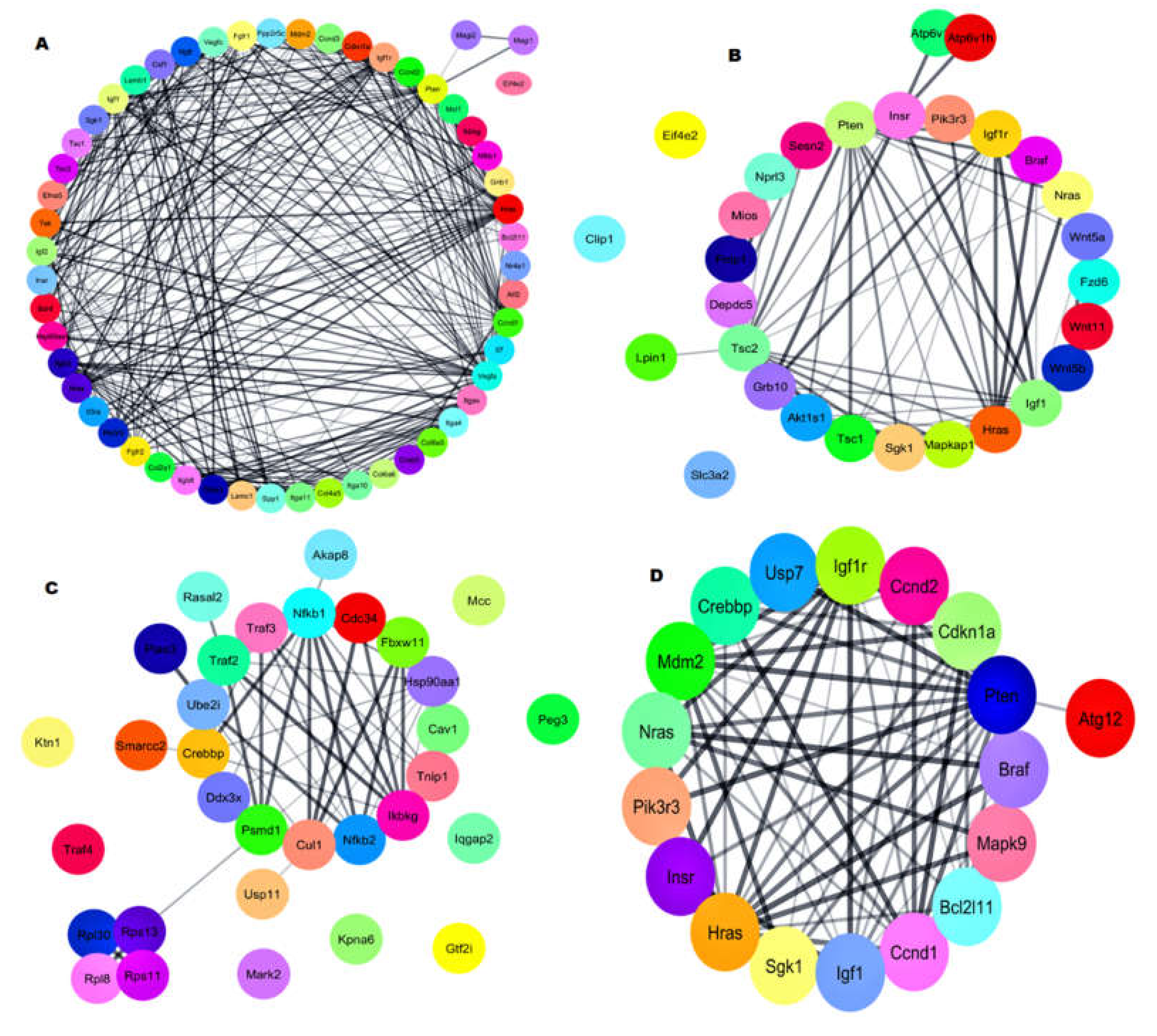
2.5. Gene–Gene Network with Pathway Enrichment Analysis
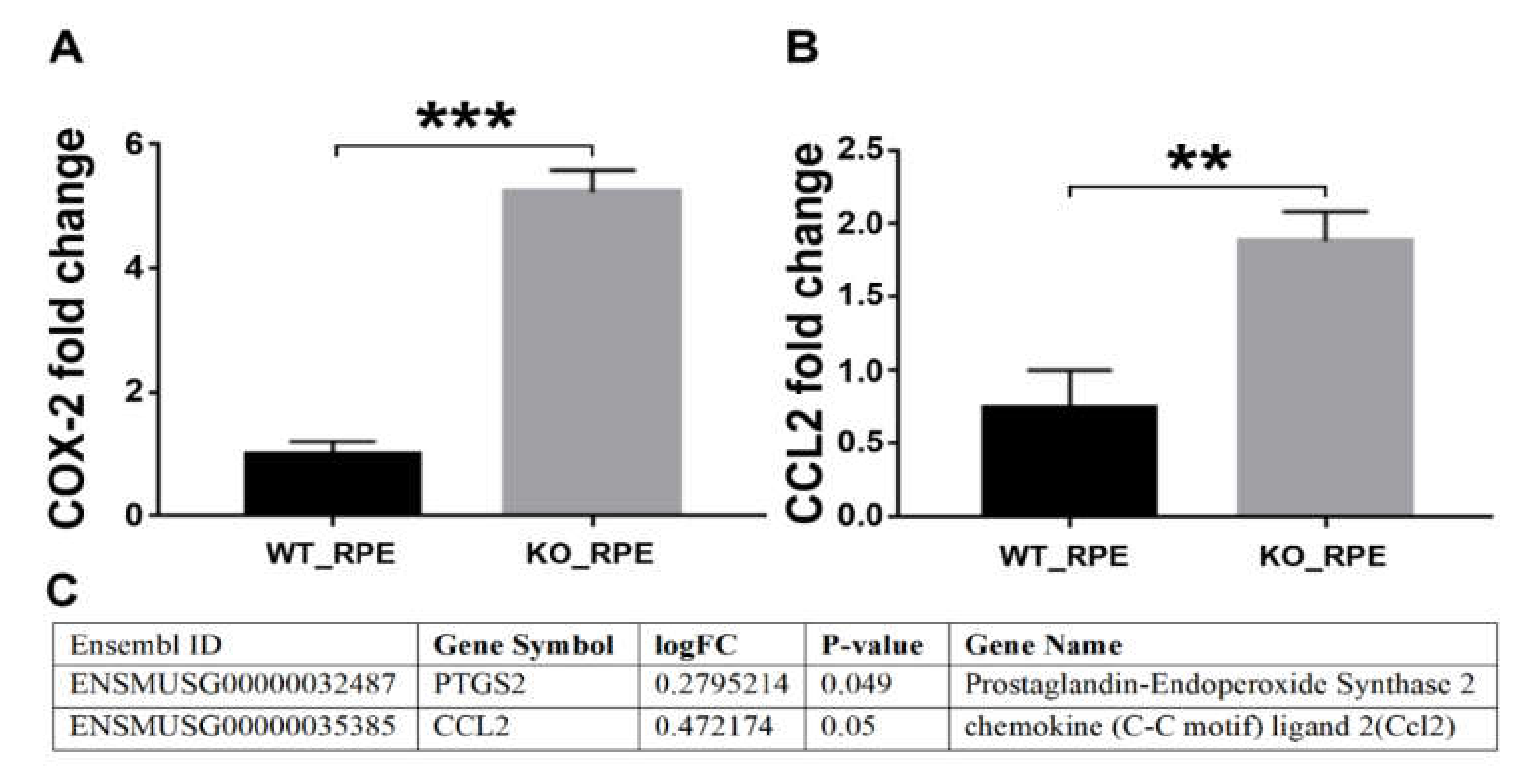
2.6. RT-PCR Validation of the RNA Seq Results
3. Discussion
4. Materials and Methods
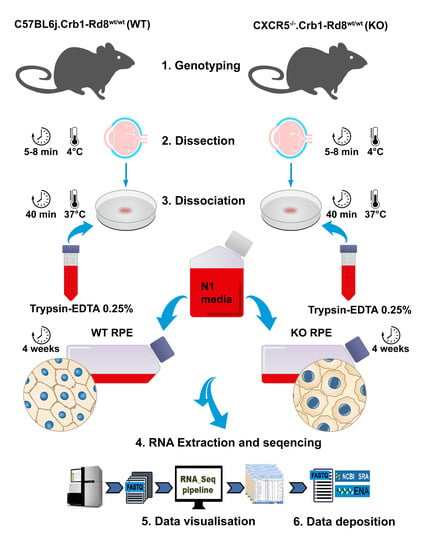
4.1. Animals
4.2. Primary RPE Cell Cultures and Treatment
4.3. Immunofluorescence Staining
4.4. Imaging
4.5. RNA Isolation and Sequencing
4.6. Bioinformatics Analysis
4.7. Gene Expression Analysis
4.8. Functional Annotation
4.9. Quantitative RT-PCR
4.10. Statistical Analysis
5. Statement of Limitations
- The small number of samples in this study limits the depth of the identified DEGs.
- Significant variability of the obtained samples, as indicated by Fig. 1G, H. The cell culture system is sensitive to various factors such as environmental factors during sample isolation, culture, and extraction. The primary cell culture has also inherited variability from the animals that have become the donors of the cells. This aspect further limits the dept of this study.
- Freshly isolated cells may be a preferable source of RNA to study the effects of CXCR5 in the RPE. However, fresh isolates of mouse RPE are technically challenging to produce due to the low number of cells available from each eye. The pooling of the significant number of mice is required to achieve levels of RNA suitable for sequencing. Mechanical isolation produces isolates of the poor purity with contamination from choroidal vasculature and other cell types. Trypsin dissociation has an issue of purity in the immediate dissociate (culture in N1 media produce positive proliferative pressure promoting growth and differentiation specifically of RPE cells but no other cell types, i.e., endothelial cells). Trypsin exposure produces its effects on various genes and pathways (i.e., activation of Protease-activated receptor 2 pathway). Primary RPE cultures could exclude indirect or paracrine effects from other retinal tissues (i.e., choriocapillaris, microglia, and photoreceptors) and rule out the systemic effect of global CXCR5 knockout.
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob Health 2014, 2, e106–e116. [Google Scholar] [CrossRef]
- Jager, R.D.; Mieler, W.F.; Miller, J.W. Age-related macular degeneration. N. Engl. J. Med. 2008, 358, 2606–2617. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.W.; Le Couter, J.; Strauss, E.C.; Ferrara, N. Vascular endothelial growth factor a in intraocular vascular disease. Ophthalmology 2013, 120, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Peng, B.; Lin, B. Fractalkine receptor regulates microglial neurotoxicity in an experimental mouse glaucoma model. Glia 2014, 62, 1943–1954. [Google Scholar] [CrossRef]
- Cao, X.; Li, W.; Liu, Y.; Huang, H.; Ye, C.H. The Anti-Inflammatory Effects of CXCR5 in the Mice Retina following Ischemia-Reperfusion Injury. Biomed Res. Int. 2019, 2019, 3487607. [Google Scholar] [CrossRef]
- Mitkin, N.A.; Hook, C.D.; Schwartz, A.M.; Biswas, S.; Kochetkov, D.V.; Muratova, A.M.; Afanasyeva, M.A.; Kravchenko, J.E.; Bhattacharyya, A.; Kuprash, D.V. p53-dependent expression of CXCR5 chemokine receptor in MCF-7 breast cancer cells. Sci. Rep. 2015, 5, 9330. [Google Scholar] [CrossRef]
- Zhang, Q.; Cao, D.L.; Zhang, Z.J.; Jiang, B.C.; Gao, Y.J. Chemokine CXCL13 mediates orofacial neuropathic pain via CXCR5/ERK pathway in the trigeminal ganglion of mice. J. Neuroinflamm. 2016, 13, 183. [Google Scholar] [CrossRef]
- Huang, H.; Liu, Y.; Wang, L.; Li, W. Age-related macular degeneration phenotypes are associated with increased tumor necrosis-alpha and subretinal immune cells in aged Cxcr5 knockout mice. PLoS ONE 2017, 12, e0173716. [Google Scholar] [CrossRef]
- Lennikov, A.; Saddala, M.S.; Mukwaya, A.; Tang, S.; Huang, H. Autoimmune-Mediated Retinopathy in CXCR5-Deficient Mice as the Result of Age-Related Macular Degeneration Associated Proteins Accumulation. Front. Immunol. 2019, 10, 1903. [Google Scholar] [CrossRef]
- Saddala, M.S.; Lennikov, A.; Mukwaya, A.; Fan, L.; Hu, Z.; Huang, H. Transcriptome-wide analysis of differentially expressed chemokine receptors, SNPs, and SSRs in the age-related macular degeneration. Human Genomics 2019, 13, 15. [Google Scholar] [CrossRef]
- Ambati, J.; Fowler, B.J. Mechanisms of age-related macular degeneration. Neuron 2012, 75, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Lopez, E.; Lopez-Colome, A.M. Thrombin activation of PI3K/PDK1/Akt signaling promotes cyclin D1 upregulation and RPE cell proliferation. Biochim. Biophys. Acta 2011, 1813, 1758–1766. [Google Scholar] [CrossRef] [PubMed]
- Ambati, J.; Anand, A.; Fernandez, S.; Sakurai, E.; Lynn, B.C.; Kuziel, W.A.; Rollins, B.J.; Ambati, B.K. An animal model of age-related macular degeneration in senescent Ccl-2- or Ccr-2-deficient mice. Nat. Med. 2003, 9, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Kamioka, Y.; Yokoi, N.; Kobayashi, T.; Hino, O.; Onodera, M.; Mochizuki, N.; Nakae, J. Interaction of FoxO1 and TSC2 induces insulin resistance through activation of the mammalian target of rapamycin/p70 S6K pathway. J. Biol. Chem. 2006, 281, 40242–40251. [Google Scholar] [CrossRef]
- Zheng, W.H.; Quirion, R. Glutamate acting on N-methyl-D-aspartate receptors attenuates insulin-like growth factor-1 receptor tyrosine phosphorylation and its survival signaling properties in rat hippocampal neurons. J. Biol. Chem. 2009, 284, 855–861. [Google Scholar] [CrossRef]
- Tanabe, K.; Torii, T.; Natsume, W.; Braesch-Andersen, S.; Watanabe, T.; Satake, M. A novel GTPase-activating protein for ARF6 directly interacts with clathrin and regulates clathrin-dependent endocytosis. Mol. Biol. Cell 2005, 16, 1617–1628. [Google Scholar] [CrossRef]
- Naslavsky, N.; Weigert, R.; Donaldson, J.G. Convergence of non-clathrin- and clathrin-derived endosomes involves Arf6 inactivation and changes in phosphoinositides. Mol. Biol. Cell 2003, 14, 417–431. [Google Scholar] [CrossRef]
- Palacios, F.; Schweitzer, J.K.; Boshans, R.L.; D’Souza-Schorey, C. ARF6-GTP recruits Nm23-H1 to facilitate dynamin-mediated endocytosis during adherens junctions disassembly. Nat. Cell Biol. 2002, 4, 929–936. [Google Scholar] [CrossRef]
- Wavre-Shapton, S.T.; Tolmachova, T.; Lopes da Silva, M.; Futter, C.E.; Seabra, M.C. Conditional ablation of the choroideremia gene causes age-related changes in mouse retinal pigment epithelium. PLoS ONE 2013, 8, e57769. [Google Scholar] [CrossRef]
- Jane-wit, D.; Surovtseva, Y.V.; Qin, L.; Li, G.; Liu, R.; Clark, P.; Manes, T.D.; Wang, C.; Kashgarian, M.; Kirkiles-Smith, N.C.; et al. Complement membrane attack complexes activate noncanonical NF-kappaB by forming an Akt+ NIK+ signalosome on Rab5+ endosomes. Proc. Natl. Acad. Sci. USA 2015, 112, 9686–9691. [Google Scholar] [CrossRef]
- Yang, p.; McKay, B.S.; Allen, J.B.; Jaffe, G.J. Effect of NF-kappa B inhibition on TNF-alpha-induced apoptosis in human RPE cells. Investig. Ophthalmol. Vis. Sci. 2004, 45, 2438–2446. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; He, S.; Worpel, V.; Ryan, S.J.; Hinton, D.R. Promotion of adhesion and migration of RPE cells to provisional extracellular matrices by TNF-alpha. Investig. Ophthalmol. Vis. Sci. 2000, 41, 4324–4332. [Google Scholar]
- Marchiando, A.M.; Shen, L.; Graham, W.V.; Weber, C.R.; Schwarz, B.T.; Austin, J.R., 2nd; Raleigh, D.R.; Guan, Y.; Watson, A.J.; Montrose, M.H.; et al. Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J. Cell Biol. 2010, 189, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.W.; Yan, X.; Li, Y.; Reddy, S.A.; Tsang, B.K. Resistance of human ovarian cancer cells to tumor necrosis factor alpha is a consequence of nuclear factor kappaB-mediated induction of Fas-associated death domain-like interleukin-1beta-converting enzyme-like inhibitory protein. Endocrinology 2003, 144, 623–630. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shinoura, N.; Yamamoto, N.; Yoshida, Y.; Fujita, T.; Saito, N.; Asai, A.; Kirino, T.; Hamada, H. Adenovirus-mediated gene transduction of IkappaB or IkappaB plus Bax gene drastically enhances tumor necrosis factor (TNF)-induced apoptosis in human gliomas. Jpn J. Cancer Res. 2000, 91, 41–51. [Google Scholar] [CrossRef]
- Takahashi, E.; Nagano, O.; Ishimoto, T.; Yae, T.; Suzuki, Y.; Shinoda, T.; Nakamura, S.; Niwa, S.; Ikeda, S.; Koga, H.; et al. Tumor necrosis factor-alpha regulates transforming growth factor-beta-dependent epithelial-mesenchymal transition by promoting hyaluronan-CD44-moesin interaction. J. Biol. Chem. 2010, 285, 4060–4073. [Google Scholar] [CrossRef]
- Oh, J.E.; Kim, R.H.; Shin, K.H.; Park, N.H.; Kang, M.K. DeltaNp63alpha protein triggers epithelial-mesenchymal transition and confers stem cell properties in normal human keratinocytes. J. Biol. Chem. 2011, 286, 38757–38767. [Google Scholar] [CrossRef]
- Lindsay, J.; McDade, S.S.; Pickard, A.; McCloskey, K.D.; McCance, D.J. Role of DeltaNp63gamma in epithelial to mesenchymal transition. J. Biol. Chem. 2011, 286, 3915–3924. [Google Scholar] [CrossRef]
- Feng, H.; Zhao, X.; Guo, Q.; Feng, Y.; Ma, M.; Guo, W.; Dong, X.; Deng, C.; Li, C.; Song, X.; et al. Autophagy resists EMT process to maintain retinal pigment epithelium homeostasis. Inter. J. Biol. Sci. 2019, 15, 507–521. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef]
- Lee, J.; Giordano, S.; Zhang, J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem. J. 2012, 441, 523–540. [Google Scholar] [CrossRef] [PubMed]
- Fullgrabe, J.; Ghislat, G.; Cho, D.H.; Rubinsztein, D.C. Transcriptional regulation of mammalian autophagy at a glance. J. Cell Sci. 2016, 129, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Heras-Sandoval, D.; Perez-Rojas, J.M.; Hernandez-Damian, J.; Pedraza-Chaverri, J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal 2014, 26, 2694–2701. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Zhao, H.; Martinez, J.; Doggett, T.A.; Kolesnikov, A.V.; Tang, P.H.; Ablonczy, Z.; Chan, C.C.; Zhou, Z.; Green, D.R.; et al. Noncanonical autophagy promotes the visual cycle. Cell 2013, 154, 365–376. [Google Scholar] [CrossRef]
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W., Jr. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 2014, 10, 1989–2005. [Google Scholar] [CrossRef] [PubMed]
- Golestaneh, N.; Chu, Y.; Xiao, Y.Y.; Stoleru, G.L.; Theos, A.C. Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death Dis. 2017, 8, e2537. [Google Scholar] [CrossRef]
- Valapala, M.; Wilson, C.; Hose, S.; Bhutto, I.A.; Grebe, R.; Dong, A.; Greenbaum, S.; Gu, L.; Sengupta, S.; Cano, M.; et al. Lysosomal-mediated waste clearance in retinal pigment epithelial cells is regulated by CRYBA1/betaA3/A1-crystallin via V-ATPase-MTORC1 signaling. Autophagy 2014, 10, 480–496. [Google Scholar] [CrossRef]
- Fernandez-Godino, R.; Bujakowska, K.M.; Pierce, E.A. Changes in extracellular matrix cause RPE cells to make basal deposits and activate the alternative complement pathway. Human Mol. Genet. 2017, 27, 147–159. [Google Scholar] [CrossRef]
- Bermudez, V.; Tenconi, P.E.; Giusto, N.M.; Mateos, M.V. Lipopolysaccharide-Induced Autophagy Mediates Retinal Pigment Epithelium Cells Survival. Modulation by the Phospholipase D Pathway. Front. Cell Neurosci. 2019, 13, 154. [Google Scholar] [CrossRef]
- Richter, M.; Weiss, M.; Weinberger, I.; Furstenberger, G.; Marian, B. Growth inhibition and induction of apoptosis in colorectal tumor cells by cyclooxygenase inhibitors. Carcinogenesis 2001, 22, 17–25. [Google Scholar] [CrossRef]
- Feng, C.; Wang, X.; Liu, T.; Zhang, M.; Xu, G.; Ni, Y. Expression of CCL2 and its receptor in activation and migration of microglia and monocytes induced by photoreceptor apoptosis. Mol. Vis. 2017, 23, 765–777. [Google Scholar]
- Fronk, A.H.; Vargis, E. Methods for culturing retinal pigment epithelial cells: A review of current protocols and future recommendations. J. Tissue Eng. 2016, 7, 2041731416650838. [Google Scholar] [CrossRef]
- Johnson, L.V.; Forest, D.L.; Banna, C.D.; Radeke, C.M.; Maloney, M.A.; Hu, J.; Spencer, C.N.; Walker, A.M.; Tsie, M.S.; Bok, D.; et al. Cell culture model that mimics drusen formation and triggers complement activation associated with age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 18277–18282. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotech. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Saddala, M.S.; Lennikov, A.; Bouras, A.; Huang, H. RNA-Seq reveals differential expression profiles and functional annotation of genes involved in retinal degeneration in Pde6c mutant Danio rerio. BMC Genom. 2020, 21, 132. [Google Scholar] [CrossRef] [PubMed]
- Saddala, M.S.; Lennikov, A.; Huang, H. Placental growth factor regulates the pentose phosphate pathway and antioxidant defense systems in human retinal endothelial cells. J. Proteom. 2020, 217, 103682. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Kir, J.; Liu, D.; Bryant, D.; Guo, Y.; Stephens, R.; Baseler, M.W.; Lane, H.C.; et al. DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007, 35, W169–W175. [Google Scholar] [CrossRef]
- Saddala, M.S.; Lennikov, A.; Grab, D.J.; Liu, G.S.; Tang, S.; Huang, H. Proteomics reveals ablation of PlGF increases antioxidant and neuroprotective proteins in the diabetic mouse retina. Sci. Rep. 2018, 8, 16728. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Rivals, I.; Personnaz, L.; Taing, L.; Potier, M.C. Enrichment or depletion of a GO category within a class of genes: Which test? Bioinformatics 2007, 23, 401–407. [Google Scholar] [CrossRef]
- Sanchez Miranda, E.; Perez Ramos, J.; Fresan Orozco, C.; Zavala Sanchez, M.A.; Perez Gutierrez, S. Anti-Inflammatory Effects of Hyptis albida Chloroform Extract on Lipopolysaccharide-Stimulated Peritoneal Macrophages. ISRN Pharmacol. 2013, 2013, 713060. [Google Scholar] [CrossRef] [PubMed]
- Schiller, K.R.; Zillhardt, M.R.; Alley, J.; Borjesson, D.L.; Beitz, A.J.; Mauro, L.J. Secretion of MCP-1 and other paracrine factors in a novel tumor-bone coculture model. BMC Cancer 2009, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Tsujita, Y.; Muraski, J.; Shiraishi, I.; Kato, T.; Kajstura, J.; Anversa, P.; Sussman, M.A. Nuclear targeting of Akt antagonizes aspects of cardiomyocyte hypertrophy. Proc. Natl. Acad. Sci. USA 2006, 103, 11946–11951. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Name | logFC 1 | p-value | FDR |
|---|---|---|---|---|
| Atf2 | Activating transcription factor 2 | −0.76676 | 0.000148 | 0.041635 |
| Bcl2l11 | BCL2-like 11 (apoptosis facilitator) | −0.76676 | 0.000148 | 0.041635 |
| Bdnf | Brain derived neurotrophic factor | 0.579128 | 0.002926 | 0.27166 |
| Ccnd1 | Cyclin D1 | 0.619593 | 0.016261 | 0.72475 |
| Ccnd2 | Cyclin D2 | 0.381798 | 0.033778 | 0.986212 |
| Ccnd3 | Cyclin D3 | −0.70571 | 0.014237 | 0.672932 |
| Cdkn1a | Cyclin-dependent kinase inhibitor 1A (P21) | 0.600435 | 0.000799 | 0.129915 |
| Col2a1 | Collagen, type II, alpha 1 | −0.60353 | 0.012355 | 0.627982 |
| Col4a5 | Collagen, type IV, alpha 5 | −0.85519 | 0.001471 | 0.187435 |
| Col6a5 | Collagen, type VI, alpha 5 | −0.58554 | 0.022181 | 0.835681 |
| Col6a6 | Collagen, type VI, alpha 6 | −1.03427 | 0.000611 | 0.108132 |
| Creb5 | cAMP responsive element binding protein 5 | −0.64907 | 0.030466 | 0.947107 |
| Csf1 | Colony-stimulating factor 1 (macrophage) | 0.515929 | 0.024112 | 0.860187 |
| Efna5 | Ephrin A5 | 0.402745 | 0.015585 | 0.709264 |
| Eif4e2 | Eukaryotic translation initiation factor 4E member 2 | 0.339331 | 0.017923 | 0.756945 |
| Fgfr1 | Fibroblast growth factor receptor 1 | −0.9058 | 0.0000018 | 0.001257 |
| Fgfr2 | Fibroblast growth factor receptor 2 | 0.641456 | 0.033897 | 0.986212 |
| Gnb1 | Guanine nucleotide binding protein (G protein), beta 1 | 0.424488 | 0.006647 | 0.453973 |
| Hras | Harvey rat sarcoma virus oncogene | 0.319113 | 0.041055 | 0.999738 |
| Hsp90aa1 | Heat shock protein 90, alpha (cytosolic), class A member 1 | 0.256992 | 0.042641 | 0.999738 |
| Igf1 | Insulin-like growth factor 1 | 0.582664 | 0.011964 | 0.618509 |
| Igf1r | Insulin-like growth factor I receptor | 0.839659 | 0.001664 | 0.199427 |
| Igf2 | Insulin-like growth factor 2 | −0.6181 | 0.035299 | 0.999738 |
| Ikbkg | Inhibitor of kappaB kinase gamma | 0.624192 | 0.00867 | 0.521805 |
| Il3ra | Interleukin 3 receptor, alpha chain | −0.70596 | 0.016436 | 0.727482 |
| Il7 | Interleukin 7 | 0.65812 | 0.030148 | 0.946044 |
| Insr | Insulin receptor | −0.50178 | 0.0276 | 0.914738 |
| Itga10 | Integrin, alpha 10 | 0.722545 | 0.005989 | 0.424382 |
| Itga11 | Integrin alpha 11 | −0.52376 | 0.040259 | 0.999738 |
| Itga4 | Integrin alpha 4 | 0.669187 | 0.021709 | 0.828055 |
| Itgav | Integrin alpha V | 0.17317 | 0.02295 | 0.84715 |
| Itgb3 | Integrin beta 3 | −0.42099 | 0.032828 | 0.976771 |
| Itgb8 | Integrin beta 8 | 0.758588 | 0.001698 | 0.200273 |
| Lamb1 | Laminin B1 | −0.43077 | 0.000041 | 0.015966 |
| Lamc1 | Laminin, gamma 1 | −0.19129 | 0.031532 | 0.958818 |
| Magi1 | Membrane-associated guanylate kinase, WW and PDZ domain containing 1 | 0.534198 | 0.030475 | 0.947107 |
| Magi2 | Membrane-associated guanylate kinase, WW and PDZ domain containing 2 | 0.852149 | 0.004337 | 0.355457 |
| Mcl1 | Myeloid cell leukemia sequence 1 | 0.239522 | 0.000743 | 0.123992 |
| Mdm2 | Transformed mouse 3T3 cell double minute 2 | 0.707868 | 0.002289 | 0.234591 |
| Nfkb1 | Nuclear factor of kappa light polypeptide gene enhancer in B cells 1, p105 | −0.6222 | 0.000512 | 0.096041 |
| Ngfr | Nerve growth factor receptor (TNFR superfamily, member 16) | −1.14937 | 0.00000013 | 0.000158 |
| Nr4a1 | Nuclear receptor subfamily 4, group A, member 1 | 0.533577 | 0.034039 | 0.988161 |
| Nras | Neuroblastoma ras oncogene | -0.98599 | 0.000281 | 0.064468 |
| Pik3r3 | Phosphoinositide-3-kinase regulatory subunit 3 | 0.655702 | 0.005241 | 0.393197 |
| Ppp2r5c | Protein phosphatase 2, regulatory subunit B’, gamma | −0.4741 | 0.013059 | 0.642713 |
| Pten | Phosphatase and tensin homolog | 0.207488 | 0.029398 | 0.933135 |
| Sgk1 | Serum/glucocorticoid regulated kinase 1 | 0.331703 | 3.24 × 10−5 | 0.012944 |
| Spp1 | Secreted phosphoprotein 1 | 0.640272 | 0.003407 | 0.3029 |
| Tek | TEK receptor tyrosine kinase | −0.59019 | 0.030625 | 0.949013 |
| Thbs1 | Thrombospondin 1 | 0.62293 | 0.000441 | 0.086938 |
| Tsc1 | Tuberous sclerosis 1 | 0.727053 | 0.003532 | 0.307612 |
| Tsc2 | Tuberous sclerosis 2 | −0.65896 | 0.029703 | 0.937812 |
| Vegfa | Vascular endothelial growth factor A | −0.50133 | 8.23 × 10−5 | 0.027091 |
| Vegfc | Vascular endothelial growth factor C | 0.251123 | 0.000599 | 0.106649 |
| Gene Symbol | Gene Name | logFC 1 | p-value | FDR |
|---|---|---|---|---|
| Akap8 | A kinase (PRKA) anchor protein 8 | 1.175046 | 0.000030 | 0.012441 |
| Cav1 | Caveolin 1, caveolae protein | 0.79122 | 0.009721 | 0.554537 |
| Cdc34 | Cell division cycle 34 | 1.35894 | 0.00000051 | 0.000446 |
| Crebbp | CREB binding protein | 0.289525 | 0.015087 | 0.698369 |
| Cul1 | Cullin 1 | 0.815439 | 0.007716 | 0.49339 |
| Ddx3x | DEAD/H (Asp-Glu-Ala-Asp/His) box Polypeptide 3, X-linked | 0.54556 | 0.005442 | 0.401934 |
| Fbxw11 | F-box and WD-40 domain protein 11 | 0.824636 | 0.00000084 | 0.000016 |
| Gtf2i | General transcription factor II I | 0.752596 | 0.002818 | 0.265882 |
| Hsp90aa1 | Heat shock protein 90, alpha (cytosolic), class A member 1 | 0.256992 | 0.042641 | 0.999738 |
| Ikbkg | Inhibitor of kappaB kinase gamma | 0.624192 | 0.00867 | 0.521805 |
| Iqgap2 | IQ motif containing GTPase activating protein 2 | −0.4761 | 0.007342 | 0.478612 |
| Kpna6 | Karyopherin (importin) alpha 6 | 0.160461 | 0.04708 | 0.999738 |
| Ktn1 | kinectin 1 | 0.793005 | 0.001688 | 0.199531 |
| Mark2 | MAP/microtubule affinity regulating kinase 2 | −0.82436 | 0.001453 | 0.186526 |
| Mcc | Mutated in colorectal cancers | -0.45668 | 0.003974 | 0.334461 |
| Nfkb1 | Nuclear factor of kappa light polypeptide gene enhancer in B cells 1, p105 | −0.6222 | 0.000512 | 0.096041 |
| Nfkb2 | Nuclear factor of kappa light polypeptide gene enhancer in B cells 2, p49/p100 | 0.305921 | 0.015427 | 0.708003 |
| Peg3 | Paternally expressed 3 | −0.76858 | 0.001785 | 0.206978 |
| Pias3 | Protein inhibitor of activated STAT 3 | −0.63029 | 0.035361 | 0.999738 |
| Psmd1 | Proteasome (prosome, macropain) 26S subunit, non-ATPase, 1 | 0.213848 | 0.002695 | 0.258467 |
| Rasal2 | RAS protein activator like 2 | 0.89088 | 0.000071 | 0.024327 |
| Rpl30 | Ribosomal protein L30 | −0.34108 | 0.000105 | 0.032262 |
| Rpl8 | Ribosomal protein L8 | −0.25082 | 0.027523 | 0.912855 |
| Rps11 | Ribosomal protein S11 | −0.2783 | 0.023677 | 0.856335 |
| Rps13 | Ribosomal protein S13 | −0.31715 | 0.000249 | 0.059475 |
| Smarcc2 | SWI/SNF-related, matrix associated, actin-dependent regulator of chromatin, subfamily c, member 2 | −0.39488 | 0.008876 | 0.52407 |
| Tnip1 | TNFAIP3 interacting protein 1 | −0.71759 | 0.018253 | 0.762899 |
| Traf2 | TNF receptor-associated factor 2 | −0.31858 | 0.027154 | 0.906937 |
| Traf3 | TNF receptor-associated factor 3 | 0.302541 | 0.025658 | 0.886335 |
| Traf4 | TNF receptor associated factor 4 | 0.663643 | 0.02293 | 0.84715 |
| Ube2i | Ubiquitin-conjugating enzyme E2I | 0.161077 | 0.021286 | 0.815485 |
| Usp11 | Ubiquitin specific peptidase 11 | −0.82071 | 0.000582 | 0.105223 |
| Dataset | WT_RPE-1 | RPE_WT-1 | RPE_WT-1 | KO_RPE-1 | KO_RPE-2 | KO_RPE-3 |
|---|---|---|---|---|---|---|
| ENSMUSG00000047880 (CXCR5) Number of transcripts per 106 reads | 109 | 341 | 316 | 0 | 0 | 0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saddala, M.S.; Lennikov, A.; Mukwaya, A.; Huang, H. Transcriptome-Wide Analysis of CXCR5 Deficient Retinal Pigment Epithelial (RPE) Cells Reveals Molecular Signatures of RPE Homeostasis. Biomedicines 2020, 8, 147. https://doi.org/10.3390/biomedicines8060147
Saddala MS, Lennikov A, Mukwaya A, Huang H. Transcriptome-Wide Analysis of CXCR5 Deficient Retinal Pigment Epithelial (RPE) Cells Reveals Molecular Signatures of RPE Homeostasis. Biomedicines. 2020; 8(6):147. https://doi.org/10.3390/biomedicines8060147
Chicago/Turabian StyleSaddala, Madhu Sudhana, Anton Lennikov, Anthony Mukwaya, and Hu Huang. 2020. "Transcriptome-Wide Analysis of CXCR5 Deficient Retinal Pigment Epithelial (RPE) Cells Reveals Molecular Signatures of RPE Homeostasis" Biomedicines 8, no. 6: 147. https://doi.org/10.3390/biomedicines8060147
APA StyleSaddala, M. S., Lennikov, A., Mukwaya, A., & Huang, H. (2020). Transcriptome-Wide Analysis of CXCR5 Deficient Retinal Pigment Epithelial (RPE) Cells Reveals Molecular Signatures of RPE Homeostasis. Biomedicines, 8(6), 147. https://doi.org/10.3390/biomedicines8060147