Novel MRI Techniques Identifying Vascular Leak and Paravascular Flow Reduction in Early Alzheimer Disease

Abstract
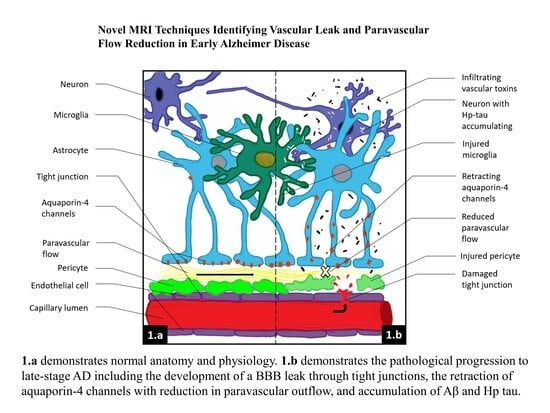
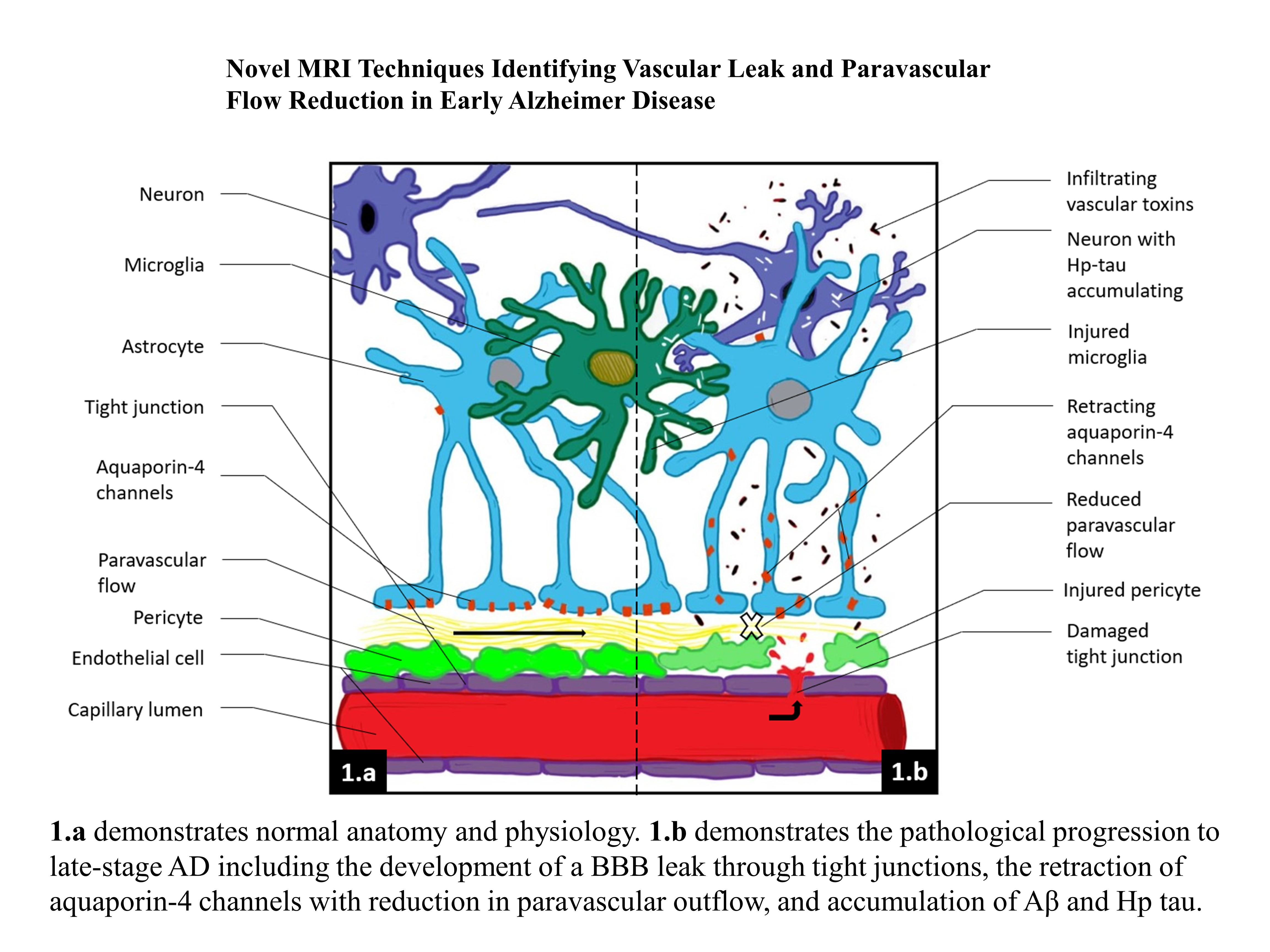
:
1. Introduction
2. Regulation of Fluid and Metabolite Inflow and Outflow
3. Blood–Brain Barrier
4. Glymphatic System
5. Microglia
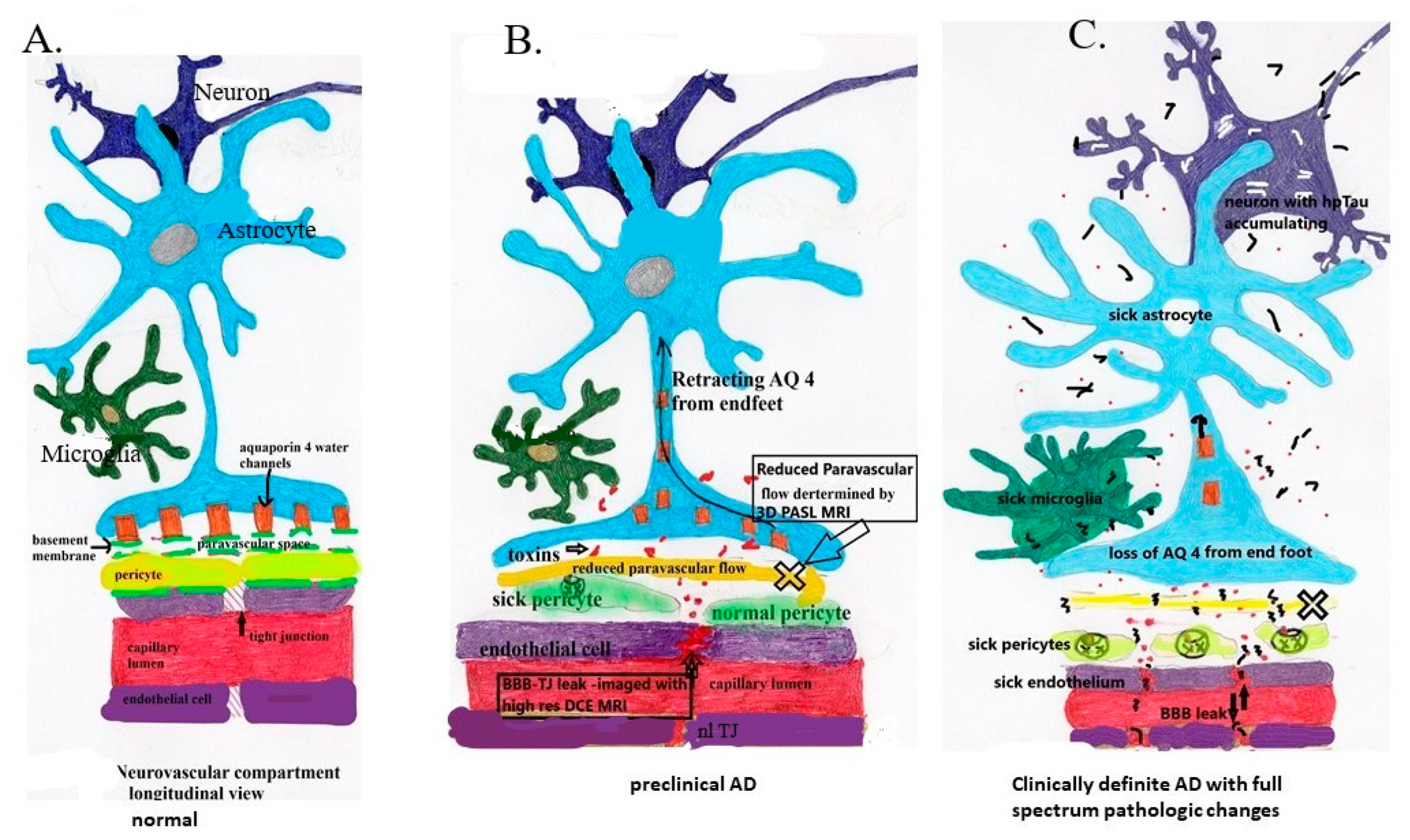
6. Loss of BBB Integrity in Early AD
7. The Evidence Demonstrating BBB Dysfunction in Early AD
8. Investigational Imaging Modalities for Preclinical AD Identification
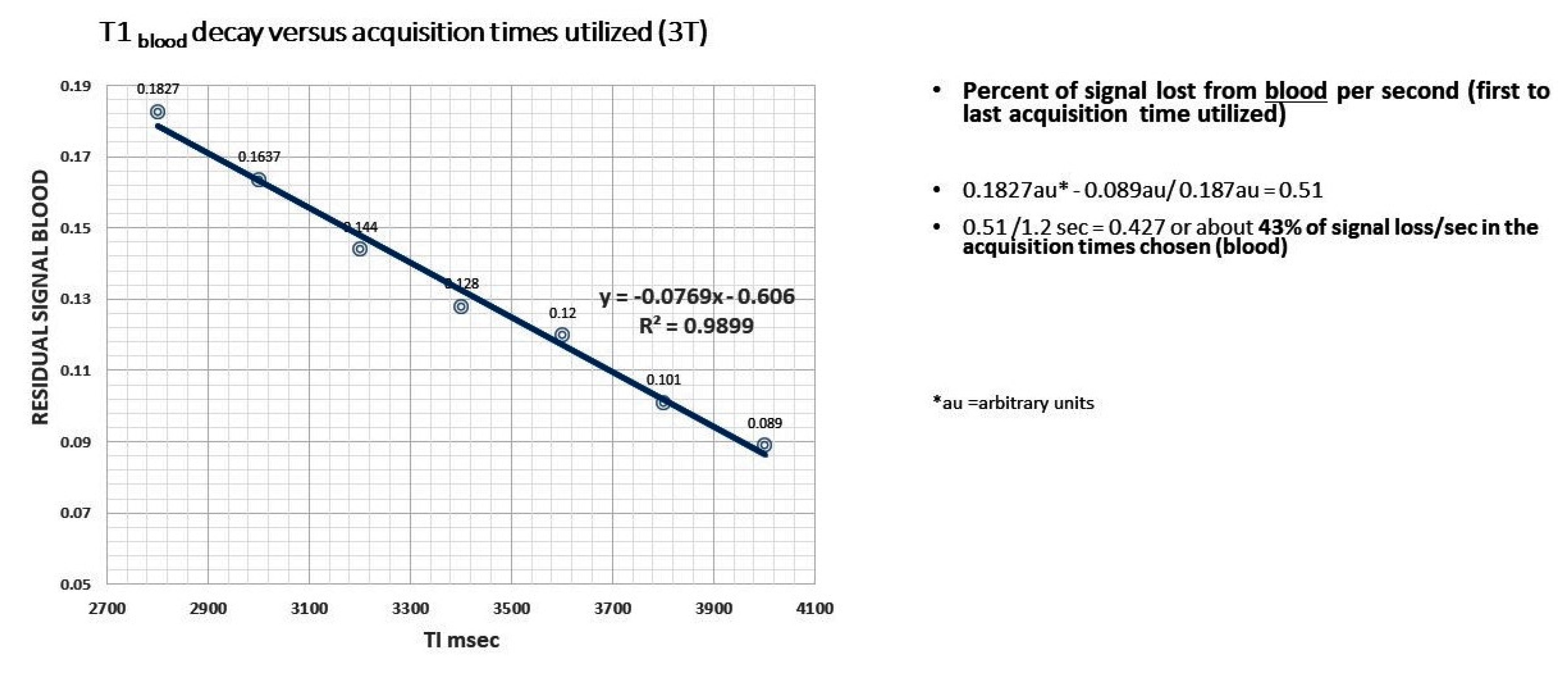
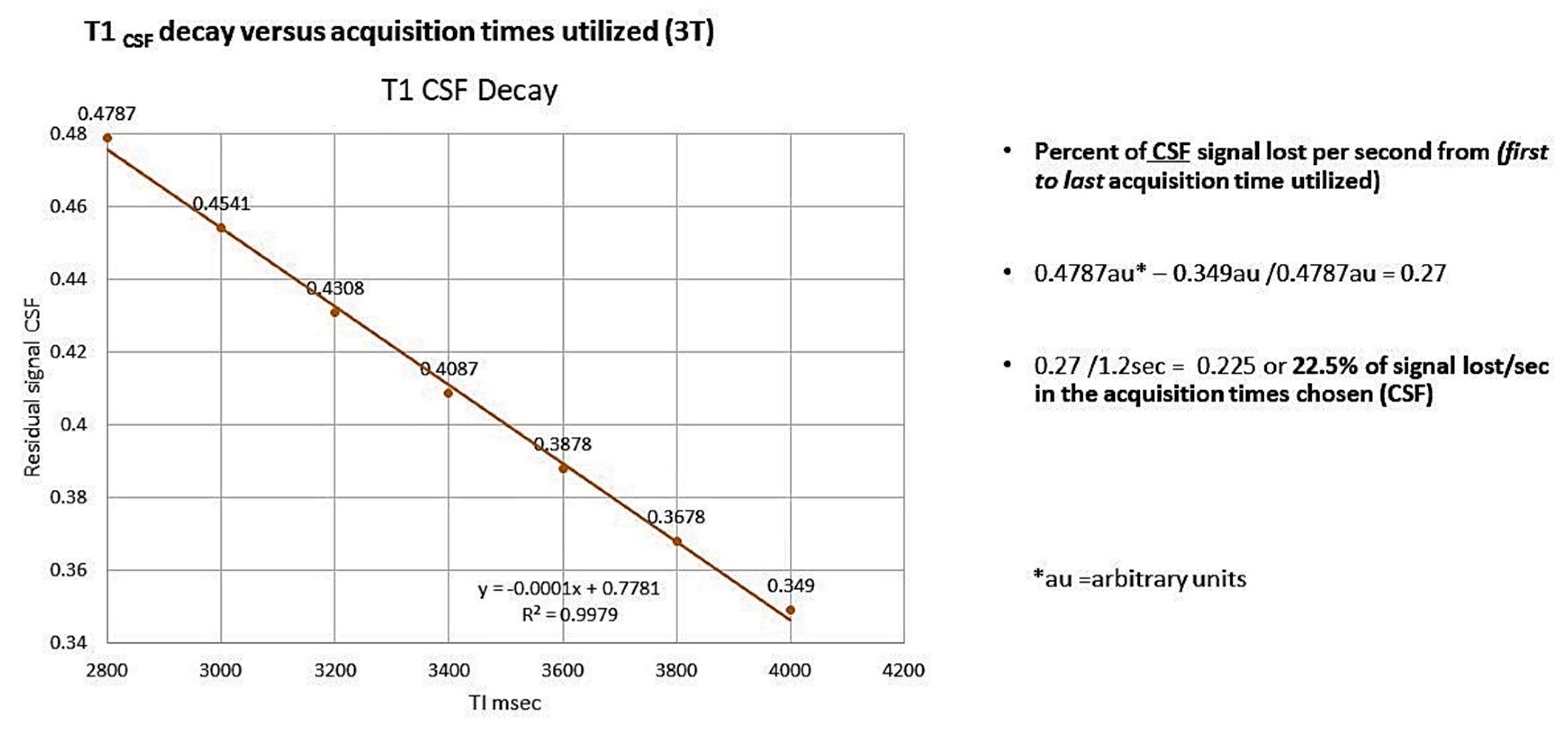
9. High-Resolution Dynamic Contrast Enhanced MRI
10. Arterial Spin Labeling (ASL) Technique
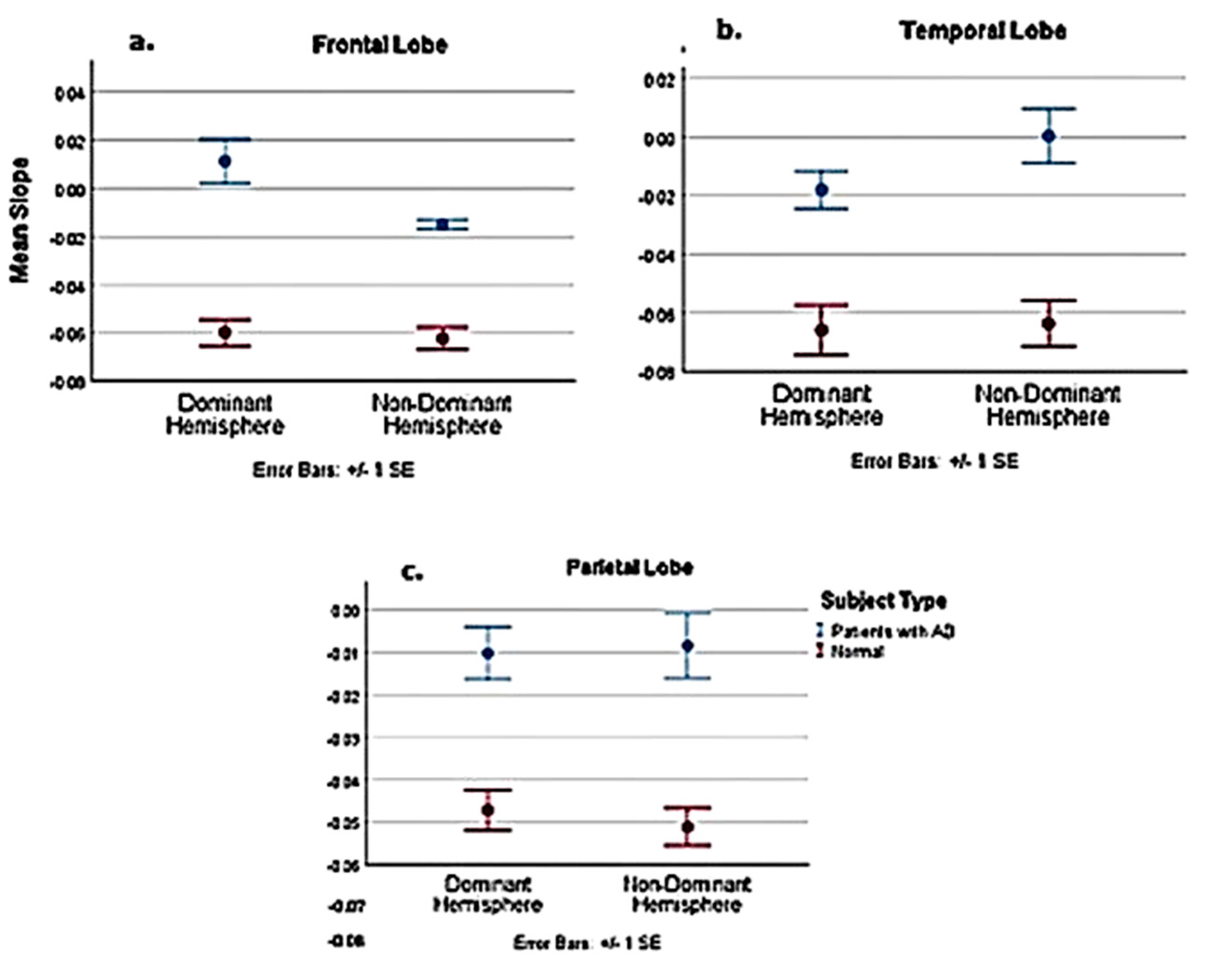
11. ASL Perfusion
12. 3—D PASL MRI Glymphatic Clearance
13. Combined Methodology for Identification of Preclinical AD
14. Current Treatment Trials and the Future
15. Conclusions
Funding
Acknowledgments
Conflicts of Interest
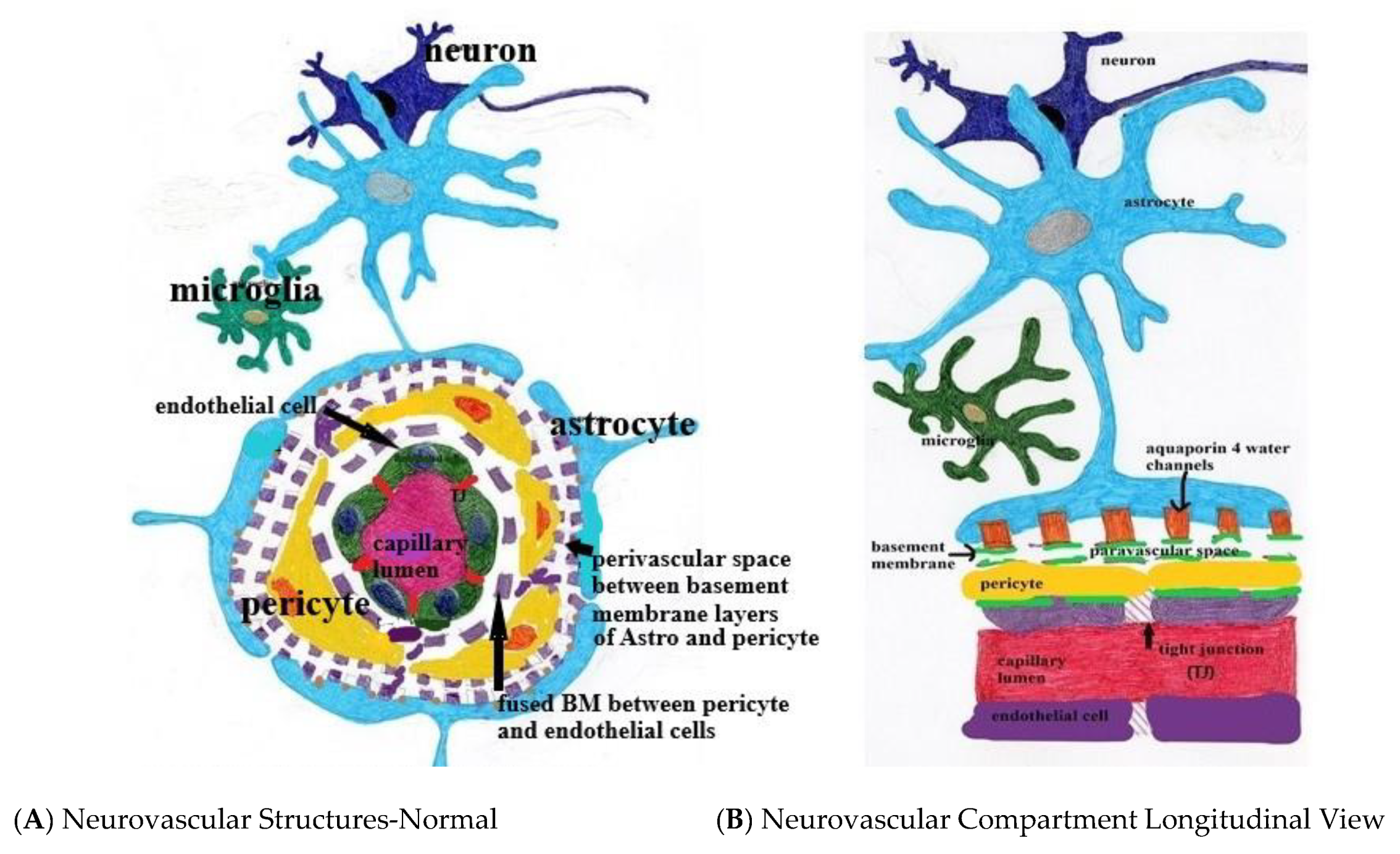
Appendix A

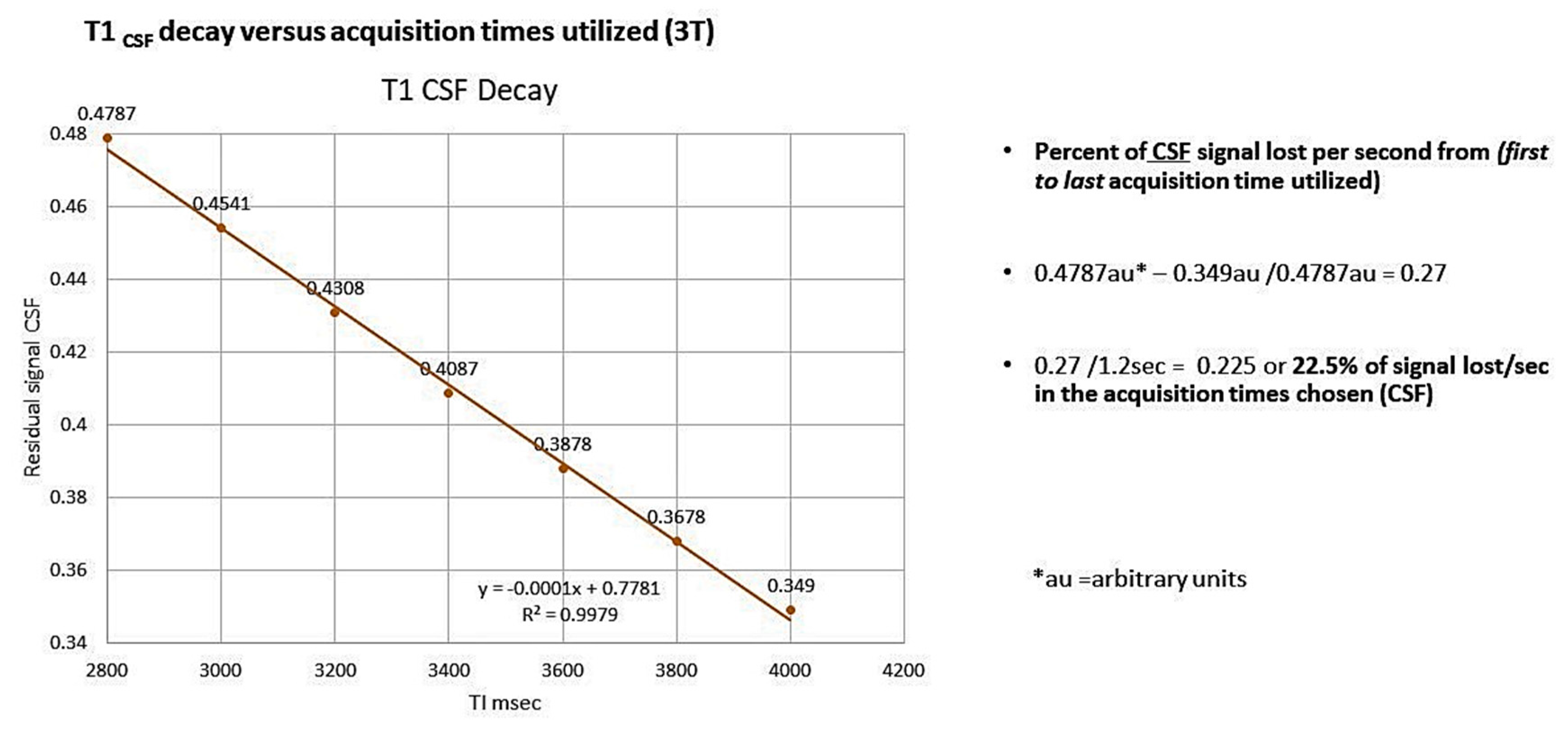
Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
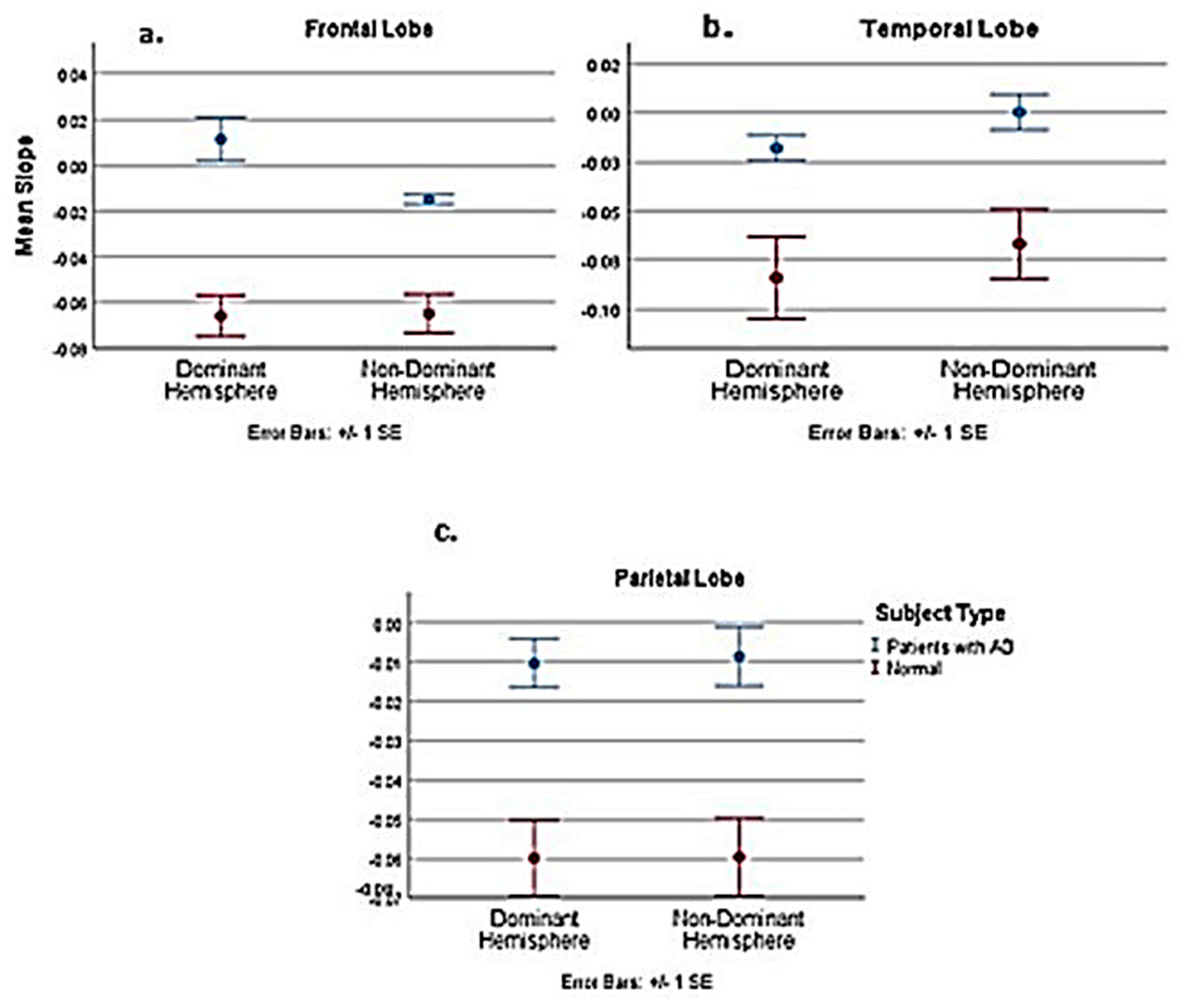
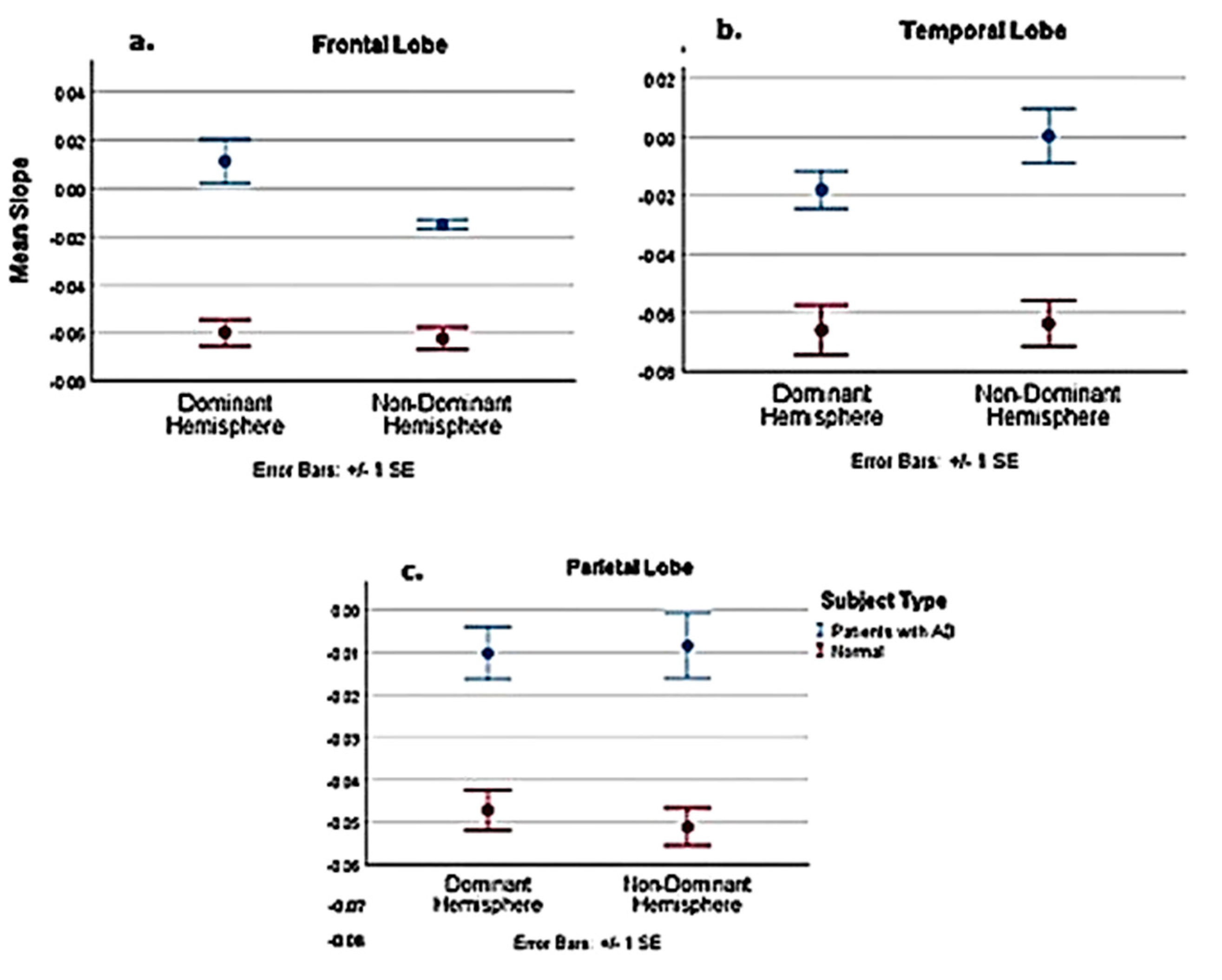
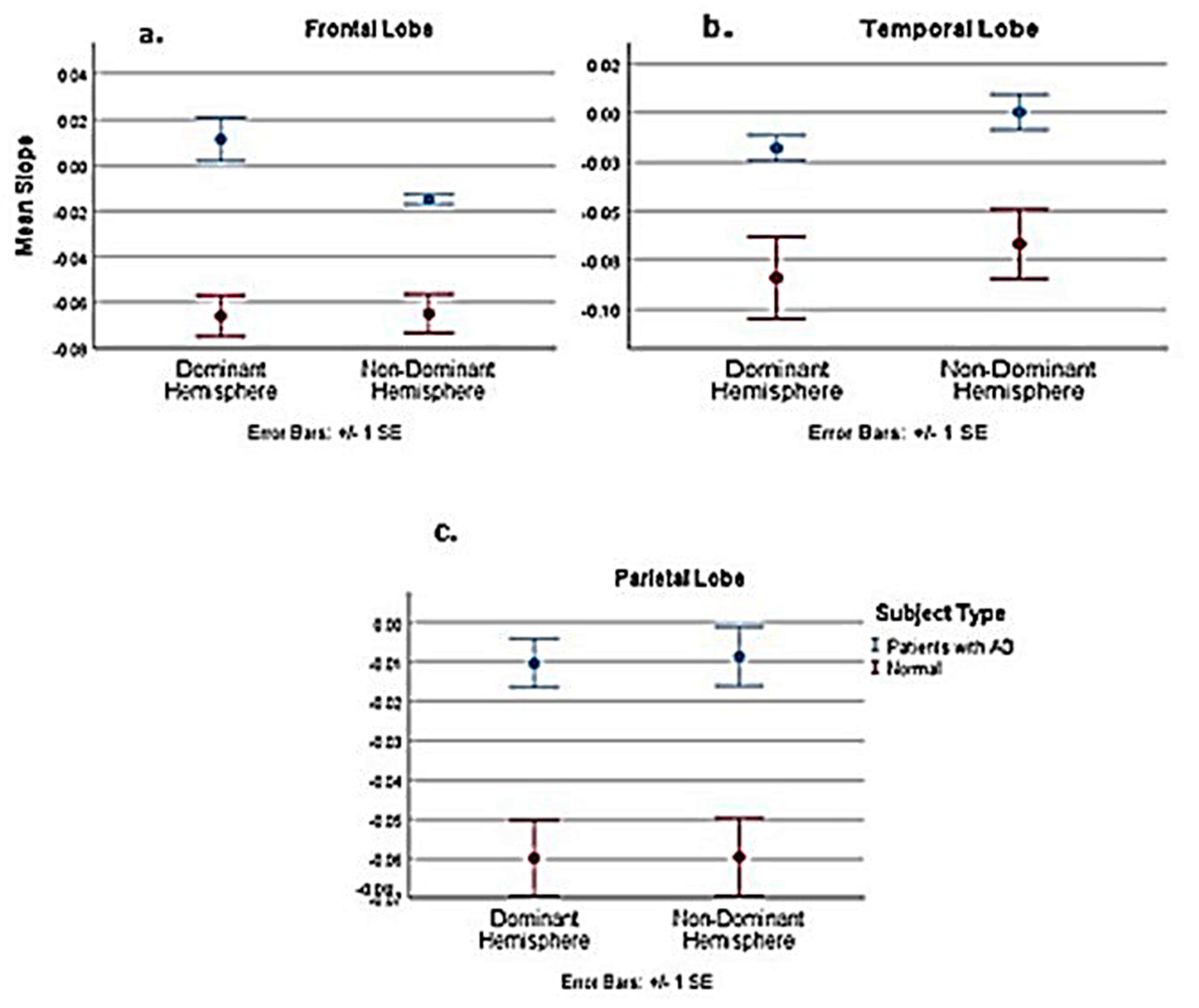
| Comparison of Pooled Normal with AD Patients | Dominant | Non–Dominant | ||||
|---|---|---|---|---|---|---|
| Frontal | Parietal | Temporal | Frontal | Parietal | Temporal | |
| t | −5.101 | −3.056 | −2.231 | −9.426 | −3.776 | −3.208 |
| Df | 19 | 19 | 19 | 18.116 | 19 | 19 |
| P-value | <0.001 | 0.007 | 0.038 | <0.001 | 0.001 | 0.005 |
| Est. Difference between AD and normal slope | −0.0711 | −0.0369 | −0.0479 | −0.0474 | −0.0426 | −0.06 |
| Lower 95% confidence interval | −0.1003 | −0.0621 | −0.0929 | −0.058 | −0.0662 | −0.1058 |
| Upper 95% confidence interval | −0.0419 | −0.0116 | −0.003 | −0.0368 | −0.019 | −0.023 |
| Comparison of normal 51–70 age group with AD patients | ||||||
| t | −5.279 | −3.383 | −2.154 | −5.78 | −3.358 | −2.534 |
| Df | 7 | 7 | 7 | 5.589 | 7 | 7 |
| P-value | 0.001 | 0.012 | 0.068 | 0.001 | 0.012 | 0.039 |
| Est. Difference between AD and normal slope | −0.0771 | −0.0496 | −0.0661 | −0.0499 | −0.0511 | −0.0671 |
| Lower 95% confidence interval | −0.1116 | −0.0842 | −0.1386 | −0.0714 | −0.0871 | −0.1297 |
| Upper 95% confidence interval | −0.0426 | −0.0871 | 0.0064 | −0.0284 | −0.0151 | −0.0045 |
References
- Girouard, H.; Iadecola, C. Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J. Appl. Physiol. 2006, 100, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The Blood-Brain Barrier in Health and Chronic Neurodegenerative Disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardridge, W.; Pardridge, W.M. Targeted delivery of protein and gene medicines through the blood-brain barrier. Clin. Pharmacol. Ther. 2014, 97, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J. Inflammatory mediators and modulation of blood-brain barrier permeability. Cell. Mol. Neurobiol. 2000, 20, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Feldman, H.H.; Frisoni, G.B.; Hampel, H.; Jagust, W.J.; Johnson, K.A.; Knopman, D.S.; et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016, 87, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Khoury, R.; Ghossoub, E. Diagnostic biomarkers of Alzheimer’s disease: A state-of-the-art review. Biomarkers Neuropsychiatry 2019, 1, 100005. [Google Scholar] [CrossRef]
- Molinuevo, J.L.; Blennow, K.; Dubois, B.; Engelborghs, S.; Lewczuk, P.; Perret-Liaudet, A.; Teunissen, C.E.; Parnetti, L. The clinical use of cerebrospinal fluid biomarker testing for Alzheimer’s disease diagnosis: A consensus paper from the Alzheimer’s Biomarkers Standardization Initiative. Alzheimer’s Dement. 2014, 10, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Begley, D.J.; Brightman, M.W. Structural and functional aspects of the blood-brain barrier. Prog. Drug Res. 2003, 61, 39–78. [Google Scholar] [CrossRef]
- Hawkins, B.T.; Davis, T.P. The Blood-Brain Barrier/Neurovascular Unit in Health and Disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef]
- Nagasawa, K.; Chiba, H.; Fujita, H.; Kojima, T.; Saito, T.; Endo, T.; Sawada, N. Possible involvement of gap junctions in the barrier function of tight junctions of brain and lung endothelial cells. J. Cell. Physiol. 2006, 208, 123–132. [Google Scholar] [CrossRef]
- Kovacs, G.G. Molecular Pathological Classification of Neurodegenerative Diseases: Turning towards Precision Medicine. Int. J. Mol. Sci. 2016, 17, 189. [Google Scholar] [CrossRef] [PubMed]
- Jucker, M.; Walker, L. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nisbet, R.; Polanco, J.C.; Ittner, L.M.; Götz, J. Tau aggregation and its interplay with amyloid-β. Acta Neuropathol. 2014, 129, 207–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, K.; Liu, F.; Gong, C.-X. Tau and neurodegenerative disease: The story so far. Nat. Rev. Neurol. 2015, 12, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Knopman, D.S.; Parisi, J.E.; Salviati, A.; Floriach-Robert, M.; Boeve, B.F.; Ivnik, R.J.; Smith, G.; Dickson, D.W.; Johnson, K.A.; Petersen, L.E.; et al. Neuropathology of cognitively normal elderly. J. Neuropathol. Exp. Neurol. 2003, 62, 1087–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, J.L.; Davis, P.; Morris, J.C.; White, D. The distribution of tangles, plaques and related immunohistochemical markers in healthy aging and Alzheimer’s disease. Neurobiol. Aging 1991, 12, 295–312. [Google Scholar] [CrossRef]
- Bennett, D.A.; Schneider, J.A.; Arvanitakis, Z.; Kelly, J.F.; Aggarwal, N.T.; Shah, R.C.; Wilson, R.S. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 2006, 66, 1837–1844. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Qian, J.; Monsell, S.E.; Blacker, D.; Gómez-Isla, T.; Betensky, R.A.; Growdon, J.H.; Johnson, K.A.; Frosch, M.P.; Sperling, R.A.; et al. Mild to moderate Alzheimer dementia with insufficient neuropathological changes. Ann. Neurol. 2014, 75, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Head, E.; Schmitt, F.A.; Davis, P.R.; Neltner, J.H.; Jicha, G.A.; Abner, E.L.; Smith, C.D.; Van Eldik, L.J.; Kryscio, R.J.; et al. Alzheimer’s disease is not “brain aging”: Neuropathological, genetic, and epidemiological human studies. Acta Neuropathol. 2011, 121, 571–587. [Google Scholar] [CrossRef] [Green Version]
- Montagne, A.; Zhao, Z.; Zlokovic, B.V. Alzheimer’s disease: A matter of blood–brain barrier dysfunction? J. Exp. Med. 2017, 214, 3151–3169. [Google Scholar] [CrossRef] [PubMed]
- Lok, J.; Gupta, P.; Guo, S.; Kim, W.J.; Whalen, M.J.; Van Leyen, K.; Lo, E.H. Cell–cell Signaling in the Neurovascular Unit. Neurochem. Res. 2007, 32, 2032–2045. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A Paravascular Pathway Facilitates CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tietz, S.; Engelhardt, B. Brain barriers: Crosstalk between complex tight junctions and adherens junctions. J. Cell Boil. 2015, 209, 493–506. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, M.S.; Routhe, L.J.; Moos, T. The vascular basement membrane in the healthy and pathological brain. Br. J. Pharmacol. 2017, 37, 3300–3317. [Google Scholar] [CrossRef]
- Yang, Y.; Estrada, E.Y.; Thompson, J.F.; Liu, W.; Rosenberg, G. Matrix Metalloproteinase-Mediated Disruption of Tight Junction Proteins in Cerebral Vessels is Reversed by Synthetic Matrix Metalloproteinase Inhibitor in Focal Ischemia in Rat. Br. J. Pharmacol. 2006, 27, 697–709. [Google Scholar] [CrossRef]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood–brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, M.; Ayyadurai, S.; Zlokovic, B.V. Pericytes of the neurovascular unit: Key functions and signaling pathways. Nat. Neurosci. 2016, 19, 771–783. [Google Scholar] [CrossRef]
- Bell, R.D.; Winkler, E.A.; Sagare, A.P.; Singh, I.; LaRue, B.; Deane, R.; Zlokovic, B.V. Pericytes Control Key Neurovascular Functions and Neuronal Phenotype in the Adult Brain and during Brain Aging. Neuron 2010, 68, 409–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacVicar, B.A.; Newman, E.A. Astrocyte Regulation of Blood Flow in the Brain. Cold Spring Harb. Perspect. Boil. 2015, 7, a020388. [Google Scholar] [CrossRef]
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sállström, J.; et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 2012, 485, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Peppiatt-Wildman, C.; Howarth, C.; Mobbs, P.; Attwell, D. Bidirectional control of CNS capillary diameter by pericytes. Nature 2006, 443, 700–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazzoni, G.; Dejana, E. Endothelial Cell-to-Cell Junctions: Molecular Organization and Role in Vascular Homeostasis. Physiol. Rev. 2004, 84, 869–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandopadhyay, R.; Orte, C.; Lawrenson, J.G.; Reid, A.; De Silva, S.; Allt, G. Contractile proteins in pericytes at the blood-brain and blood-retinal barriers. J. Neurocytol. 2001, 30, 35–44. [Google Scholar] [CrossRef]
- Donahue, J.E.; Flaherty, S.L.; Johanson, C.E.; Duncan, J.A.; Silverberg, G.D.; Miller, M.C.; Tavares, R.; Yang, W.; Wu, Q.; Sabo, E.; et al. RAGE, LRP-1, and amyloid-beta protein in Alzheimer’s disease. Acta Neuropathol. 2006, 112, 405–415. [Google Scholar] [CrossRef]
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Zlokovic, B.V. Role of A β Transport and Clearance in the Pathogenesis and Treatment of Alzheimer’s Disease. In Membrane Biogenesis; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2007; pp. 179–198. [Google Scholar]
- Mader, S.; Brimberg, L. Aquaporin-4 Water Channel in the Brain and Its Implication for Health and Disease. Cells 2019, 8, 90. [Google Scholar] [CrossRef] [Green Version]
- Bakker, E.N.T.P.; Bacskai, B.J.; Arbel-Ornath, M.; Aldea, R.; Bedussi, B.; Morris, A.W.J.; Weller, R.O.; Carare, R.O. Lymphatic Clearance of the Brain: Perivascular, Paravascular and Significance for Neurodegenerative Diseases. Cell. Mol. Neurobiol. 2016, 36, 181–194. [Google Scholar] [CrossRef] [Green Version]
- Keep, R.F.; Jones, H.C.; Drewes, L.R. The year in review: Progress in brain barriers and brain fluid research in 2018. Fluids Barriers CNS 2019, 16, 1–12. [Google Scholar] [CrossRef]
- Louveau, A.; Plog, B.A.; Antila, S.; Alitalo, K.; Nedergaard, M.; Kipnis, J. Understanding the functions and relationships of the glymphatic system and meningeal lymphatics. J. Clin. Investig. 2017, 127, 3210–3219. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.J.; Verkman, A.S. The “glymphatic” mechanism for solute clearance in Alzheimer’s disease: Game changer or unproven speculation? FASEB J. 2018, 32, 543–551. [Google Scholar] [CrossRef] [Green Version]
- Plog, B.A.; Nedergaard, M. The Glymphatic System in Central Nervous System Health and Disease: Past, Present, and Future. Annu. Rev. Pathol. Mech. Dis. 2018, 13, 379–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokkers, R.P.; Bremmer, J.P.; Van Berckel, B.N.; Lammertsma, A.A.; Hendrikse, J.; Pluim, J.P.; Kappelle, L.J.; Boellaard, R.; Klijn, C.J. Arterial Spin Labeling Perfusion MRI at Multiple Delay Times: A Correlative Study with H215O Positron Emission Tomography in Patients with Symptomatic Carotid Artery Occlusion. Br. J. Pharmacol. 2009, 30, 222–229. [Google Scholar] [CrossRef] [Green Version]
- Joseph, C.R.; Benhatzel, C.M.; Stern, L.J.; Hopper, O.M.; Lockwood, M.D. Pilot study utilizing MRI 3D TGSE PASL (arterial spin labeling) differentiating clearance rates of labeled protons in the CNS of patients with early Alzheimer disease from normal subjects. Magn. Reson. Mater. Phys. Boil. Med. 2020, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Khoshbouei, H.; Bechmann, I. Dystrophic microglia in late-onset Alzheimer’s disease. Glia 2020, 68, 845–854. [Google Scholar] [CrossRef]
- Johnson, E.C.B.; Dammer, E.B.; Duong, D.M.; Ping, L.; Zhou, M.; Yin, L.; Higginbotham, L.A.; Guajardo, A.; White, B.; Troncoso, J.C.; et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 2020, 26, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Arellano, J.J.R.; Verkhratsky, A. Neuroglial Roots of Neurodegenerative Diseases? Mol. Neurobiol. 2010, 43, 87–96. [Google Scholar] [CrossRef]
- Kettenmann, H.; Verkhratsky, A. Neuroglia, der lebende Nervenkitt. Fortschritte der Neurol. Psychiatr. 2011, 79, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Ercan-Herbst, E.; Ehrig, J.; Schöndorf, D.C.; Behrendt, A.; Klaus, B.; Ramos, B.G.; Oriol, N.P.; Weber, C.; Ehrnhoefer, D.E. A post-translational modification signature defines changes in soluble tau correlating with oligomerization in early stage Alzheimer’s disease brain. Acta Neuropathol. Commun. 2019, 7, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Gong, C.-X.; Liu, F.; Iqbal, K. O-GlcNAcylation: A regulator of tau pathology and neurodegeneration. Alzheimer’s Dement. 2016, 12, 1078–1089. [Google Scholar] [CrossRef]
- Ittner, L.M.; Götz, J. Amyloid-β and tau — A toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2010, 12, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Tosun, D.; Landau, S.; Aisen, P.S.; Petersen, R.C.; Mintun, M.; Jagust, W.; Weiner, M.W.; Initiative, F.T.A.D.N. Association between tau deposition and antecedent amyloid-β accumulation rates in normal and early symptomatic individuals. Brain 2017, 140, 1499–1512. [Google Scholar] [CrossRef] [PubMed]
- Stratmann, K.; Heinsen, H.; Korf, H.-W.; Del Turco, D.; Ghebremedhin, E.; Seidel, K.; Bouzrou, M.; Grinberg, L.T.; Bohl, J.; Wharton, S.B.; et al. Precortical Phase of Alzheimer’s Disease (AD)-Related Tau Cytoskeletal Pathology. Brain Pathol. 2015, 26, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Hultman, K.; Strickland, S.; Norris, E.H. TheAPOEε4/ε4 Genotype Potentiates Vascular Fibrin(Ogen) Deposition in Amyloid-Laden Vessels in the Brains of Alzheimer’s Disease Patients. Br. J. Pharmacol. 2013, 33, 1251–1258. [Google Scholar] [CrossRef] [Green Version]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2016, 107, 41–56. [Google Scholar] [CrossRef] [Green Version]
- Zenaro, E.; Pietronigro, E.; Della Bianca, V.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S.; et al. Neutrophils promote Alzheimer’s disease–like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 2015, 21, 880–886. [Google Scholar] [CrossRef]
- Montagne, A.; Barnes, S.; Sweeney, M.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Shams, S.; Wahlund, L.-O. Cerebral microbleeds as a biomarker in Alzheimer’s disease? A review in the field. Biomarkers Med. 2016, 10, 9–18. [Google Scholar] [CrossRef]
- Brundel, M.; De Bresser, J.; Van Dillen, J.J.; Kappelle, L.J.; Biessels, G.J. Cerebral Microinfarcts: A Systematic Review of Neuropathological Studies. Br. J. Pharmacol. 2012, 32, 425–436. [Google Scholar] [CrossRef] [Green Version]
- Heringa, S.M.; Reijmer, Y.D.; Leemans, A.; Koek, H.L.; Kappelle, L.J.; Biessels, G.J.; on behalf of the Utrecht Vascular Cognitive Impairment (VCI) study group. Multiple Microbleeds are Related to Cerebral Network Disruptions in Patients with Early Alzheimer’s Disease. J. Alzheimer’s Dis. 2013, 38, 211–221. [Google Scholar] [CrossRef]
- Poliakova, T.; Levin, O.; Arablinskiy, A.; Vasenina, E.; Zerr, I. Cerebral microbleeds in early Alzheimer’s disease. J. Neurol. 2016, 263, 1961–1968. [Google Scholar] [CrossRef]
- Pettersen, J.A.; Sathiyamoorthy, G.; Gao, F.-Q.; Szilagyi, G.; Nadkarni, N.K.; George-Hyslop, P.S.; Rogaeva, E.; Black, S.E. Microbleed Topography, Leukoaraiosis, and Cognition in Probable Alzheimer Disease from the Sunnybrook Dementia Study. Arch. Neurol. 2008, 65, 790–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaasch, J.A.; Lockman, P.R.; Geldenhuys, W.J.; Allen, D.D.; Van Der Schyf, C.J. Brain Iron Toxicity: Differential Responses of Astrocytes, Neurons, and Endothelial Cells. Neurochem. Res. 2007, 32, 1196–1208. [Google Scholar] [CrossRef] [PubMed]
- Olmedo-Díaz, S.; Estévez-Silva, H.; Orädd, G.; Bjerkén, S.A.; Marcellino, D.; Virel, A.; Sonia, O.-D.; Héctor, E.-S.; Greger, O.; Sara, A.B.; et al. An altered blood–brain barrier contributes to brain iron accumulation and neuroinflammation in the 6-OHDA rat model of Parkinson’s disease. Neuroscience 2017, 362, 141–151. [Google Scholar] [CrossRef]
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 2015, 18, 521–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farkas, E.; Luiten, P.G. Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog. Neurobiol. 2001, 64, 575–611. [Google Scholar] [CrossRef] [Green Version]
- Sagare, A.P.; Bell, R.D.; Zlokovic, B.V. Neurovascular Dysfunction and Faulty Amyloid β-Peptide Clearance in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a011452. [Google Scholar] [CrossRef] [Green Version]
- Park, L.; Anrather, J.; Zhou, P.; Frys, K.; Pitstick, R.; Younkin, S.; Carlson, G.A.; Iadecola, C.; Katada, S.; Hirokawa, T.; et al. NADPH Oxidase-Derived Reactive Oxygen Species Mediate the Cerebrovascular Dysfunction Induced by the Amyloid β Peptide. J. Neurosci. 2005, 25, 1769–1777. [Google Scholar] [CrossRef]
- Biron, K.E.; Dickstein, D.L.; Gopaul, R.; Jefferies, W.A. Amyloid Triggers Extensive Cerebral Angiogenesis Causing Blood Brain Barrier Permeability and Hypervascularity in Alzheimer’s Disease. PLoS ONE 2011, 6, e23789. [Google Scholar] [CrossRef] [Green Version]
- Paul, J.; Strickland, S.; Melchor, J.P. Fibrin deposition accelerates neurovascular damage and neuroinflammation in mouse models of Alzheimer’s disease. J. Exp. Med. 2007, 204, 1999–2008. [Google Scholar] [CrossRef]
- Sagare, A.P.; Bell, R.D.; Zhao, Z.; Ma, Q.; Winkler, E.A.; Ramanathan, A.; Zlokovic, B.V. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat. Commun. 2013, 4, 2932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ujiie, M.; Dickstein, D.L.; Carlow, D.A.; Jefferies, W.A. Blood-Brain Barrier Permeability Precedes Senile Plaque Formation in an Alzheimer Disease Model. Microcirculation 2003, 10, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Zlokovic, B.V. Neurovascular mechanisms and blood–brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 103–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kook, S.-Y.; Hong, H.S.; Moon, M.; Ha, C.M.; Chang, S.; Mook-Jung, I. Aβ1–42-RAGE Interaction Disrupts Tight Junctions of the Blood–Brain Barrier Via Ca2+-Calcineurin Signaling. J. Neurosci. 2012, 32, 8845–8854. [Google Scholar] [CrossRef]
- Barnes, S.; Ng, T.S.C.; Montagne, A.; Law, M.; Zlokovic, B.V.; Jacobs, R. Optimal acquisition and modeling parameters for accurate assessment of low Ktrans blood-brain barrier permeability using dynamic contrast-enhanced MRI. Magn. Reson. Med. 2015, 75, 1967–1977. [Google Scholar] [CrossRef] [Green Version]
- Günther, M.; Oshio, K.; Feinberg, D.A. Single-shot 3D imaging techniques improve arterial spin labeling perfusion measurements. Magn. Reson. Med. 2005, 54, 491–498. [Google Scholar] [CrossRef]
- Hafezi-Moghadam, A.; Thomas, K.L.; Wagner, D.D. ApoE deficiency leads to a progressive age-dependent blood-brain barrier leakage. Am. J. Physiol. Physiol. 2007, 292, C1256–C1262. [Google Scholar] [CrossRef] [Green Version]
- Barnes, S.; Ng, T.S.C.; Santa-Maria, N.; Montagne, A.; Zlokovic, B.V.; Jacobs, R. ROCKETSHIP: A flexible and modular software tool for the planning, processing and analysis of dynamic MRI studies. BMC Med. Imaging 2015, 15, 19. [Google Scholar] [CrossRef] [Green Version]
- Van De Haar, H.J.; Burgmans, S.; Jansen, J.F.; Van Osch, M.J.P.; Van Buchem, M.A.; Muller, M.; Hofman, P.A.; Verhey, F.R.J.; Backes, W.H. Blood-Brain Barrier Leakage in Patients with Early Alzheimer Disease. Radiology 2016, 281, 527–535. [Google Scholar] [CrossRef]
- Grade, M.; Tamames, J.A.H.; Pizzini, F.B.; Achten, E.; Golay, X.; Smits, M. A neuroradiologist’s guide to arterial spin labeling MRI in clinical practice. Neuroradiol. 2015, 57, 1181–1202. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Clingman, C.; Golay, X.; Van Zijl, P.C. Determining the longitudinal relaxation time (T1) of blood at 3.0 Tesla. Magn. Reson. Med. 2004, 52, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Stanisz, G.J.; Odrobina, E.E.; Pun, J.; Escaravage, M.; Graham, S.J.; Bronskill, M.J.; Henkelman, R.M. T1, T2 relaxation and magnetization transfer in tissue at 3T. Magn. Reson. Med. 2005, 54, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, M.E.; Berman, A.; Mazerolle, E.L.; Williams, R.J.; Pike, G.B. Modeling hyperoxia-induced BOLD signal dynamics to estimate cerebral blood flow, volume and mean transit time. NeuroImage 2018, 178, 461–474. [Google Scholar] [CrossRef] [PubMed]
- Alsop, D.C.; Detre, J.A.; Golay, X.; Günther, M.; Hendrikse, J.; Hernandez-Garcia, L.; Lu, H.; MacIntosh, B.J.; Parkes, L.M.; Smits, M.; et al. Recommended implementation of arterial spin-labeled perfusion MRI for clinical applications: A consensus of the ISMRM perfusion study group and the European consortium for ASL in dementia. Magn. Reson. Med. 2014, 73, 102–116. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Liu, C.Y.; Smith, R.X.; Jog, M.; Langham, M.; Krasileva, K.; Chen, Y.; Ringman, J.M.; Wang, D.J. Assessing intracranial vascular compliance using dynamic arterial spin labeling. NeuroImage 2015, 124, 433–441. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, D.A.; Beckett, A.; Chen, L. Arterial spin labeling with simultaneous multi-slice echo planar imaging. Magn. Reson. Med. 2013, 70, 1500–1506. [Google Scholar] [CrossRef] [Green Version]
- Chao, L.L.; Buckley, S.T.; Kornak, J.; Schuff, N.; Madison, C.; Yaffe, K.; Miller, B.L.; Kramer, J.H.; Weiner, M.W. ASL Perfusion MRI Predicts Cognitive Decline and Conversion from MCI to Dementia. Alzheimer Dis. Assoc. Disord. 2010, 24, 19–27. [Google Scholar] [CrossRef]
- Mosconi, L.; De Santi, S.; Li, J.; Tsui, W.H.; Li, Y.; Boppana, M.; Laska, E.; Rusinek, H.; De Leon, M.J. Hippocampal hypometabolism predicts cognitive decline from normal aging. Neurobiol. Aging 2008, 29, 676–692. [Google Scholar] [CrossRef] [Green Version]
- Montagne, A.; Nation, D.A.; Pa, J.; Sweeney, M.; Toga, A.W.; Zlokovic, B.V. Brain imaging of neurovascular dysfunction in Alzheimer’s disease. Acta Neuropathol. 2016, 131, 687–707. [Google Scholar] [CrossRef]
- Yang, Y.; Engelien, W.; Xu, S.; Gu, H.; Silbersweig, D.A.; Stern, E. Transit time, trailing time, and cerebral blood flow during brain activation: Measurement using multislice, pulsed spin-labeling perfusion imaging. Magn. Reson. Med. 2000, 44, 680–685. [Google Scholar] [CrossRef]
- Ai, H. Layer-by-layer capsules for magnetic resonance imaging and drug delivery. Adv. Drug Deliv. Rev. 2011, 63, 772–788. [Google Scholar] [CrossRef] [PubMed]

 Aβ,
Aβ,  infiltrating vascular toxins,
infiltrating vascular toxins,  Hp tau.
Aβ, infiltrating vascular toxins, Hp tau.
Hp tau.
Aβ, infiltrating vascular toxins, Hp tau.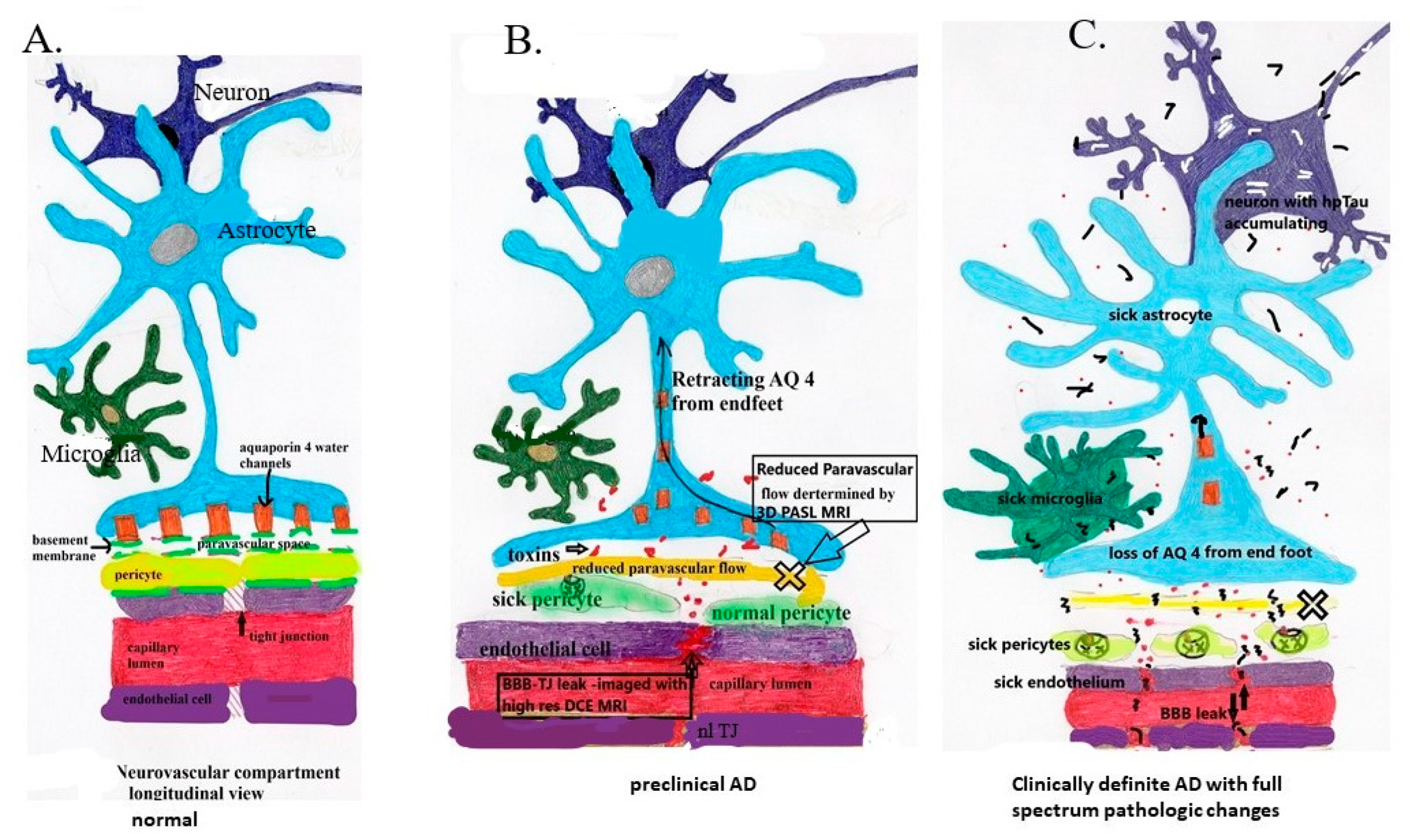
| A = Aggregated Amyloid Aβ | T = Aggregated Tau | N = Neurodegeneration |
|---|---|---|
| CSF Aβ42 or Aβ42/ Aβ40 ratio | CSF phosphorylated tau | Anatomic MRI -atrophy |
| Amyloid PET | Tau Pet | FDG PET |
| Test | Preclinical Stage | Early Clinical (MCI) | Definite AD (Late) |
|---|---|---|---|
| DCE | + | + | + |
| ASL flow | ? | + | + |
| CSF Aβ42 or Aβ42/ Aβ40 ratio | ± | + | + |
| CSF phosphorylated tau | - | +/-± | + |
| Amyloid PET | ± | + | + |
| Tau Pet | - | +/-± | + |
| Anatomic MRI -atrophy | - | - | + |
| FDG PET | - | ± | + |
| CSF total tau | - | - | + |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joseph, C.R. Novel MRI Techniques Identifying Vascular Leak and Paravascular Flow Reduction in Early Alzheimer Disease. Biomedicines 2020, 8, 228. https://doi.org/10.3390/biomedicines8070228
Joseph CR. Novel MRI Techniques Identifying Vascular Leak and Paravascular Flow Reduction in Early Alzheimer Disease. Biomedicines. 2020; 8(7):228. https://doi.org/10.3390/biomedicines8070228
Chicago/Turabian StyleJoseph, Charles R. 2020. "Novel MRI Techniques Identifying Vascular Leak and Paravascular Flow Reduction in Early Alzheimer Disease" Biomedicines 8, no. 7: 228. https://doi.org/10.3390/biomedicines8070228
APA StyleJoseph, C. R. (2020). Novel MRI Techniques Identifying Vascular Leak and Paravascular Flow Reduction in Early Alzheimer Disease. Biomedicines, 8(7), 228. https://doi.org/10.3390/biomedicines8070228