Antiplatelet Therapy for Acute Respiratory Distress Syndrome




Abstract
1. Introduction
1.1. Definition and Epidemiology of ARDS
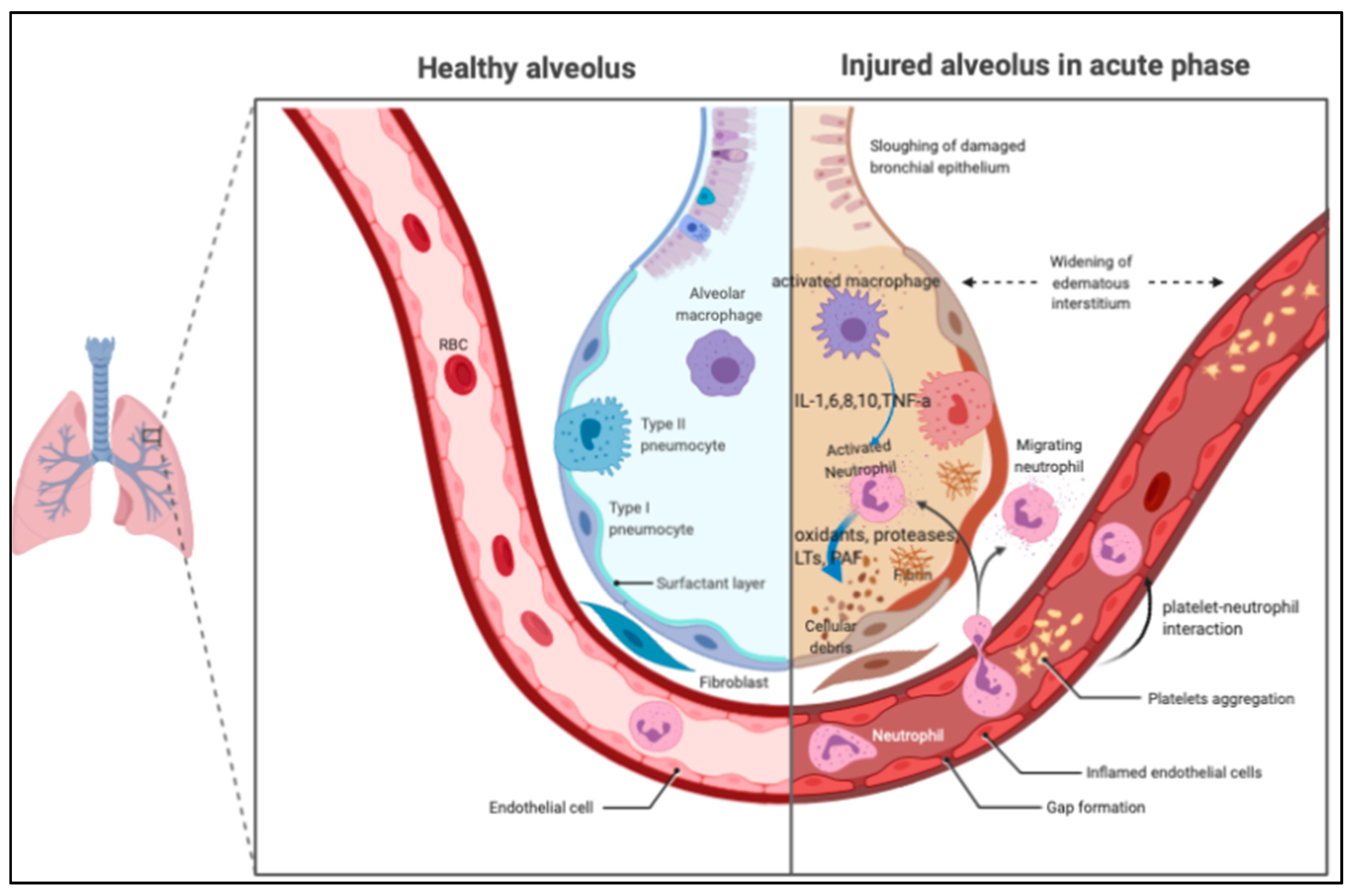
1.2. Pathogenesis of ARDS
2. Mechanisms of Platelet in Lung Inflammation
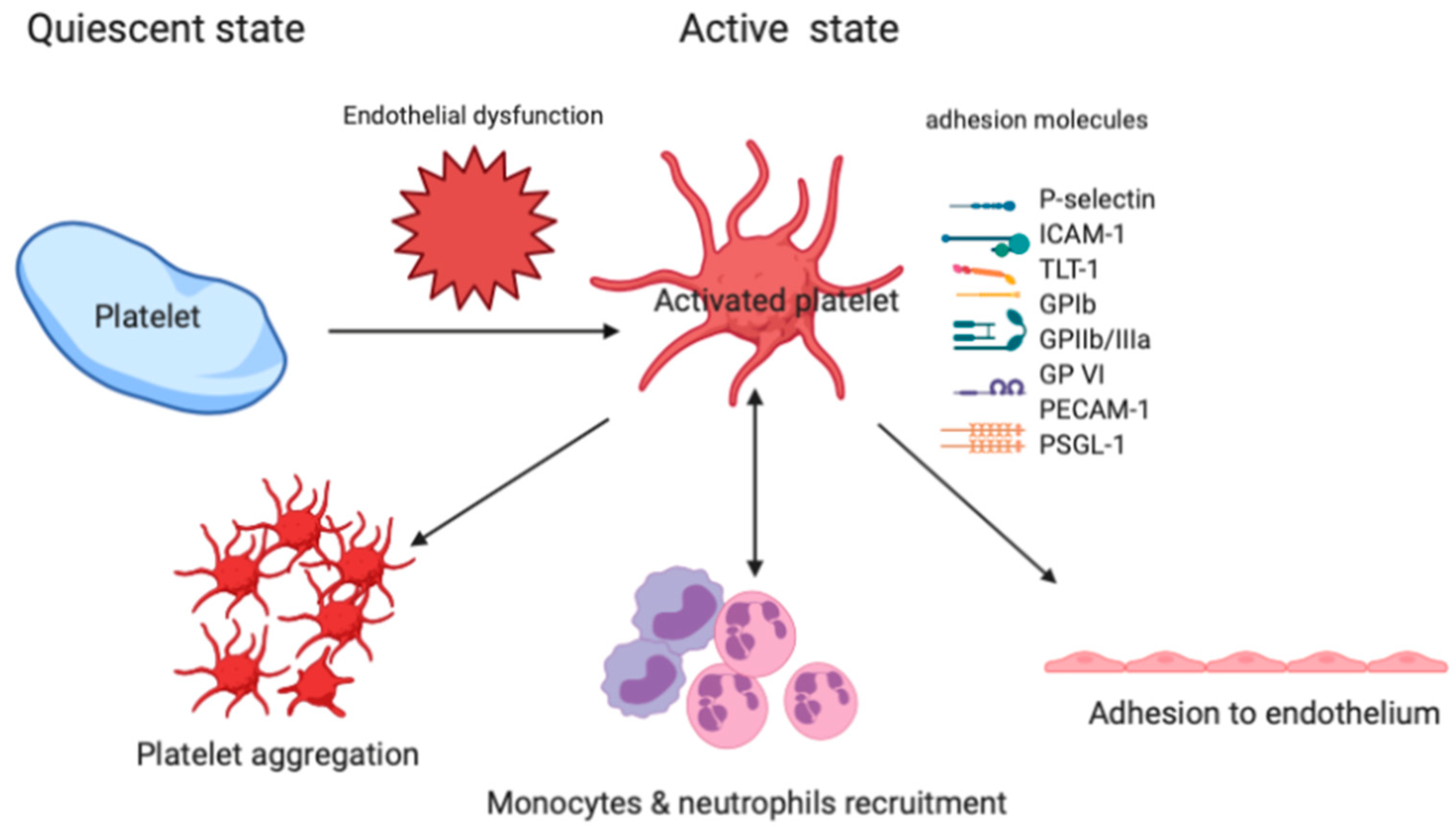
2.1. The Role of Platelet in the ARDS
2.2. Platelet Adhesion to the Injured Vessel Wall
2.3. Platelet-Leukocyte-Endothelium Interactions
2.4. Lipid Mediators and Platelet Aggregation
2.5. Neutrophil Extracellular Traps (NETs)
3. Antiplatelet Agents in Experimental Studies
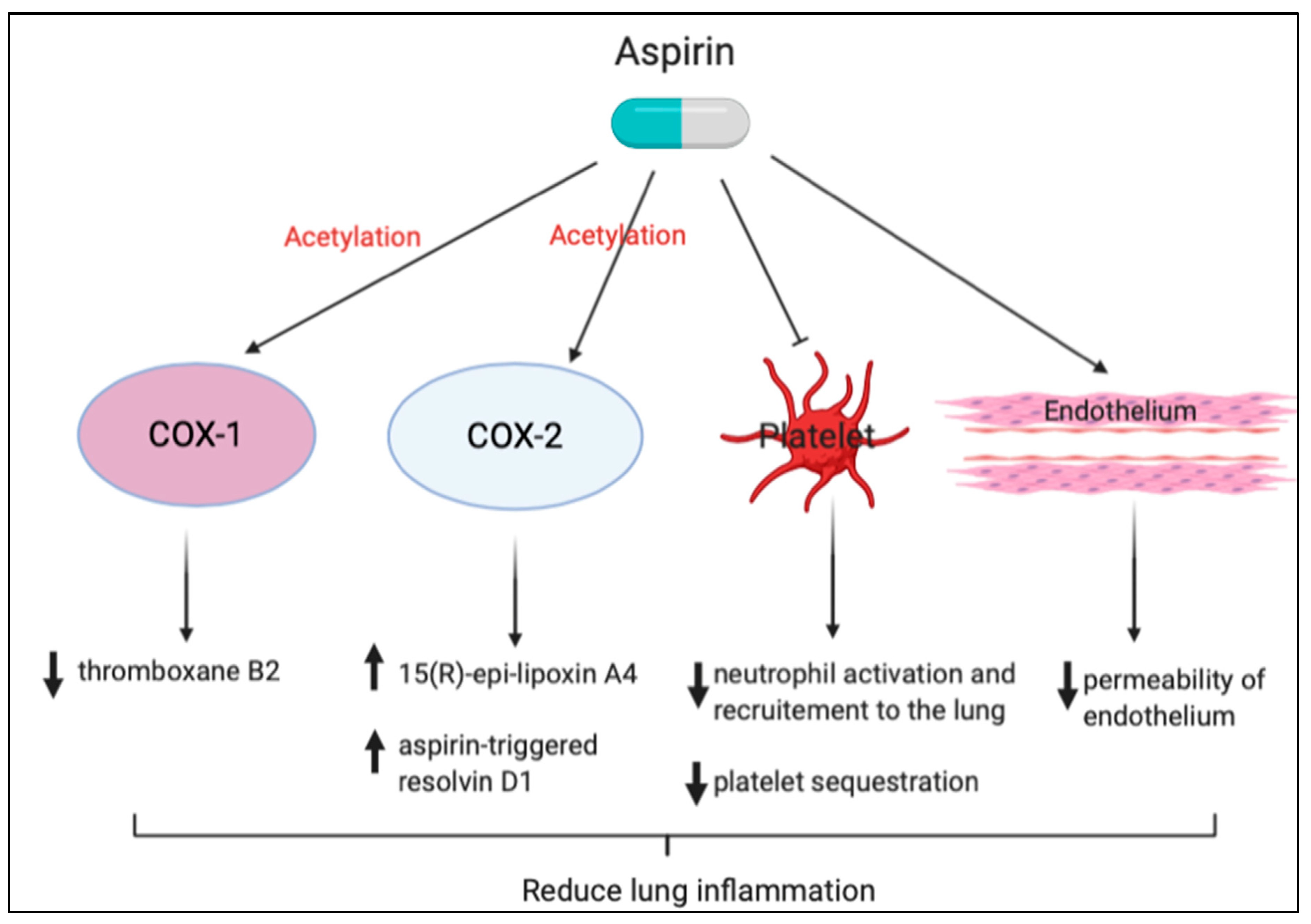
3.1. Aspirin
3.2. GP IIb/IIIa Antagonists
3.3. P2Y12 Inhibitors
4. Antiplatelet Agents in Clinical Studies
5. Meta-Analysis of Clinical Studies
6. Conclusions
Funding
Conflicts of Interest
References
- Force, A.D.T.; Ranieri, V.M.; Rubenfeld, G.D.; Thompson, B.T.; Ferguson, N.D.; Caldwell, E.; Fan, E.; Camporota, L.; Slutsky, A.S. Acute respiratory distress syndrome: The Berlin Definition. JAMA 2012, 307, 2526–2533. [Google Scholar]
- Bernard, G.R.; Artigas, A.; Brigham, K.L.; Carlet, J.; Falke, K.; Hudson, L.; Lamy, M.; Legall, J.R.; Morris, A.; Spragg, R. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am. J. Respir. Crit. Care Med. 1994, 149, 818–824. [Google Scholar] [CrossRef] [PubMed]
- Ware, L.B.; Matthay, M.A. The acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1334–1349. [Google Scholar] [CrossRef] [PubMed]
- Rubenfeld, G.D.; Caldwell, E.; Peabody, E.; Weaver, J.; Martin, D.P.; Neff, M.; Stern, E.J.; Hudson, L.D. Incidence and outcomes of acute lung injury. N. Engl. J. Med. 2005, 353, 1685–1693. [Google Scholar] [CrossRef]
- Chen, W.; Chen, Y.Y.; Tsai, C.F.; Chen, S.C.; Lin, M.S.; Ware, L.B.; Chen, C.M. Incidence and Outcomes of Acute Respiratory Distress Syndrome: A Nationwide Registry-Based Study in Taiwan, 1997 to 2011. Medicine 2015, 94, e1849. [Google Scholar] [CrossRef]
- Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N. Engl. J. Med. 2000, 342, 1301–1308. [Google Scholar] [CrossRef]
- Jepsen, S.; Herlevsen, P.; Knudsen, P.; Bud, M.I.; Klausen, N.O. Antioxidant treatment with N-acetylcysteine during adult respiratory distress syndrome: A prospective, randomized, placebo-controlled study. Crit. Care Med. 1992, 20, 918–923. [Google Scholar] [CrossRef] [PubMed]
- Ketoconazole for early treatment of acute lung injury and acute respiratory distress syndrome: A randomized controlled trial. The ARDS Network. JAMA 2000, 283, 1995–2002.
- Meade, M.O.; Jacka, M.J.; Cook, D.J.; Dodek, P.; Griffith, L.; Guyatt, G.H.; Canadian Critical Care Trials, G. Survey of interventions for the prevention and treatment of acute respiratory distress syndrome. Crit. Care Med. 2004, 32, 946–954. [Google Scholar] [CrossRef]
- Avecillas, J.F.; Freire, A.X.; Arroliga, A.C. Clinical epidemiology of acute lung injury and acute respiratory distress syndrome: Incidence, diagnosis, and outcomes. Clin. Chest Med. 2006, 27, 549–557. [Google Scholar] [CrossRef]
- Caser, E.B.; Zandonade, E.; Pereira, E.; Gama, A.M.; Barbas, C.S. Impact of distinct definitions of acute lung injury on its incidence and outcomes in Brazilian ICUs: Prospective evaluation of 7,133 patients*. Crit. Care Med. 2014, 42, 574–582. [Google Scholar] [CrossRef]
- Herridge, M.S.; Cheung, A.M.; Tansey, C.M.; Matte-Martyn, A.; Diaz-Granados, N.; Al-Saidi, F.; Cooper, A.B.; Guest, C.B.; Mazer, C.D.; Mehta, S.; et al. One-year outcomes in survivors of the acute respiratory distress syndrome. N. Engl. J. Med. 2003, 348, 683–693. [Google Scholar] [CrossRef]
- Herridge, M.S.; Tansey, C.M.; Matte, A.; Tomlinson, G.; Diaz-Granados, N.; Cooper, A.; Guest, C.B.; Mazer, C.D.; Mehta, S.; Stewart, T.E.; et al. Functional disability 5 years after acute respiratory distress syndrome. N. Engl. J. Med. 2011, 364, 1293–1304. [Google Scholar] [CrossRef]
- Sasannejad, C.; Ely, E.W.; Lahiri, S. Long-term cognitive impairment after acute respiratory distress syndrome: A review of clinical impact and pathophysiological mechanisms. Crit Care 2019, 23, 352. [Google Scholar] [CrossRef]
- Bein, T.; Weber-Carstens, S.; Apfelbacher, C.; Brandstetter, S.; Blecha, S.; Dodoo-Schittko, F.; Brandl, M.; Quintel, M.; Kluge, S.; Putensen, C.; et al. The quality of acute intensive care and the incidence of critical events have an impact on health-related quality of life in survivors of the acute respiratory distress syndrome—A nationwide prospective multicenter observational study. Ger. Med. Sci. 2020, 18, Doc01. [Google Scholar] [PubMed]
- Bachofen, M.; Weibel, E.R. Alterations of the gas exchange apparatus in adult respiratory insufficiency associated with septicemia. Am. Rev. Respir. Dis. 1977, 116, 589–615. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Zemans, R.L. The acute respiratory distress syndrome: Pathogenesis and treatment. Annu. Rev. Pathol. 2011, 6, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Huppert, L.A.; Matthay, M.A.; Ware, L.B. Pathogenesis of Acute Respiratory Distress Syndrome. Semin. Respir. Crit. Care Med. 2019, 40, 31–39. [Google Scholar] [CrossRef]
- Kim, T.H.; Hong, S.B.; Lim, C.M.; Koh, Y.; Jang, E.Y.; Huh, J.W. The Role of Exosomes in Bronchoalveloar Lavage from Patients with Acute Respiratory Distress Syndrome. J. Clin. Med. 2019, 8, 1148. [Google Scholar] [CrossRef]
- Pugin, J.; Verghese, G.; Widmer, M.C.; Matthay, M.A. The alveolar space is the site of intense inflammatory and profibrotic reactions in the early phase of acute respiratory distress syndrome. Crit. Care Med. 1999, 27, 304–312. [Google Scholar] [CrossRef]
- Yang, C.Y.; Chen, C.S.; Yiang, G.T.; Cheng, Y.L.; Yong, S.B.; Wu, M.Y.; Li, C.J. New Insights into the Immune Molecular Regulation of the Pathogenesis of Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2018, 19, 588. [Google Scholar] [CrossRef] [PubMed]
- Ware, L.B.; Herridge, M. Acute lung injury. Semin. Respir. Crit. Care Med. 2013, 34, 439–440. [Google Scholar] [CrossRef]
- Weiss, S.J. Tissue destruction by neutrophils. N. Engl. J. Med. 1989, 320, 365–376. [Google Scholar] [PubMed]
- Babior, B.M.; Takeuchi, C.; Ruedi, J.; Gutierrez, A.; Wentworth, P., Jr. Investigating antibody-catalyzed ozone generation by human neutrophils. Proc. Natl. Acad. Sci. USA 2003, 100, 3031–3034. [Google Scholar] [CrossRef] [PubMed]
- Stormann, P.; Becker, N.; Vollrath, J.T.; Kohler, K.; Janicova, A.; Wutzler, S.; Hildebrand, F.; Marzi, I.; Relja, B. Early Local Inhibition of Club Cell Protein 16 Following Chest Trauma Reduces Late Sepsis-Induced Acute Lung Injury. J. Clin. Med. 2019, 8, 896. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.C. Acute Respiratory Distress Syndrome as an Organ Phenotype of Vascular Microthrombotic Disease: Based on Hemostatic Theory and Endothelial Molecular Pathogenesis. Clin. Appl. Thromb. Hemost. 2019, 25, 1076029619887437. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Wiener-Kronish, J.P. Intact epithelial barrier function is critical for the resolution of alveolar edema in humans. Am. Rev. Respir. Dis. 1990, 142, 1250–1257. [Google Scholar] [CrossRef]
- Perkins, G.D.; Nathani, N.; McAuley, D.F.; Gao, F.; Thickett, D.R. In vitro and in vivo effects of salbutamol on neutrophil function in acute lung injury. Thorax 2007, 62, 36–42. [Google Scholar] [CrossRef]
- Abraham, E. Neutrophils and acute lung injury. Crit. Care Med. 2003, 31, S195–S199. [Google Scholar] [CrossRef]
- Frank, J.A.; Wray, C.M.; McAuley, D.F.; Schwendener, R.; Matthay, M.A. Alveolar macrophages contribute to alveolar barrier dysfunction in ventilator-induced lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L1191–L1198. [Google Scholar] [CrossRef]
- Matthay, M.A.; Zimmerman, G.A.; Esmon, C.; Bhattacharya, J.; Coller, B.; Doerschuk, C.M.; Floros, J.; Gimbrone, M.A., Jr.; Hoffman, E.; Hubmayr, R.D.; et al. Future research directions in acute lung injury: Summary of a National Heart, Lung, and Blood Institute working group. Am. J. Respir. Crit. Care Med. 2003, 167, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Zimmerman, G.A. Acute lung injury and the acute respiratory distress syndrome: Four decades of inquiry into pathogenesis and rational management. Am. J.Respir. Cell Mol. Biol. 2005, 33, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Gadek, J.E. Adverse effects of neutrophils on the lung. Am. J. Med. 1992, 92, 27S–31S. [Google Scholar] [CrossRef]
- Grimminger, F.; Menger, M.; Becker, G.; Seeger, W. Potentiation of leukotriene production following sequestration of neutrophils in isolated lungs: Indirect evidence for intercellular leukotriene A4 transfer. Blood 1988, 72, 1687–1692. [Google Scholar] [CrossRef]
- Fantone, J.C.; Ward, P.A. Role of oxygen-derived free radicals and metabolites in leukocyte-dependent inflammatory reactions. Am. J. Pathol. 1982, 107, 395–418. [Google Scholar]
- Matthay, M.A.; Ware, L.B.; Zimmerman, G.A. The acute respiratory distress syndrome. J. Clin. Investig. 2012, 122, 2731–2740. [Google Scholar] [CrossRef]
- Zarbock, A.; Polanowska-Grabowska, R.K.; Ley, K. Platelet-neutrophil-interactions: Linking hemostasis and inflammation. Blood Rev. 2007, 21, 99–111. [Google Scholar] [CrossRef]
- Privratsky, J.R.; Paddock, C.M.; Florey, O.; Newman, D.K.; Muller, W.A.; Newman, P.J. Relative contribution of PECAM-1 adhesion and signaling to the maintenance of vascular integrity. J. Cell Sci. 2011, 124, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Villar, J.; Zhang, H.; Slutsky, A.S. Lung Repair and Regeneration in ARDS: Role of PECAM1 and Wnt Signaling. Chest 2019, 155, 587–594. [Google Scholar] [CrossRef]
- Zarbock, A.; Singbartl, K.; Ley, K. Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation. J. Clin. Investig. 2006, 116, 3211–3219. [Google Scholar] [CrossRef]
- Kiefmann, R.; Heckel, K.; Schenkat, S.; Dorger, M.; Wesierska-Gadek, J.; Goetz, A.E. Platelet-endothelial cell interaction in pulmonary micro-circulation: The role of PARS. Thromb. Haemost. 2004, 91, 761–770. [Google Scholar] [CrossRef]
- Kiefmann, R.; Heckel, K.; Schenkat, S.; Dorger, M.; Goetz, A.E. Role of p-selectin in platelet sequestration in pulmonary capillaries during endotoxemia. J. Vasc. Res. 2006, 43, 473–481. [Google Scholar] [CrossRef]
- Wang, Y.; Zhong, M.; Wang, Z.; Song, J.; Wu, W.; Zhu, D. The preventive effect of antiplatelet therapy in acute respiratory distress syndrome: A meta-analysis. Crit. Care 2018, 22, 60. [Google Scholar] [CrossRef]
- Panka, B.A.; de Grooth, H.J.; Spoelstra-de Man, A.M.; Looney, M.R.; Tuinman, P.R. Prevention or Treatment of Ards With Aspirin: A Review of Preclinical Models and Meta-Analysis of Clinical Studies. Shock 2017, 47, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Hamid, U.; Krasnodembskaya, A.; Fitzgerald, M.; Shyamsundar, M.; Kissenpfennig, A.; Scott, C.; Lefrancais, E.; Looney, M.R.; Verghis, R.; Scott, J.; et al. Aspirin reduces lipopolysaccharide-induced pulmonary inflammation in human models of ARDS. Thorax 2017, 72, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Frantzeskaki, F.; Armaganidis, A.; Orfanos, S.E. Immunothrombosis in Acute Respiratory Distress Syndrome: Cross Talks between Inflammation and Coagulation. Respiration 2017, 93, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Zarbock, A.; Ley, K. The role of platelets in acute lung injury (ALI). Front. Biosci. 2009, 14, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, Z.M. Von Willebrand factor. Curr. Opin. Hematol. 2003, 10, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, Z.M.; Dent, J.A.; Saldivar, E. Contribution of distinct adhesive interactions to platelet aggregation in flowing blood. Blood 1999, 94, 172–178. [Google Scholar] [CrossRef]
- Jurk, K.; Clemetson, K.J.; de Groot, P.G.; Brodde, M.F.; Steiner, M.; Savion, N.; Varon, D.; Sixma, J.J.; Van Aken, H.; Kehrel, B.E. Thrombospondin-1 mediates platelet adhesion at high shear via glycoprotein Ib (GPIb): An alternative/backup mechanism to von Willebrand factor. FASEB J. 2003, 17, 1490–1492. [Google Scholar] [CrossRef]
- Savage, B.; Almus-Jacobs, F.; Ruggeri, Z.M. Specific synergy of multiple substrate-receptor interactions in platelet thrombus formation under flow. Cell 1998, 94, 657–666. [Google Scholar] [CrossRef]
- Kasirer-Friede, A.; Kahn, M.L.; Shattil, S.J. Platelet integrins and immunoreceptors. Immunol. Rev. 2007, 218, 247–264. [Google Scholar] [CrossRef]
- Washington, A.V.; Esponda, O.; Gibson, A. Platelet biology of the rapidly failing lung. Br. J. Haematol. 2020, 188, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Lax, S.; Rayes, J.; Wichaiyo, S.; Haining, E.J.; Lowe, K.; Grygielska, B.; Laloo, R.; Flodby, P.; Borok, Z.; Crandall, E.D.; et al. Platelet CLEC-2 protects against lung injury via effects of its ligand podoplanin on inflammatory alveolar macrophages in the mouse. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L1016–L1029. [Google Scholar] [CrossRef] [PubMed]
- Nieswandt, B.; Echtenacher, B.; Wachs, F.P.; Schroder, J.; Gessner, J.E.; Schmidt, R.E.; Grau, G.E.; Mannel, D.N. Acute systemic reaction and lung alterations induced by an antiplatelet integrin gpIIb/IIIa antibody in mice. Blood 1999, 94, 684–693. [Google Scholar] [CrossRef]
- Koedam, J.A.; Cramer, E.M.; Briend, E.; Furie, B.; Furie, B.C.; Wagner, D.D. P-selectin, a granule membrane protein of platelets and endothelial cells, follows the regulated secretory pathway in AtT-20 cells. J. Cell Biol. 1992, 116, 617–625. [Google Scholar] [CrossRef]
- Tabuchi, A.; Kuebler, W.M. Endothelium-platelet interactions in inflammatory lung disease. Vasc. Pharmacol. 2008, 49, 141–150. [Google Scholar] [CrossRef]
- Urzainqui, A.; Serrador, J.M.; Viedma, F.; Yanez-Mo, M.; Rodriguez, A.; Corbi, A.L.; Alonso-Lebrero, J.L.; Luque, A.; Deckert, M.; Vazquez, J.; et al. ITAM-based interaction of ERM proteins with Syk mediates signaling by the leukocyte adhesion receptor PSGL-1. Immunity 2002, 17, 401–412. [Google Scholar] [CrossRef]
- Haselmayer, P.; Grosse-Hovest, L.; von Landenberg, P.; Schild, H.; Radsak, M.P. TREM-1 ligand expression on platelets enhances neutrophil activation. Blood 2007, 110, 1029–1035. [Google Scholar] [CrossRef]
- Zhao, C.; Su, E.M.; Yang, X.; Gao, Z.; Li, L.; Wu, H.; Jiang, Y.; Su, X. Important role of platelets in modulating endotoxin-induced lung inflammation in CFTR-deficient mice. PLoS ONE 2013, 8, e82683. [Google Scholar] [CrossRef]
- Morales-Ortiz, J.; Deal, V.; Reyes, F.; Maldonado-Martinez, G.; Ledesma, N.; Staback, F.; Croft, C.; Pacheco, A.; Ortiz-Zuazaga, H.; Yost, C.C.; et al. Platelet-derived TLT-1 is a prognostic indicator in ALI/ARDS and prevents tissue damage in the lungs in a mouse model. Blood 2018, 132, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Caudrillier, A.; Kessenbrock, K.; Gilliss, B.M.; Nguyen, J.X.; Marques, M.B.; Monestier, M.; Toy, P.; Werb, Z.; Looney, M.R. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J. Clin. Investig. 2012, 122, 2661–2671. [Google Scholar] [CrossRef] [PubMed]
- Vieira-de-Abreu, A.; Campbell, R.A.; Weyrich, A.S.; Zimmerman, G.A. Platelets: Versatile effector cells in hemostasis, inflammation, and the immune continuum. Semin. Immunopathol. 2012, 34, 5–30. [Google Scholar] [CrossRef] [PubMed]
- Cerletti, C.; Tamburrelli, C.; Izzi, B.; Gianfagna, F.; de Gaetano, G. Platelet-leukocyte interactions in thrombosis. Thromb. Res. 2012, 129, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Zarbock, A.; Bishop, J.; Muller, H.; Schmolke, M.; Buschmann, K.; Van Aken, H.; Singbartl, K. Chemokine homeostasis vs. chemokine presentation during severe acute lung injury: The other side of the Duffy antigen receptor for chemokines. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 298, L462–L471. [Google Scholar] [CrossRef]
- Ortiz-Munoz, G.; Mallavia, B.; Bins, A.; Headley, M.; Krummel, M.F.; Looney, M.R. Aspirin-triggered 15-epi-lipoxin A4 regulates neutrophil-platelet aggregation and attenuates acute lung injury in mice. Blood 2014, 124, 2625–2634. [Google Scholar] [CrossRef]
- Goldman, G.; Welbourn, R.; Kobzik, L.; Valeri, C.R.; Shepro, D.; Hechtman, H.B. Synergism between leukotriene B4 and thromboxane A2 in mediating acid-aspiration injury. Surgery 1992, 111, 55–61. [Google Scholar]
- Wahn, H.; Hammerschmidt, S. Influence of cyclooxygenase and lipoxygenase inhibitors on oxidative stress-induced lung injury. Crit. Care Med. 2001, 29, 802–807. [Google Scholar] [CrossRef]
- Looney, M.R.; Nguyen, J.X.; Hu, Y.; Van Ziffle, J.A.; Lowell, C.A.; Matthay, M.A. Platelet depletion and aspirin treatment protect mice in a two-event model of transfusion-related acute lung injury. J. Clin. Investig. 2009, 119, 3450–3461. [Google Scholar] [CrossRef]
- Nagase, T.; Uozumi, N.; Ishii, S.; Kume, K.; Izumi, T.; Ouchi, Y.; Shimizu, T. Acute lung injury by sepsis and acid aspiration: A key role for cytosolic phospholipase A2. Nat. Immunol. 2000, 1, 42–46. [Google Scholar] [CrossRef]
- Kostopanagiotou, G.; Avgerinos, E.; Costopanagiotou, C.; Arkadopoulos, N.; Andreadou, I.; Diamantopoulou, K.; Lekka, M.; Smyrniotis, V.; Nakos, G. Acute lung injury in a rat model of intestinal ischemia-reperfusion: The potential time depended role of phospholipases A(2). J. Surg. Res. 2008, 147, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Pittet, J.F.; Mackersie, R.C.; Martin, T.R.; Matthay, M.A. Biological markers of acute lung injury: Prognostic and pathogenetic significance. Am. J. Respir. Crit. Care Med. 1997, 155, 1187–1205. [Google Scholar] [CrossRef]
- Lv, W.; Wang, S.; Wang, L.; Wu, Z.; Jiang, Y.; Chen, X.; Gao, R. G994T polymorphism in exon 9 of plasma platelet-activating factor acetylhydrolase gene and lung ultrasound score as prognostic markers in evaluating the outcome of acute respiratory distress syndrome. Exp. Ther. Med. 2019, 17, 3174–3180. [Google Scholar] [CrossRef] [PubMed]
- Hudson, L.D.; Milberg, J.A.; Anardi, D.; Maunder, R.J. Clinical risks for development of the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1995, 151, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, S.C.; Haslett, C. Cellular mechanisms of acute lung injury: Implications for future treatment in the adult respiratory distress syndrome. Thorax 1992, 47, 260–263. [Google Scholar] [CrossRef]
- Semple, J.W.; Italiano, J.E., Jr.; Freedman, J. Platelets and the immune continuum. Nat. Rev. Immunol. 2011, 11, 264–274. [Google Scholar] [CrossRef]
- Brinkmann, V.; Zychlinsky, A. Beneficial suicide: Why neutrophils die to make NETs. Nat. Rev. Microbiol. 2007, 5, 577–582. [Google Scholar] [CrossRef]
- Gupta, A.K.; Hasler, P.; Holzgreve, W.; Gebhardt, S.; Hahn, S. Induction of neutrophil extracellular DNA lattices by placental microparticles and IL-8 and their presence in preeclampsia. Hum. Immunol. 2005, 66, 1146–1154. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Krumbholz, M.; Schonermarck, U.; Back, W.; Gross, W.L.; Werb, Z.; Grone, H.J.; Brinkmann, V.; Jenne, D.E. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 2009, 15, 623–625. [Google Scholar] [CrossRef]
- Lande, R.; Ganguly, D.; Facchinetti, V.; Frasca, L.; Conrad, C.; Gregorio, J.; Meller, S.; Chamilos, G.; Sebasigari, R.; Riccieri, V.; et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci. Transl. Med. 2011, 3, 73ra19. [Google Scholar] [CrossRef]
- Chang, Y.W.; Tseng, C.P.; Lee, C.H.; Hwang, T.L.; Chen, Y.L.; Su, M.T.; Chong, K.Y.; Lan, Y.W.; Wu, C.C.; Chen, K.J.; et al. beta-Nitrostyrene derivatives attenuate LPS-mediated acute lung injury via the inhibition of neutrophil-platelet interactions and NET release. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L654–L669. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.T.; Li, S.L.; Cai, E.Q.; Wu, W.L.; Jin, J.S.; Zhu, B. LPS induces pulmonary intravascular macrophages producing inflammatory mediators via activating NF-kappaB. J. Cell. Biochem. 2003, 89, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- Tuinman, P.R.; Muller, M.C.; Jongsma, G.; Hegeman, M.A.; Juffermans, N.P. High-dose acetylsalicylic acid is superior to low-dose as well as to clopidogrel in preventing lipopolysaccharide-induced lung injury in mice. Shock 2013, 40, 334–338. [Google Scholar] [CrossRef]
- Kario, K.; Eguchi, K.; Hoshide, S.; Hoshide, Y.; Umeda, Y.; Mitsuhashi, T.; Shimada, K. U-curve relationship between orthostatic blood pressure change and silent cerebrovascular disease in elderly hypertensives: Orthostatic hypertension as a new cardiovascular risk factor. J. Am. Coll. Cardiol. 2002, 40, 133–141. [Google Scholar] [CrossRef]
- Eickmeier, O.; Seki, H.; Haworth, O.; Hilberath, J.N.; Gao, F.; Uddin, M.; Croze, R.H.; Carlo, T.; Pfeffer, M.A.; Levy, B.D. Aspirin-triggered resolvin D1 reduces mucosal inflammation and promotes resolution in a murine model of acute lung injury. Mucosal Immunol. 2013, 6, 256–266. [Google Scholar] [CrossRef]
- Bates, J.J.; Watson, R.W.; Glynn, C.M.; O’Neill, A.J.; Fitzpatrick, J.M.; Buggy, D.J. Aspirin preserves neutrophil apoptosis after cardiopulmonary bypass. Shock 2004, 21, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Claria, J.; Serhan, C.N. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc. Natl. Acad. Sci. USA 1995, 92, 9475–9479. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukot. Essent. Fat. Acids 2005, 73, 141–162. [Google Scholar] [CrossRef] [PubMed]
- Levy, B.D.; Clish, C.B.; Schmidt, B.; Gronert, K.; Serhan, C.N. Lipid mediator class switching during acute inflammation: Signals in resolution. Nat. Immunol. 2001, 2, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Resolution phase of inflammation: Novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu. Rev. Immunol. 2007, 25, 101–137. [Google Scholar] [CrossRef] [PubMed]
- Matute-Bello, G.; Liles, W.C.; Radella, F., 2nd; Steinberg, K.P.; Ruzinski, J.T.; Jonas, M.; Chi, E.Y.; Hudson, L.D.; Martin, T.R. Neutrophil apoptosis in the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1997, 156, 1969–1977. [Google Scholar] [CrossRef] [PubMed]
- El Kebir, D.; Jozsef, L.; Pan, W.; Wang, L.; Petasis, N.A.; Serhan, C.N.; Filep, J.G. 15-epi-lipoxin A4 inhibits myeloperoxidase signaling and enhances resolution of acute lung injury. Am. J. Respir. Crit. Care Med. 2009, 180, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.P.; Oh, S.F.; Uddin, J.; Yang, R.; Gotlinger, K.; Campbell, E.; Colgan, S.P.; Petasis, N.A.; Serhan, C.N. Resolvin D1 and its aspirin-triggered 17R epimer. Stereochemical assignments, anti-inflammatory properties, and enzymatic inactivation. J. Biol. Chem. 2007, 282, 9323–9334. [Google Scholar] [CrossRef]
- Tang, H.; Liu, Y.; Yan, C.; Petasis, N.A.; Serhan, C.N.; Gao, H. Protective actions of aspirin-triggered (17R) resolvin D1 and its analogue, 17R-hydroxy-19-para-fluorophenoxy-resolvin D1 methyl ester, in C5a-dependent IgG immune complex-induced inflammation and lung injury. J. Immunol. 2014, 193, 3769–3778. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.M.; Tung, Y.T.; Wei, C.H.; Lee, P.Y.; Chen, W. Anti-Inflammatory and Reactive Oxygen Species Suppression through Aspirin Pretreatment to Treat Hyperoxia-Induced Acute Lung Injury in NF-kappaB-Luciferase Inducible Transgenic Mice. Antioxidants 2020, 9, 429. [Google Scholar] [CrossRef]
- Cox, R., Jr.; Phillips, O.; Fukumoto, J.; Fukumoto, I.; Tamarapu Parthasarathy, P.; Arias, S.; Cho, Y.; Lockey, R.F.; Kolliputi, N. Aspirin-Triggered Resolvin D1 Treatment Enhances Resolution of Hyperoxic Acute Lung Injury. Am. J. Respir. Cell Mol. Biol. 2015, 53, 422–435. [Google Scholar] [CrossRef] [PubMed]
- Schmitt-Sody, M.; Metz, P.; Gottschalk, O.; Zysk, S.; Birkenmaier, C.; Goebl, M.; von Schulze Pellengahr, C.; Veihelmann, A.; Jansson, V. Selective inhibition of platelets by the GPIIb/IIIa receptor antagonist Tirofiban reduces leukocyte-endothelial cell interaction in murine antigen-induced arthritis. Inflamm. Res. 2007, 56, 414–420. [Google Scholar] [CrossRef]
- Le, V.B.; Schneider, J.G.; Boergeling, Y.; Berri, F.; Ducatez, M.; Guerin, J.L.; Adrian, I.; Errazuriz-Cerda, E.; Frasquilho, S.; Antunes, L.; et al. Platelet activation and aggregation promote lung inflammation and influenza virus pathogenesis. Am. J. Respir. Crit. Care Med. 2015, 191, 804–819. [Google Scholar] [CrossRef]
- Sharron, M.; Hoptay, C.E.; Wiles, A.A.; Garvin, L.M.; Geha, M.; Benton, A.S.; Nagaraju, K.; Freishtat, R.J. Platelets induce apoptosis during sepsis in a contact-dependent manner that is inhibited by GPIIb/IIIa blockade. PLoS ONE 2012, 7, e41549. [Google Scholar] [CrossRef]
- Dorsam, R.T.; Kunapuli, S.P. Central role of the P2Y12 receptor in platelet activation. J. Clin. Investig. 2004, 113, 340–345. [Google Scholar] [CrossRef]
- Muhlestein, J.B. Effect of antiplatelet therapy on inflammatory markers in atherothrombotic patients. Thromb. Haemost. 2010, 103, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Harr, J.N.; Moore, E.E.; Wohlauer, M.V.; Fragoso, M.; Gamboni, F.; Liang, X.; Banerjee, A.; Silliman, C.C. Activated platelets in heparinized shed blood: The “second hit” of acute lung injury in trauma/hemorrhagic shock models. Shock 2011, 36, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Erlich, J.M.; Talmor, D.S.; Cartin-Ceba, R.; Gajic, O.; Kor, D.J. Prehospitalization antiplatelet therapy is associated with a reduced incidence of acute lung injury: A population-based cohort study. Chest 2011, 139, 289–295. [Google Scholar] [CrossRef] [PubMed]
- O’Neal, H.R., Jr.; Koyama, T.; Koehler, E.A.; Siew, E.; Curtis, B.R.; Fremont, R.D.; May, A.K.; Bernard, G.R.; Ware, L.B. Prehospital statin and aspirin use and the prevalence of severe sepsis and acute lung injury/acute respiratory distress syndrome. Crit. Care Med. 2011, 39, 1343–1350. [Google Scholar] [CrossRef]
- Kor, D.J.; Erlich, J.; Gong, M.N.; Malinchoc, M.; Carter, R.E.; Gajic, O.; Talmor, D.S.; Illness, U.S.C.; Injury Trials Group: Lung Injury Prevention Study Investigators. Association of prehospitalization aspirin therapy and acute lung injury: Results of a multicenter international observational study of at-risk patients. Crit. Care Med. 2011, 39, 2393–2400. [Google Scholar] [CrossRef]
- Chen, W.; Janz, D.R.; Bastarache, J.A.; May, A.K.; O’Neal, H.R., Jr.; Bernard, G.R.; Ware, L.B. Prehospital aspirin use is associated with reduced risk of acute respiratory distress syndrome in critically ill patients: A propensity-adjusted analysis. Crit. Care Med. 2015, 43, 801–807. [Google Scholar] [CrossRef]
- Boyle, A.J.; Di Gangi, S.; Hamid, U.I.; Mottram, L.J.; McNamee, L.; White, G.; Cross, L.J.; McNamee, J.J.; O’Kane, C.M.; McAuley, D.F. Aspirin therapy in patients with acute respiratory distress syndrome (ARDS) is associated with reduced intensive care unit mortality: A prospective analysis. Crit. Care 2015, 19, 109. [Google Scholar] [CrossRef] [PubMed]
- Kor, D.J.; Talmor, D.S.; Banner-Goodspeed, V.M.; Carter, R.E.; Hinds, R.; Park, P.K.; Gajic, O.; Gong, M.N.; Illness, U.S.C.; Injury Trials Group: Lung Injury Prevention with Aspirin Study Group. Lung Injury Prevention with Aspirin (LIPS-A): A protocol for a multicentre randomised clinical trial in medical patients at high risk of acute lung injury. BMJ Open 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Kor, D.J.; Carter, R.E.; Park, P.K.; Festic, E.; Banner-Goodspeed, V.M.; Hinds, R.; Talmor, D.; Gajic, O.; Ware, L.B.; Gong, M.N.; et al. Effect of Aspirin on Development of ARDS in At-Risk Patients Presenting to the Emergency Department: The LIPS-A Randomized Clinical Trial. JAMA 2016, 315, 2406–2414. [Google Scholar] [CrossRef] [PubMed]
- Toner, P.; McAuley, D.F.; Shyamsundar, M. Aspirin as a potential treatment in sepsis or acute respiratory distress syndrome. Crit. Care 2015, 19, 374. [Google Scholar] [CrossRef]
- Yu, H.; Ni, Y.N.; Liang, Z.A.; Liang, B.M.; Wang, Y. The effect of aspirin in preventing the acute respiratory distress syndrome/acute lung injury: A meta-analysis. Am. J. Emerg. Med. 2018, 36, 1486–1491. [Google Scholar] [CrossRef]
- Jin, W.; Chuang, C.C.; Jin, H.; Ye, J.; Kandaswamy, E.; Wang, L.; Zuo, L. Effects of Pre-Hospital Antiplatelet Therapy on the Incidence of ARDS. Respir. Care 2020, 65, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Mohananey, D.; Sethi, J.; Villablanca, P.A.; Ali, M.S.; Kumar, R.; Baruah, A.; Bhatia, N.; Agrawal, S.; Hussain, Z.; Shamoun, F.E.; et al. Effect of antiplatelet therapy on mortality and acute lung injury in critically ill patients: A systematic review and meta-analysis. Ann. Card. Anaesth. 2016, 19, 626–637. [Google Scholar] [PubMed]
- Sinha, P.; Churpek, M.M.; Calfee, C.S. Machine Learning Classifier Models Can Identify ARDS Phenotypes Using Readily Available Clinical Data. Am. J. Respir. Crit. Care Med. 2020, 201, A1014. [Google Scholar] [CrossRef]
- Sinha, P.; Delucchi, K.L.; McAuley, D.F.; O’Kane, C.M.; Matthay, M.A.; Calfee, C.S. Development and validation of parsimonious algorithms to classify acute respiratory distress syndrome phenotypes: A secondary analysis of randomised controlled trials. Lancet Respir. Med. 2020, 8, 247–257. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Authors | Study Country | Study Design | Patient’s Inclusion Criteria | Patient No. | Medications | Dosage (mg) | Medication Given Time Point | Outcome Variables | Results |
|---|---|---|---|---|---|---|---|---|---|
| Erlich el al. [103] | Minnesota, US | Retrospective cohort | at least one major risk factor for ALI & age > 18 years | 161 | Any kind of antiplatelet medications | at the time of hospital admission |
| Reduced incidence of ALI/ARDS (OR:0.37) | |
| O’Neal Jr et al. [104] | Tennessee, US | Prospective cohort | critically ill patients admitted to the medical or surgical ICU and age >18 years | 575 | Aspirin combined with statin | 81or 365 daily | Prehospital use |
| Two combined had the lowest rates of severe sepsis, ALI/ARDS and mortality. |
| Kor et al. [105] | 20 hospitals in US & 2 in Turkey | Prospective cohort | non-surgical patients admitted to the hospital with at least one major risk factor for ALI and age >18 years | 3855 | Aspirin | No mention | at the time of hospital admission |
| No significant effect |
| Chen et al. [106] | Tennessee, US | Prospective cohort | critically ill patients (age ≥ 40) admitted to the medical or surgical ICU | 1149 | Aspirin | 81 or 365 daily | Prehospital use |
| Decreased risk of ARDS (OR: 0.66) in the entire cohort also sepsis (OR: 0.60) |
| Boyle et al. [107] | United Kingdom | Prospective cohort | patients (>16 years-old) requiring invasive mechanical ventilation admitted to the medical or surgical ICU | 202 | Aspirin | No mention | both pre-admission and during ICU stay | ICU mortality | Reduction in ICU mortality (OR: 0.38) |
| Kor et al. [109] | 16 US academic hospitals | multicenter, double-blind, RCT | patients at risk for ARDS (Lung Injury Prediction Score ≥4) | 7673 | Aspirin | 325 loading dose followed by 81 | within 24 hours of emergency department presentation |
| No significant effect |
| Hamid et al. [45] | United Kingdom | double-blind, RCT | Healthy volunteers | 33 | Aspirin | 75 or 1200 for 7 days | seven days prior to LPS inhalation | Inflammatory biomarkers | Reduced pulmonary neutrophilia and tissue damaging, reduced BAL concentrations of TNF-α and pulmonary TXB2. |
| Authors | Enrolled Types of Studies | Enrolled Study Numbers | Medications | Outcome Variables | Results | Between-Study Heterogeneity | Conclusions |
|---|---|---|---|---|---|---|---|
| Panka et al. [44] | 1 RCT, 7 OS | 8 | Aspirin | the risk of ARDS | pooled OR was 0.59 | Q = 2.44, I2 = 68% | A beneficial role for Aspirin in ARDS prevention and treatment. |
| Yu et al. [111] | 1 RCT, 5 OS | 6 | Aspirin |
| pooled OR was 0.71 | I2 = 0%, P = 0.419 | Aspirin could provide protective effect on the rate of ARDS/ALI, but it could not reduce the mortality. |
| Jin et al. [112] | 7 OS | 7 | Antiplatelet agents (aspirin 75 to 300 mg daily), (clopidogrel, 75 mg daily), and ticlo- pidine. |
| pooled OR was 0.68 | I2 = 34% | Pre-hospital antiplatelet therapy was associated with a reduced rate of ARDS but had no effect on the mortality in the subjects at high risk |
| Mohanney et al. [113] | 17 OS | 17 | Aspirin and other antiplatelet agents |
| pooled OR was 0.67 | I2 = 25% | Antiplatelet therapy had an improved survival, decreased incidence of ARDS |
| Wang et al. [43] | 2 RCT, 7 OS | 9 | Aspirin and other antiplatelet agents |
| pool OR was 0.68 from OS; but no significant difference in RCT | I2 = 0.0%, p = 0.329 for RCT I2 = 68.4%, p = 0.004 for OS | Whether antiplatelet therapy is associated with a decreased incidence of ARDS in patients at a high risk of developing the condition remains unclear. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.-M.; Lu, H.-C.; Tung, Y.-T.; Chen, W. Antiplatelet Therapy for Acute Respiratory Distress Syndrome. Biomedicines 2020, 8, 230. https://doi.org/10.3390/biomedicines8070230
Chen C-M, Lu H-C, Tung Y-T, Chen W. Antiplatelet Therapy for Acute Respiratory Distress Syndrome. Biomedicines. 2020; 8(7):230. https://doi.org/10.3390/biomedicines8070230
Chicago/Turabian StyleChen, Chuan-Mu, Hsiao-Ching Lu, Yu-Tang Tung, and Wei Chen. 2020. "Antiplatelet Therapy for Acute Respiratory Distress Syndrome" Biomedicines 8, no. 7: 230. https://doi.org/10.3390/biomedicines8070230
APA StyleChen, C.-M., Lu, H.-C., Tung, Y.-T., & Chen, W. (2020). Antiplatelet Therapy for Acute Respiratory Distress Syndrome. Biomedicines, 8(7), 230. https://doi.org/10.3390/biomedicines8070230