Dietary Flavonoids in p53—Mediated Immune Dysfunctions Linking to Cancer Prevention

 ,
, 
 and
and
Abstract
:1. Introduction
1.1. p53 as a Transcription Factor
1.2. p53 as a Regulator of Metabolic Pathways
1.3. p53 as a Drug Target for Cancer
2. p53-Mediated Inflammatory Response and Immune Signaling
2.1. Cross-Talk between p53 and MHCI Pathway
2.2. Cross-Talk between p53 and Immune Checkpoints
2.3. Cross-Talk between p53 and TLRs
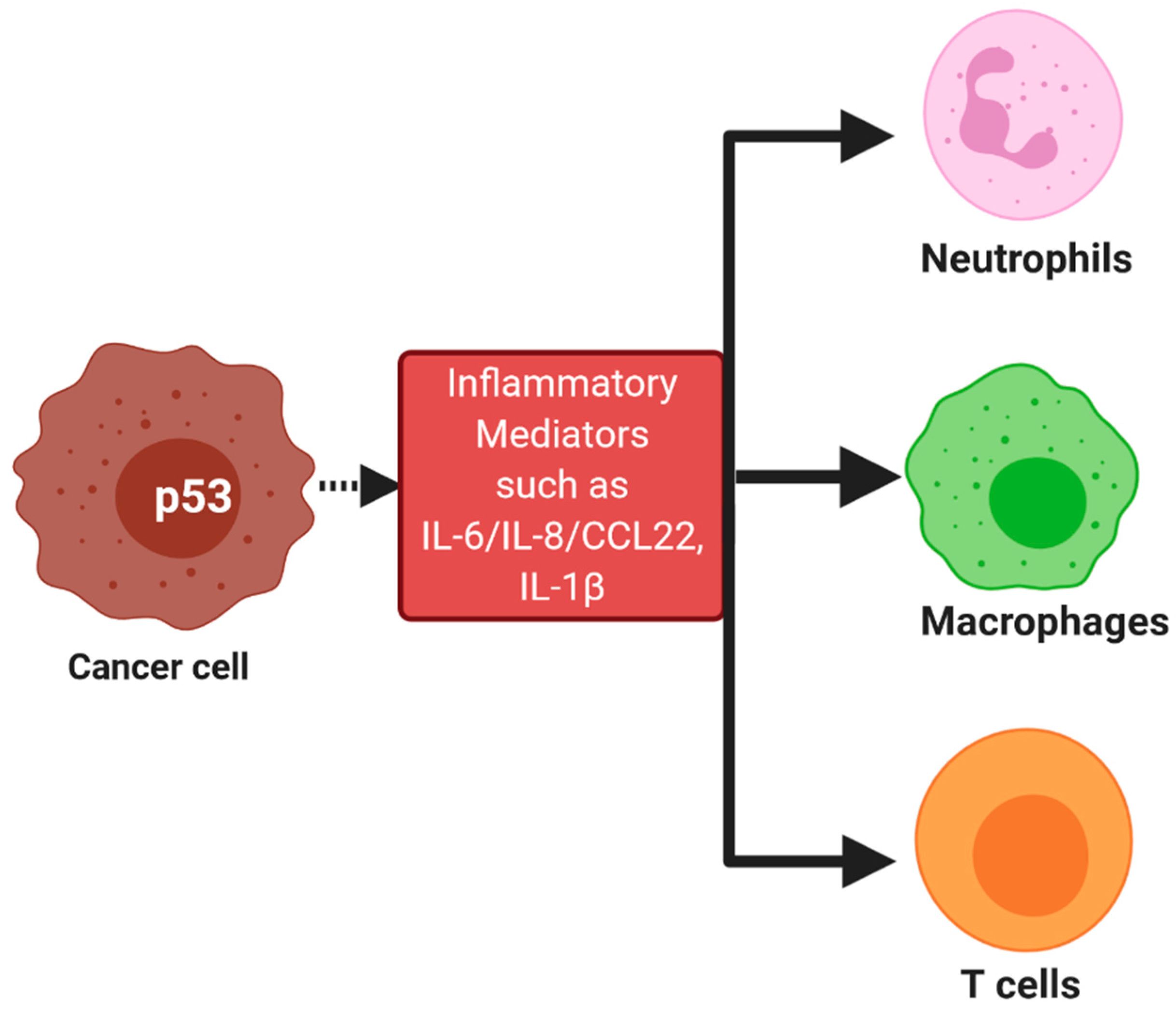
3. p53 in the Tumor Microenvironment and Cancer Immunotherapy
p53 as a Target for Cancer Immunotherapy
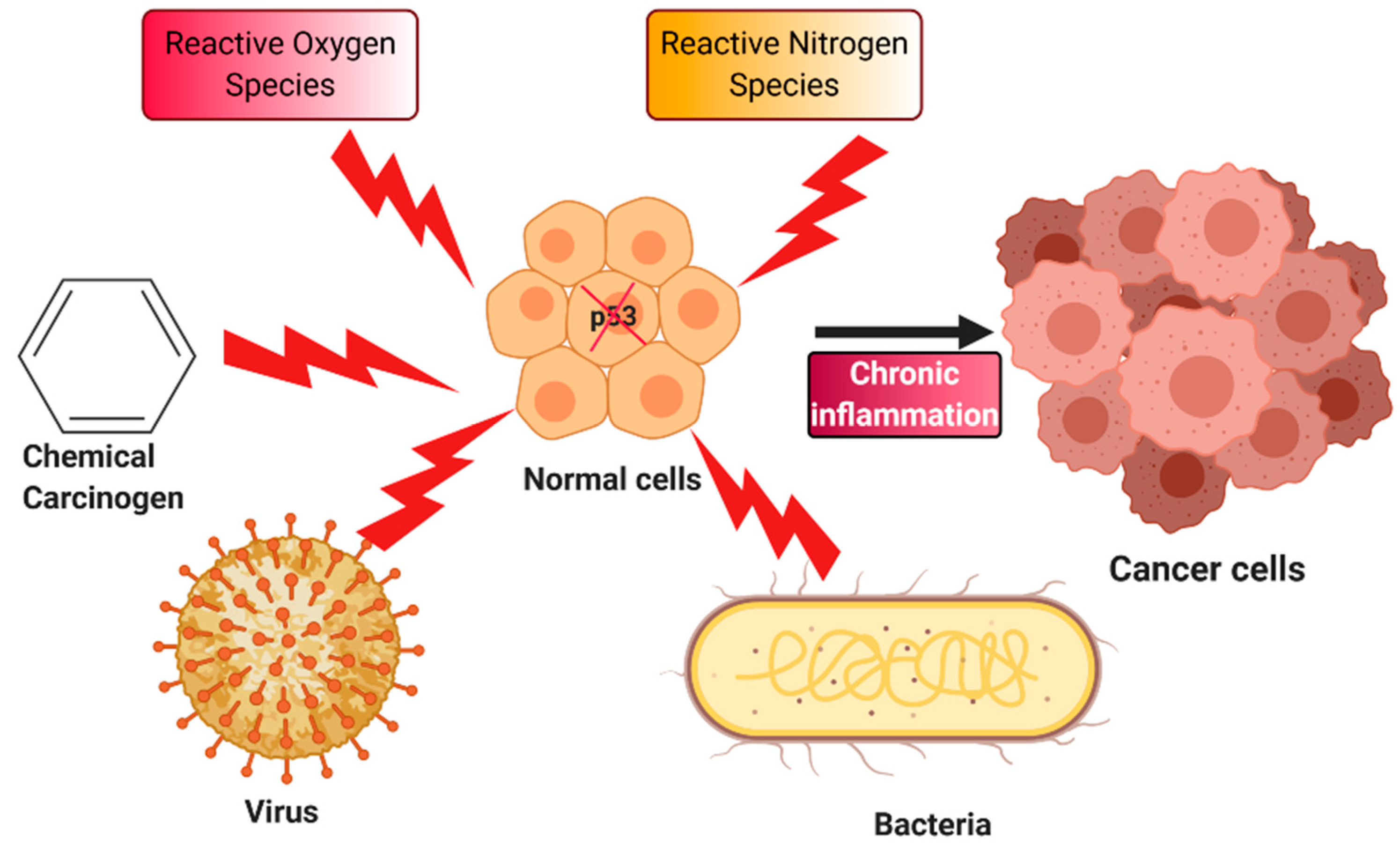
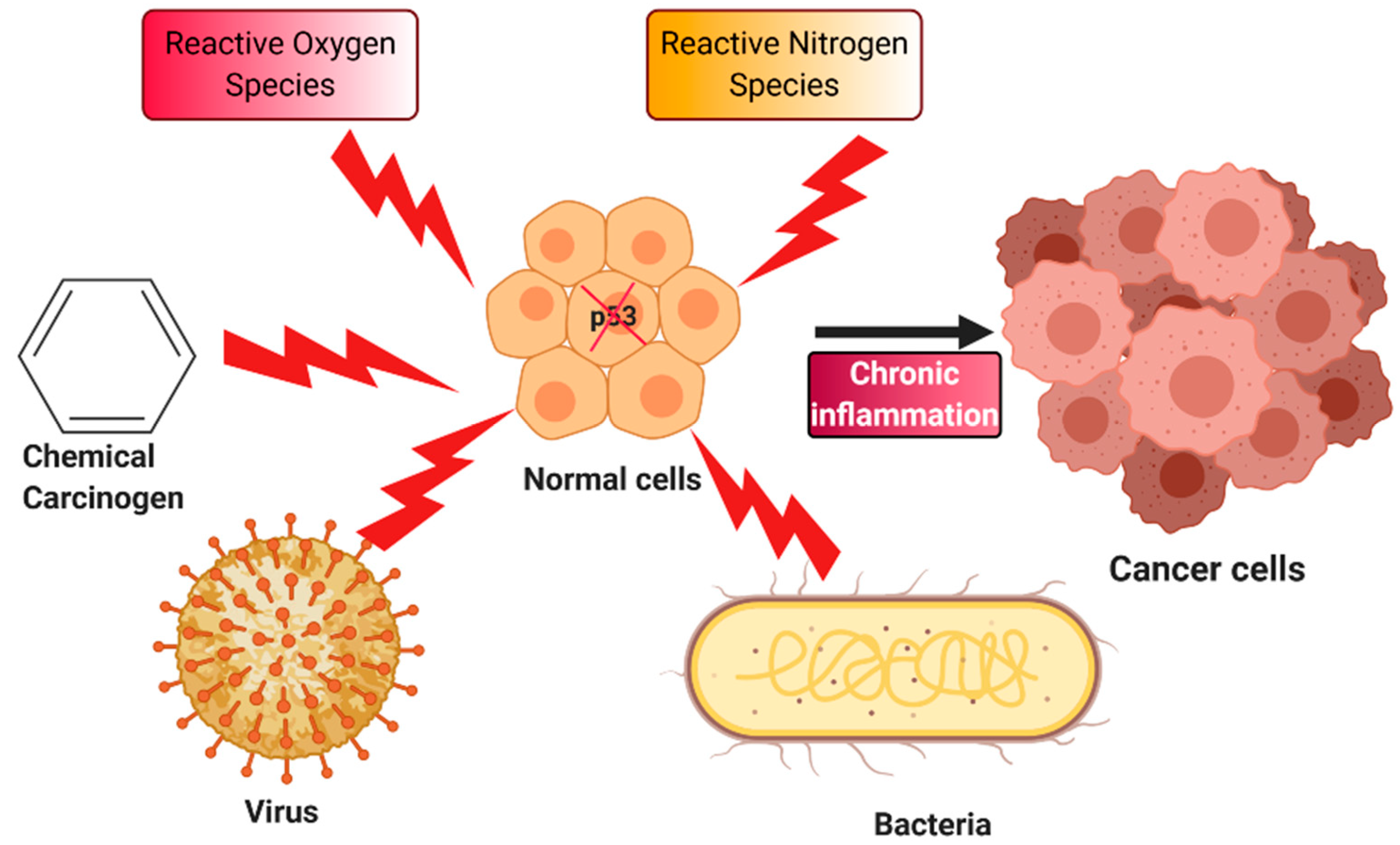
4. Chronic Inflammation and Cancer
NRF2 Pathway and Its Role in Cancer
5. Oxidative Stress, p53, and Inflammation
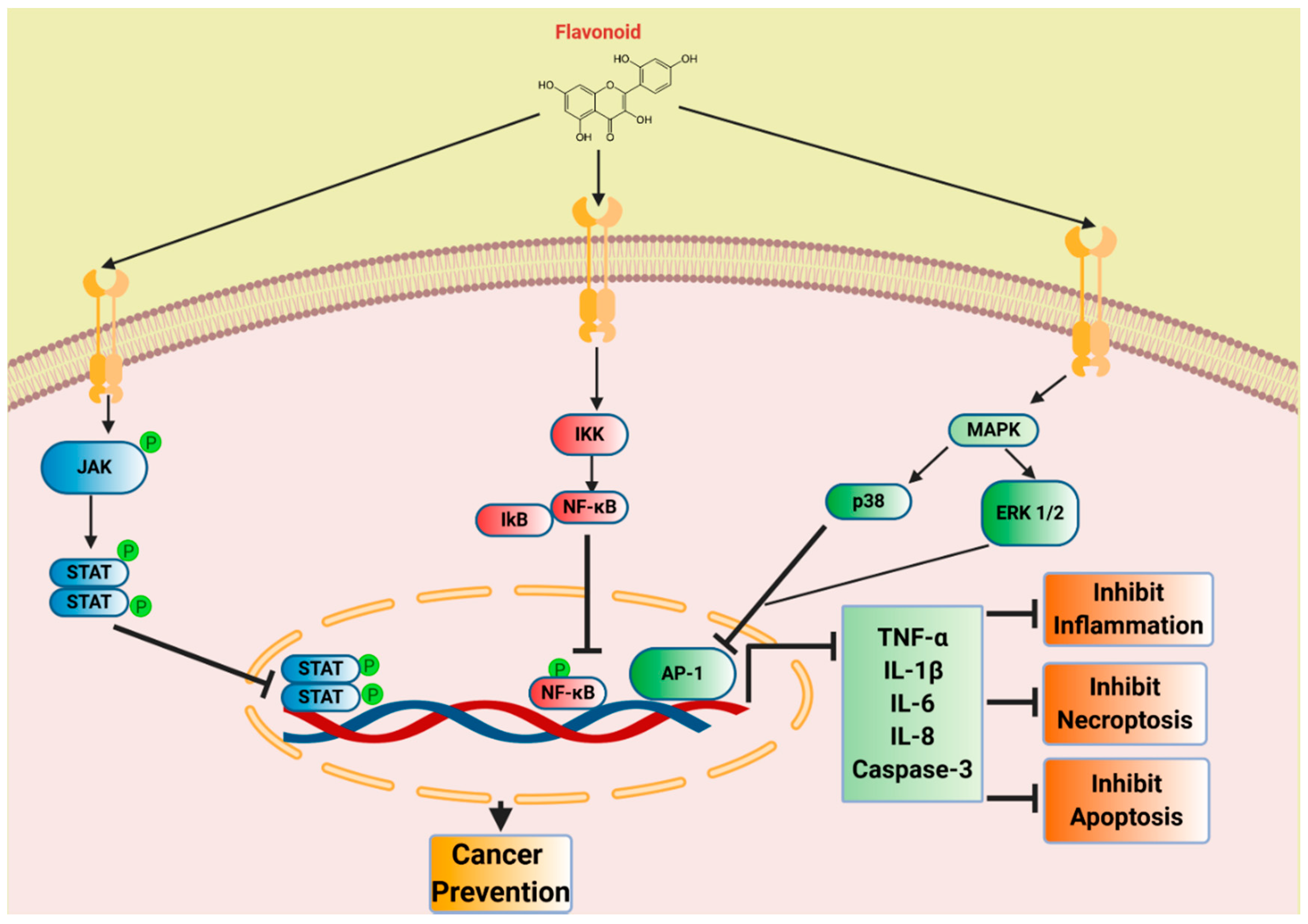
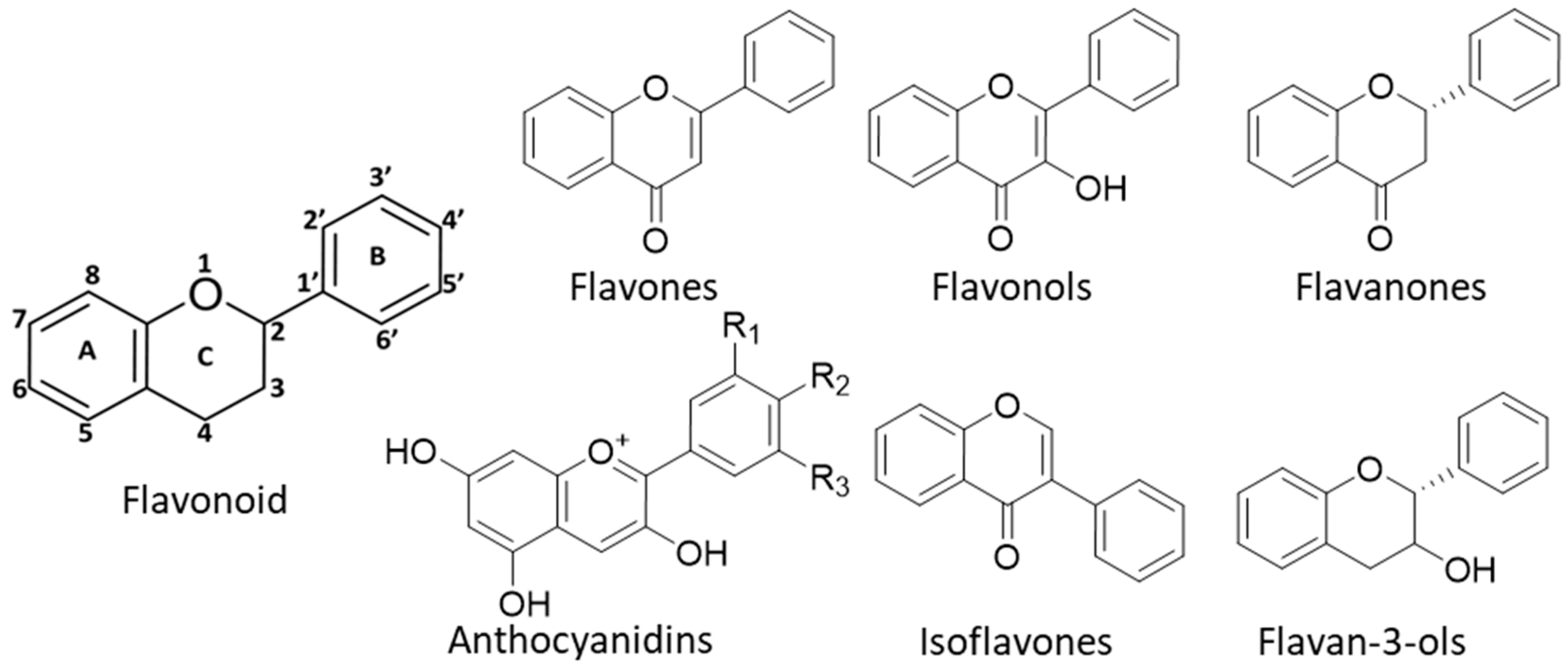
6. Plant Flavonoids as Therapeutic Agents
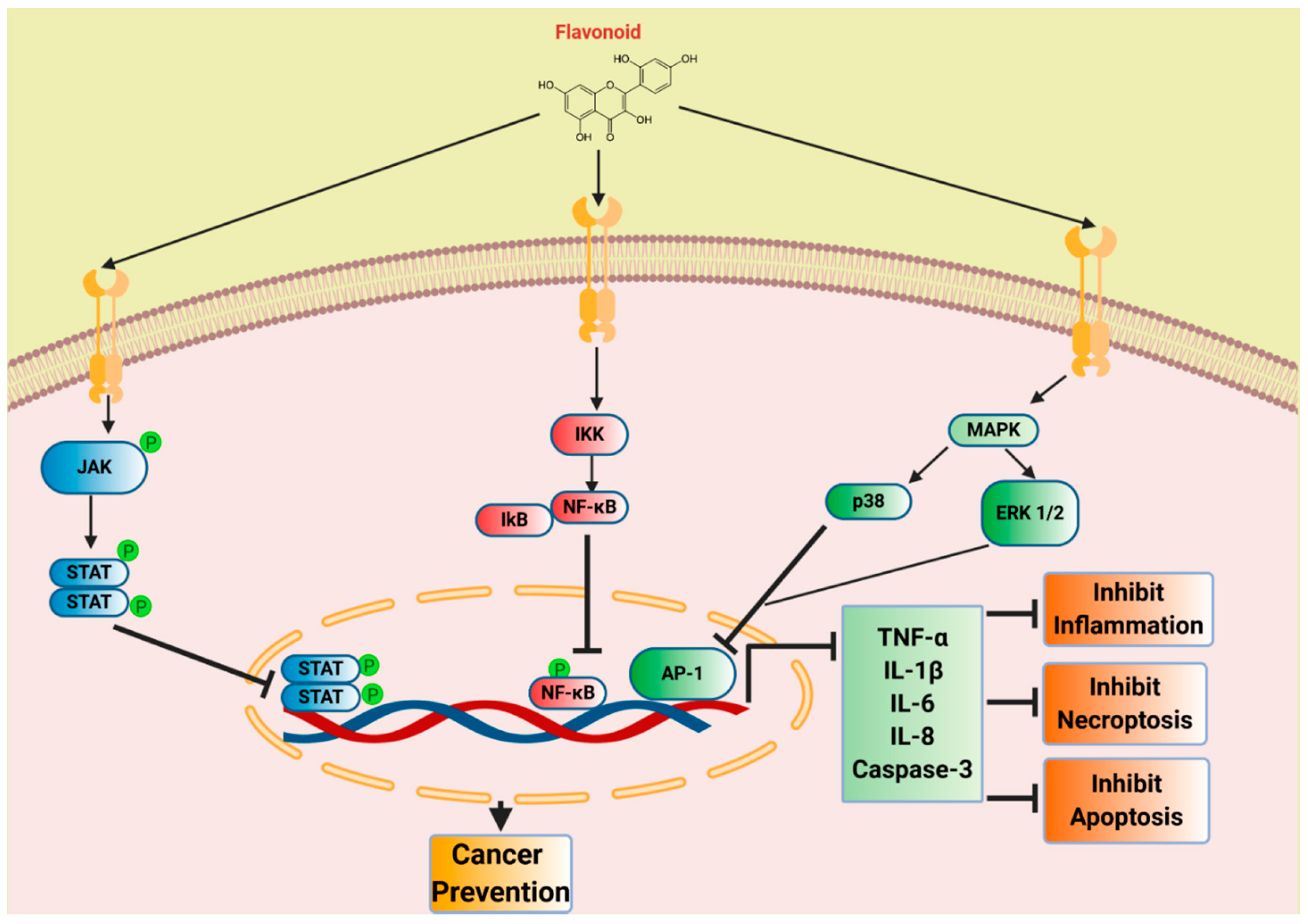
7. Flavonoids in Inflammation and Cancer Prevention
7.1. Quercetin
7.2. Luteolin
7.3. EGCG
7.4. Cyanidin
7.5. Daidzein
8. Flavonoids and Dietary Intake
9. Conclusions and Future Directions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AID | activation-induced cytidine deaminase |
| AOM | azoxymethane |
| AP1 | activator protein 1 |
| ARE | anti-oxidant response element |
| ATP | adenosine triphosphate |
| BSO | buthionine sulfoximine |
| Cag-A | cytotoxin-associated gene A |
| Cat | catalase |
| CIA | collagen-induced arthritis |
| CNS | central nervous system |
| CVD | cardiovascular diseases |
| DDR | DNA damage response |
| DMH | 1,2-dimethylhydrazine |
| EAE | experimental autoimmune encephalomyelitis |
| EGCG | epigallocatechin gallate |
| ERAP | endoplasmic reticulum aminopeptidase 1 |
| HCC | hepatocellular carcinoma |
| HFD | high fat diet |
| HIF1 | hypoxia inducible factor |
| IKK | IκB kinase |
| IPM | immune prognostic model |
| LFS | Li-Fraumeni syndrome |
| LOH | loss of heterozygosity |
| LPS | lipopolysaccharides |
| M-CSF | macrophage colony-stimulating factor |
| MHC | major histocompatibility complex |
| MMP | matrix metalloprotease |
| NF-κB | nuclear factor-κB |
| NRF2 | nuclear factor erythroid 2-related factor 2 |
| PAMPs | pathogen-associated molecular patterns |
| PPAR | peroxisome proliferator-activated receptors |
| PTEN | phosphatase and tensin homolog |
| ROS | Reactive oxygen species |
| SOCS1 | suppressors of cytokine signaling 1 |
| SOD | superoxide dismutase |
| STAT3 | signal transducer and activator of transcription 3 |
| TAM | tumor-associated macrophage |
| TAP1 | transporter associated with Antigen Processing 1 |
| TLR | toll-like receptor |
| TNBS | 2,4,6-trinitrobenzenesulfonic acid |
| UVB | ultraviolet B radiation |
| WT | wild type |
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Ben David, Y.; Prideaux, V.R.; Chow, V.; Benchimol, S.; Bernstein, A. Inactivation of the p53 oncogene by internal deletion or retroviral integration in erythroleukemic cell lines induced by Friend leukemia virus. Oncogene 1988, 3, 179–185. [Google Scholar]
- Wolf, D.; Rotter, V. Inactivation of p53 gene expression by an insertion of Moloney murine leukemia virus-like DNA sequences. Mol. Cell. Biol. 1984, 4, 1402–1410. [Google Scholar] [CrossRef] [Green Version]
- Baker, S.J.; Fearon, E.R.; Nigro, J.M.; Hamilton, S.R.; Preisinger, A.C.; Jessup, J.M.; Van Tuinen, P.; Ledbetter, D.H.; Barker, D.F.; Nakamura, Y.; et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science 1989, 244, 217–221. [Google Scholar] [CrossRef]
- Petitjean, A.; Mathe, E.; Kato, S.; Ishioka, C.; Tavtigian, S.V.; Hainaut, P.; Olivier, M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum. Mutat. 2007, 28, 622–629. [Google Scholar] [CrossRef]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef]
- Li, F.P.; Fraumeni, J.F. Rhabdomyosarcoma in children: Epidemiologic study and identification of a familial cancer syndrome. J. Natl. Cancer Inst. 1969, 43, 1365–1373. [Google Scholar] [PubMed]
- Li, F.P.; Fraumeni, J.F. Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann. Intern. Med. 1969, 71, 747–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.; Zou, Z.Q.; Pirollo, K.; Blattner, W.; Chang, E.H. Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li-Fraumeni syndrome. Nature 1990, 348, 747–749. [Google Scholar] [CrossRef] [PubMed]
- Malkin, D.; Li, F.P.; Strong, L.C.; Fraumeni, J.F.; Nelson, C.E.; Kim, D.H.; Kassel, J.; Gryka, M.A.; Bischoff, F.Z.; Tainsky, M.A.; et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990, 250, 1233–1238. [Google Scholar] [CrossRef]
- Fields, S.; Jang, S.K. Presence of a potent transcription activating sequence in the p53 protein. Science 1990, 249, 1046–1049. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, K.D.; Galbraith, M.D.; Andrysik, Z.; Espinosa, J.M. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 2018, 25, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Raycroft, L.; Wu, H.Y.; Lozano, G. Transcriptional activation by wild-type but not transforming mutants of the p53 anti-oncogene. Science 1990, 249, 1049–1051. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, M.; Arimura, Y.; Nozawa, K.; Kurumizaka, H. Linker DNA and histone contributions in nucleosome binding by p53. J. Biochem. 2020, mvva081. [Google Scholar] [CrossRef]
- Lang, Y.; Yu, C.; Tang, J.; Li, G.; Bai, R. Characterization of porcine p53 and its regulation by porcine Mdm2. Gene 2020, 748, 144699. [Google Scholar] [CrossRef]
- Popova, G.; Ladds, M.J.G.W.; Johansson, L.; Saleh, A.; Larsson, J.; Sandberg, L.; Sahlberg, S.H.; Qian, W.; Gullberg, H.; Garg, N.; et al. Optimization of Tetrahydroindazoles as Inhibitors of Human Dihydroorotate Dehydrogenase and Evaluation of Their Activity and In Vitro Metabolic Stability. J. Med. Chem. 2020, 63, 3915–3934. [Google Scholar] [CrossRef]
- Kurbegovic, A.; Trudel, M. The master regulators Myc and p53 cellular signaling and functions in polycystic kidney disease. Cell. Signal. 2020, 71, 109594. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Sheikh, M.S.; Huang, Y. ECRG2, a novel transcriptional target of p53, modulates cancer cell sensitivity to DNA damage. Cell Death Dis. 2020, 11, 543. [Google Scholar] [CrossRef] [PubMed]
- Puzio-Kuter, A.M. The Role of p53 in Metabolic Regulation. Genes Cancer 2011, 2, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Fontela, C.; Mandinova, A.; Aaronson, S.A.; Lee, S.W. Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat. Rev. Immunol. 2016, 16, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Kogan, S.; Carpizo, D. Pharmacological targeting of mutant p53. Transl. Cancer Res. 2016, 5, 698–706. [Google Scholar] [CrossRef]
- Raghavan, V.; Agrahari, M.; Gowda, D.K. Virtual screening of p53 mutants reveals Y220S as an additional rescue drug target for PhiKan083 with higher binding characteristics. Comput. Biol. Chem. 2019, 80, 398–408. [Google Scholar] [CrossRef]
- Li, X.; Zhang, X.-X.; Lin, Y.-X.; Xu, X.-M.; Li, L.; Yang, J.-B. Virtual Screening Based on Ensemble Docking Targeting Wild-Type p53 for Anticancer Drug Discovery. Chem. Biodivers. 2019, 16, e1900170. [Google Scholar] [CrossRef]
- Synnott, N.C.; Bauer, M.R.; Madden, S.; Murray, A.; Klinger, R.; O’Donovan, N.; O’Connor, D.; Gallagher, W.M.; Crown, J.; Fersht, A.R.; et al. Mutant p53 as a therapeutic target for the treatment of triple-negative breast cancer: Preclinical investigation with the anti-p53 drug, PK11007. Cancer Lett. 2018, 414, 99–106. [Google Scholar] [CrossRef]
- Multhoff, G.; Molls, M.; Radons, J. Chronic inflammation in cancer development. Front. Immunol. 2011, 2, 98. [Google Scholar] [CrossRef] [Green Version]
- Kruk, J.; Aboul-Enein, H.Y. Reactive Oxygen and Nitrogen Species in Carcinogenesis: Implications of Oxidative Stress on the Progression and Development of Several Cancer Types. Mini. Rev. Med. Chem. 2017, 17, 904–919. [Google Scholar] [CrossRef]
- Van Elsland, D.; Neefjes, J. Bacterial infections and cancer. EMBO Rep. 2018, 19, e46632. [Google Scholar] [CrossRef] [PubMed]
- Uehara, I.; Tanaka, N. Role of p53 in the Regulation of the Inflammatory Tumor Microenvironment and Tumor Suppression. Cancers 2018, 10, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuda, Y.; Okuda, M.; Bernard, C.C.A. Regulatory role of p53 in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2003, 135, 29–37. [Google Scholar] [CrossRef]
- Yamanishi, Y.; Boyle, D.L.; Pinkoski, M.J.; Mahboubi, A.; Lin, T.; Han, Z.; Zvaifler, N.J.; Green, D.R.; Firestein, G.S. Regulation of joint destruction and inflammation by p53 in collagen-induced arthritis. Am. J. Pathol. 2002, 160, 123–130. [Google Scholar] [CrossRef] [Green Version]
- Davidson, L.A.; Callaway, E.S.; Kim, E.; Weeks, B.R.; Fan, Y.-Y.; Allred, C.D.; Chapkin, R.S. Targeted Deletion of p53 in Lgr5-Expressing Intestinal Stem Cells Promotes Colon Tumorigenesis in a Preclinical Model of Colitis-Associated Cancer. Cancer Res. 2015, 75, 5392–5397. [Google Scholar] [CrossRef] [Green Version]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. Activated p53 induces NF-kappaB DNA binding but suppresses its transcriptional activation. Biochem. Biophys. Res. Commun. 2008, 372, 137–141. [Google Scholar] [CrossRef]
- Son, D.-S.; Kabir, S.M.; Dong, Y.-L.; Lee, E.; Adunyah, S.E. Inhibitory effect of tumor suppressor p53 on proinflammatory chemokine expression in ovarian cancer cells by reducing proteasomal degradation of IκB. PLoS ONE 2012, 7, e51116. [Google Scholar] [CrossRef]
- Blagih, J.; Buck, M.D.; Vousden, K.H. P53, cancer and the immune response. J. Cell. Sci. 2020, 133, jcs237453. [Google Scholar] [CrossRef] [Green Version]
- Keates, S.; Hitti, Y.S.; Upton, M.; Kelly, C.P. Helicobacter pylori infection activates NF-kappa B in gastric epithelial cells. Gastroenterology 1997, 113, 1099–1109. [Google Scholar] [CrossRef]
- Uemura, N.; Okamoto, S.; Yamamoto, S.; Matsumura, N.; Yamaguchi, S.; Yamakido, M.; Taniyama, K.; Sasaki, N.; Schlemper, R.J. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 2001, 345, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Marusawa, H.; Kinoshita, K.; Endo, Y.; Kou, T.; Morisawa, T.; Azuma, T.; Okazaki, I.-M.; Honjo, T.; Chiba, T. Helicobacter pylori infection triggers aberrant expression of activation-induced cytidine deaminase in gastric epithelium. Nat. Med. 2007, 13, 470–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butin-Israeli, V.; Bui, T.M.; Wiesolek, H.L.; Mascarenhas, L.; Lee, J.J.; Mehl, L.C.; Knutson, K.R.; Adam, S.A.; Goldman, R.D.; Beyder, A.; et al. Neutrophil-induced genomic instability impedes resolution of inflammation and wound healing. J. Clin. Invest. 2019, 129, 712–726. [Google Scholar] [CrossRef] [PubMed]
- Brazil, J.C.; Louis, N.A.; Parkos, C.A. The role of polymorphonuclear leukocyte trafficking in the perpetuation of inflammation during inflammatory bowel disease. Inflamm. Bowel Dis. 2013, 19, 1556–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Álvaro-Benito, M.; Stolzenberg, S.; Noé, F.; Freund, C. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front. Immunol. 2017, 8, 292. [Google Scholar] [CrossRef] [Green Version]
- Griffioen, M.; Steegenga, W.T.; Ouwerkerk, I.J.; Peltenburg, L.T.; Jochemsen, A.G.; Schrier, P.I. Repression of the minimal HLA-B promoter by c-myc and p53 occurs through independent mechanisms. Mol. Immunol. 1998, 35, 829–835. [Google Scholar] [CrossRef]
- Wang, B.; Niu, D.; Lai, L.; Ren, E.C. P53 increases MHC class I expression by upregulating the endoplasmic reticulum aminopeptidase ERAP1. Nat. Commun. 2013, 4, 2359. [Google Scholar] [CrossRef]
- Zhu, K.; Wang, J.; Zhu, J.; Jiang, J.; Shou, J.; Chen, X. P53 induces TAP1 and enhances the transport of MHC class I peptides. Oncogene 1999, 18, 7740–7747. [Google Scholar] [CrossRef] [Green Version]
- Garancher, A.; Suzuki, H.; Haricharan, S.; Chau, L.Q.; Masihi, M.B.; Rusert, J.M.; Norris, P.S.; Carrette, F.; Romero, M.M.; Morrissy, S.A.; et al. Tumor necrosis factor overcomes immune evasion in p53-mutant medulloblastoma. Nat. Neurosci. 2020, 23, 842–853. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [Green Version]
- Cortez, M.A.; Ivan, C.; Valdecanas, D.; Wang, X.; Peltier, H.J.; Ye, Y.; Araujo, L.; Carbone, D.P.; Shilo, K.; Giri, D.K.; et al. PDL1 Regulation by p53 via miR-34. J. Natl. Cancer Inst. 2016, 108, djv303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamoto, K.; Hatae, R.; Honjo, T. Current issues and perspectives in PD-1 blockade cancer immunotherapy. Int. J. Clin. Oncol. 2020, 25, 790–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, L.; Cai, S.-Y.; Shao, J.-Z.; Chen, J. Toll-Like Receptors, Associated Biological Roles, and Signaling Networks in Non-Mammals. Front. Immunol. 2018, 9, 1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urban-Wojciuk, Z.; Khan, M.M.; Oyler, B.L.; Fåhraeus, R.; Marek-Trzonkowska, N.; Nita-Lazar, A.; Hupp, T.R.; Goodlett, D.R. The Role of TLRs in Anti-cancer Immunity and Tumor Rejection. Front. Immunol. 2019, 10, 2388. [Google Scholar] [CrossRef]
- Menendez, D.; Lowe, J.M.; Snipe, J.; Resnick, M.A. Ligand dependent restoration of human TLR3 signaling and death in p53 mutant cells. Oncotarget 2016, 7, 61630–61642. [Google Scholar] [CrossRef] [Green Version]
- Shatz, M.; Shats, I.; Menendez, D.; Resnick, M.A. P53 amplifies Toll-like receptor 5 response in human primary and cancer cells through interaction with multiple signal transduction pathways. Oncotarget 2015, 6, 16963–16980. [Google Scholar] [CrossRef] [Green Version]
- Haricharan, S.; Brown, P. TLR4 has a TP53-dependent dual role in regulating breast cancer cell growth. Proc. Natl. Acad. Sci. USA 2015, 112, E3216–E3225. [Google Scholar] [CrossRef] [Green Version]
- Menendez, D.; Snipe, J.; Marzec, J.; Innes, C.L.; Polack, F.P.; Caballero, M.T.; Schurman, S.H.; Kleeberger, S.R.; Resnick, M.A. P53-responsive TLR8 SNP enhances human innate immune response to respiratory syncytial virus. J. Clin. Invest. 2019, 129, 4875–4884. [Google Scholar] [CrossRef] [Green Version]
- Rusanen, P.; Marttila, E.; Uittamo, J.; Hagström, J.; Salo, T.; Rautemaa-Richardson, R. TLR1-10, NF-κB and p53 expression is increased in oral lichenoid disease. PLoS ONE 2017, 12, e0181361. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xia, G.; Zhang, Y.; Liu, J.; Liu, X.; Li, W.; Lv, Y.; Wei, S.; Liu, J.; Quan, J. Palmitate induces VSMC apoptosis via toll like receptor (TLR)4/ROS/p53 pathway. Atherosclerosis 2017, 263, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-W.; Chang, S.-H.; Mu, S.-W.; Jiang, H.-Y.; Wang, S.-T.; Kao, J.-K.; Huang, J.-L.; Wu, C.-Y.; Chen, Y.-J.; Shieh, J.-J. Imiquimod activates p53-dependent apoptosis in a human basal cell carcinoma cell line. J. Dermatol. Sci. 2016, 81, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Wellenstein, M.D.; Coffelt, S.B.; Duits, D.E.M.; Van Miltenburg, M.H.; Slagter, M.; De Rink, I.; Henneman, L.; Kas, S.M.; Prekovic, S.; Hau, C.-S.; et al. Loss of p53 triggers WNT-dependent systemic inflammation to drive breast cancer metastasis. Nature 2019, 572, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Bezzi, M.; Seitzer, N.; Ishikawa, T.; Reschke, M.; Chen, M.; Wang, G.; Mitchell, C.; Ng, C.; Katon, J.; Lunardi, A.; et al. Diverse genetic-driven immune landscapes dictate tumor progression through distinct mechanisms. Nat. Med. 2018, 24, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Walton, J.; Blagih, J.; Ennis, D.; Leung, E.; Dowson, S.; Farquharson, M.; Tookman, L.A.; Orange, C.; Athineos, D.; Mason, S.; et al. CRISPR/Cas9-Mediated Trp53 and Brca2 Knockout to Generate Improved Murine Models of Ovarian High-Grade Serous Carcinoma. Cancer Res. 2016, 76, 6118–6129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruddell, A.; Kelly-Spratt, K.S.; Furuya, M.; Parghi, S.S.; Kemp, C.J. P19/Arf and p53 suppress sentinel lymph node lymphangiogenesis and carcinoma metastasis. Oncogene 2008, 27, 3145–3155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blagih, J.; Zani, F.; Chakravarty, P.; Hennequart, M.; Pilley, S.; Hobor, S.; Hock, A.K.; Walton, J.B.; Morton, J.P.; Gronroos, E.; et al. Cancer-Specific Loss of p53 Leads to a Modulation of Myeloid and T Cell Responses. Cell Rep. 2020, 30, 481–496. [Google Scholar] [CrossRef]
- Li, D.; Bentley, C.; Anderson, A.; Wiblin, S.; Cleary, K.L.S.; Koustoulidou, S.; Hassanali, T.; Yates, J.; Greig, J.; Nordkamp, M.O.; et al. Development of a T-cell Receptor Mimic Antibody against Wild-Type p53 for Cancer Immunotherapy. Cancer Res. 2017, 77, 2699–2711. [Google Scholar] [CrossRef] [Green Version]
- Low, L.; Goh, A.; Koh, J.; Lim, S.; Wang, C.-I. Targeting mutant p53-expressing tumours with a T cell receptor-like antibody specific for a wild-type antigen. Nat. Commun. 2019, 10, 5382. [Google Scholar] [CrossRef] [Green Version]
- Kortylewski, M.; Yu, H. Stat3 as a Potential Target for Cancer Immunotherapy. J. Immunother. 2007, 30, 131–139. [Google Scholar] [CrossRef]
- Wörmann, S.M.; Song, L.; Ai, J.; Diakopoulos, K.N.; Kurkowski, M.U.; Görgülü, K.; Ruess, D.; Campbell, A.; Doglioni, C.; Jodrell, D.; et al. Loss of P53 Function Activates JAK2-STAT3 Signaling to Promote Pancreatic Tumor Growth, Stroma Modification, and Gemcitabine Resistance in Mice and Is Associated With Patient Survival. Gastroenterology 2016, 151, 180–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak, D.G.; Cho, H.; Herzka, T.; Watrud, K.; DeMarco, D.V.; Wang, V.M.Y.; Senturk, S.; Fellmann, C.; Ding, D.; Beinortas, T.; et al. MYC Drives Pten/Trp53-Deficient Proliferation and Metastasis due to IL6 Secretion and AKT Suppression via PHLPP2. Cancer Discov. 2015, 5, 636–651. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, V.; Mallette, F.A.; Deschênes-Simard, X.; Ramanathan, S.; Gagnon, J.; Moores, A.; Ilangumaran, S.; Ferbeyre, G. SOCS1 links cytokine signaling to p53 and senescence. Mol. Cell 2009, 36, 754–767. [Google Scholar] [CrossRef] [PubMed]
- Cooks, T.; Pateras, I.S.; Tarcic, O.; Solomon, H.; Schetter, A.J.; Wilder, S.; Lozano, G.; Pikarsky, E.; Forshew, T.; Rosenfeld, N.; et al. Mutant p53 prolongs NF-κB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell 2013, 23, 634–646. [Google Scholar] [CrossRef] [Green Version]
- Rahnamoun, H.; Lu, H.; Duttke, S.H.; Benner, C.; Glass, C.K.; Lauberth, S.M. Mutant p53 shapes the enhancer landscape of cancer cells in response to chronic immune signaling. Nat. Commun. 2017, 8, 754. [Google Scholar] [CrossRef]
- Ubertini, V.; Norelli, G.; D’Arcangelo, D.; Gurtner, A.; Cesareo, E.; Baldari, S.; Gentileschi, M.P.; Piaggio, G.; Nisticò, P.; Soddu, S.; et al. Mutant p53 gains new function in promoting inflammatory signals by repression of the secreted interleukin-1 receptor antagonist. Oncogene 2015, 34, 2493–2504. [Google Scholar] [CrossRef]
- Headland, S.E.; Norling, L.V. The resolution of inflammation: Principles and challenges. Semin. Immunol. 2015, 27, 149–160. [Google Scholar] [CrossRef]
- Singh, N.; Baby, D.; Rajguru, J.; Patil, P.; Thakkannavar, S.; Pujari, V. Inflammation and cancer. Ann. Afr. Med. 2019, 18, 121. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.; Ding, A. Nonresolving Inflammation. Cell 2010, 140, 871–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamp, D.W.; Shacter, E.; Weitzman, S.A. Chronic inflammation and cancer: The role of the mitochondria. Oncology (Williston Park, N.Y.) 2011, 25, 400–410, 413. [Google Scholar]
- Maeda, H.; Akaike, T. Nitric oxide and oxygen radicals in infection, inflammation, and cancer. Biochem. Mosc. 1998, 63, 854–865. [Google Scholar]
- Kawanishi, S.; Ohnishi, S.; Ma, N.; Hiraku, Y.; Oikawa, S.; Murata, M. Nitrative and oxidative DNA damage in infection-related carcinogenesis in relation to cancer stem cells. Genes Environ. 2016, 38, 26. [Google Scholar] [CrossRef] [PubMed]
- Souici, A.-C. Transition mutation in codon 248 of the p53 tumor suppressor gene induced by reactive oxygen species and a nitric oxide-releasing compound. Carcinogenesis 2000, 21, 281–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Varki, A.; Schnaar, R.L.; Crocker, P.R. I-Type Lectins. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015. [Google Scholar]
- Siddiqui, S.S.; Matar, R.; Merheb, M.; Hodeify, R.; Vazhappilly, C.G.; Marton, J.; Shamsuddin, S.A.; Al Zouabi, H. Siglecs in Brain Function and Neurological Disorders. Cells 2019, 8, 1125. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, S.; Schwarz, F.; Springer, S.; Khedri, Z.; Yu, H.; Deng, L.; Verhagen, A.; Naito-Matsui, Y.; Jiang, W.; Kim, D.; et al. Studies on the Detection, Expression, Glycosylation, Dimerization, and Ligand Binding Properties of Mouse Siglec-E. J. Biol. Chem. 2017, 292, 1029–1037. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, S.S.; Springer, S.A.; Verhagen, A.; Sundaramurthy, V.; Alisson-Silva, F.; Jiang, W.; Ghosh, P.; Varki, A. The Alzheimer’s disease-protective CD33 splice variant mediates adaptive loss of function via diversion to an intracellular pool. J. Biol. Chem. 2017, 292, 15312–15320. [Google Scholar] [CrossRef] [Green Version]
- Läubli, H.; Pearce, O.M.T.; Schwarz, F.; Siddiqui, S.S.; Deng, L.; Stanczak, M.A.; Deng, L.; Verhagen, A.; Secrest, P.; Lusk, C.; et al. Engagement of myelomonocytic Siglecs by tumor-associated ligands modulates the innate immune response to cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 14211–14216. [Google Scholar] [CrossRef] [Green Version]
- Läubli, H.; Alisson-Silva, F.; Stanczak, M.A.; Siddiqui, S.S.; Deng, L.; Verhagen, A.; Varki, N.; Varki, A. Lectin galactoside-binding soluble 3 binding protein (LGALS3BP) is a tumor-associated immunomodulatory ligand for CD33-related Siglecs. J. Biol. Chem. 2014, 289, 33481–33491. [Google Scholar] [CrossRef] [Green Version]
- Stanczak, M.A.; Siddiqui, S.S.; Trefny, M.P.; Thommen, D.S.; Boligan, K.F.; Von Gunten, S.; Tzankov, A.; Tietze, L.; Lardinois, D.; Heinzelmann-Schwarz, V.; et al. Self-associated molecular patterns mediate cancer immune evasion by engaging Siglecs on T cells. J. Clin. Invest. 2018, 128, 4912–4923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchiyama, S.; Sun, J.; Fukahori, K.; Ando, N.; Wu, M.; Schwarz, F.; Siddiqui, S.S.; Varki, A.; Marth, J.D.; Nizet, V. Dual actions of group B Streptococcus capsular sialic acid provide resistance to platelet-mediated antimicrobial killing. Proc. Natl. Acad. Sci. USA 2019, 116, 7465–7470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, S.; Siddiqui, S.S.; Khan, N.; Verhagen, A.; Jiang, W.; Springer, S.; Ghosh, P.; Varki, A. Controversies about the subcellular localization and mechanisms of action of the Alzheimer’s disease-protective CD33 splice variant. Acta Neuropathol. 2019, 138, 671–672. [Google Scholar] [CrossRef]
- Schwarz, F.; Landig, C.S.; Siddiqui, S.; Secundino, I.; Olson, J.; Varki, N.; Nizet, V.; Varki, A. Paired Siglec receptors generate opposite inflammatory responses to a human-specific pathogen. EMBO J. 2017, 36, 751–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Vega, M.R.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef]
- Ahmed, S.M.U.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. et Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef]
- Tu, W.; Wang, H.; Li, S.; Liu, Q.; Sha, H. The Anti-Inflammatory and Anti-Oxidant Mechanisms of the Keap1/Nrf2/ARE Signaling Pathway in Chronic Diseases. Aging Dis. 2019, 10, 637. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Sun, Z.; Wang, X.-J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct Interaction between Nrf2 and p21Cip1/WAF1 Upregulates the Nrf2-Mediated Antioxidant Response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Zimta, A.-A.; Cenariu, D.; Irimie, A.; Magdo, L.; Nabavi, S.M.; Atanasov, A.G.; Berindan-Neagoe, I. The Role of Nrf2 Activity in Cancer Development and Progression. Cancers 2019, 11, 1755. [Google Scholar] [CrossRef] [Green Version]
- Menegon, S.; Columbano, A.; Giordano, S. The Dual Roles of NRF2 in Cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef] [Green Version]
- Hussain, T.; Tan, B.; Yin, Y.; Blachier, F.; Tossou, M.C.B.; Rahu, N. Oxidative Stress and Inflammation: What Polyphenols Can Do for Us? Oxid. Med. Cell. Longev. 2016, 2016, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. et Biophys. Acta (BBA) Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Chen, X.; Song, X.; Muhammad, A.; Jia, R.; Zou, Y.; Yin, L.; Li, L.; He, C.; Ye, G.; et al. The immune-adjuvant activity and the mechanism of resveratrol on pseudorabies virus vaccine in a mouse model. Int. Immunopharmacol. 2019, 76, 105876. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Jia, H.; Xu, Q.; Zhao, C.; Xu, C. Lycopene alleviates H2O2-induced oxidative stress, inflammation and apoptosis in bovine mammary epithelial cells via the NFE2L2 signaling pathway. Food Funct. 2019, 10, 6276–6285. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, A.; Ahmad, S.F.; Al-Harbi, N.O.; Attia, S.M.; Alshammari, M.A.; Alzahrani, K.S.; Bakheet, S.A. Increased oxidative stress in the cerebellum and peripheral immune cells leads to exaggerated autism-like repetitive behavior due to deficiency of antioxidant response in BTBR T + tf/J mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 89, 245–253. [Google Scholar] [CrossRef]
- Solleiro-Villavicencio, H.; Rivas-Arancibia, S. Effect of Chronic Oxidative Stress on Neuroinflammatory Response Mediated by CD4+T Cells in Neurodegenerative Diseases. Front. Cell. Neurosci. 2018, 12, 114. [Google Scholar] [CrossRef] [Green Version]
- Karin, M. NF-B as a Critical Link Between Inflammation and Cancer. Cold Spring Harb. Perspect. Biol. 2009, 1, a000141. [Google Scholar] [CrossRef]
- Plewka, D.; Plewka, A.; Miskiewicz, A.; Morek, M.; Bogunia, E. Nuclear factor-kappa B as potential therapeutic target in human colon cancer. J. Cancer Res. Ther. 2018, 14, 516. [Google Scholar] [CrossRef]
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef] [PubMed]
- Cordani, M.; Butera, G.; Pacchiana, R.; Masetto, F.; Mullappilly, N.; Riganti, C.; Donadelli, M. Mutant p53-Associated Molecular Mechanisms of ROS Regulation in Cancer Cells. Biomolecules 2020, 10, 361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Liu, K.; Shi, Y.; Shao, C. The tango of ROS and p53 in tissue stem cells. Cell Death Differ. 2018, 25, 639–641. [Google Scholar] [CrossRef] [Green Version]
- Olivos, D.; Mayo, L. Emerging Non-Canonical Functions and Regulation by p53: P53 and Stemness. Int. J. Mol. Sci. 2016, 17, 1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernando, W.; Rupasinghe, H.P.V.; Hoskin, D.W. Dietary phytochemicals with anti-oxidant and pro-oxidant activities: A double-edged sword in relation to adjuvant chemotherapy and radiotherapy? Cancer Lett. 2019, 452, 168–177. [Google Scholar] [CrossRef]
- Wu, Y.; Lee, S.; Bobadilla, S.; Duan, S.Z.; Liu, X. High glucose-induced p53 phosphorylation contributes to impairment of endothelial antioxidant system. Biochim. et Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 2355–2362. [Google Scholar] [CrossRef]
- D’Angelo, S.; Martino, E.; Ilisso, C.P.; Bagarolo, M.L.; Porcelli, M.; Cacciapuoti, G. Pro-oxidant and pro-apoptotic activity of polyphenol extract from Annurca apple and its underlying mechanisms in human breast cancer cells. Int. J. Oncol. 2017, 51, 939–948. [Google Scholar] [CrossRef] [Green Version]
- Sablina, A.A.; Budanov, A.V.; Ilyinskaya, G.V.; Agapova, L.S.; Kravchenko, J.E.; Chumakov, P.M. The antioxidant function of the p53 tumor suppressor. Nat. Med. 2005, 11, 1306–1313. [Google Scholar] [CrossRef] [Green Version]
- Vazhappilly, C.G.; Ansari, S.A.; Al-Jaleeli, R.; Al-Azawi, A.M.; Ramadan, W.S.; Menon, V.; Hodeify, R.; Siddiqui, S.S.; Merheb, M.; Matar, R.; et al. Role of flavonoids in thrombotic, cardiovascular, and inflammatory diseases. Inflammopharmacol 2019, 27, 863–869. [Google Scholar] [CrossRef]
- Gunathilake, K.; Ranaweera, K.; Rupasinghe, H. In Vitro Anti-Inflammatory Properties of Selected Green Leafy Vegetables. Biomedicines 2018, 6, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, V.C.; Vijesh, V.V.; Amararathna, D.I.M.; Lakshmi, C.A.; Anbarasu, K.; Kumar, D.R.N.; Ethiraj, K.R.; Kumar, R.A.; Rupasinghe, H.P.V. Mechanism of Action of Flavonoids in Prevention of Inflammation-Associated Skin Cancer. CMC 2016, 23, 3697–3716. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, M.; Andruniów, T.; Sroka, Z. Flavones’ and Flavonols’ Antiradical Structure–Activity Relationship—A Quantum Chemical Study. Antioxidants 2020, 9, 461. [Google Scholar] [CrossRef] [PubMed]
- Sarian, M.N.; Ahmed, Q.U.; Mat So’ad, S.Z.; Alhassan, A.M.; Murugesu, S.; Perumal, V.; Syed Mohamad, S.N.A.; Khatib, A.; Latip, J. Antioxidant and Antidiabetic Effects of Flavonoids: A Structure-Activity Relationship Based Study. BioMed Res. Int. 2017, 2017, 8386065. [Google Scholar] [CrossRef] [PubMed]
- Sordon, S.; Popłoński, J.; Milczarek, M.; Stachowicz, M.; Tronina, T.; Kucharska, A.Z.; Wietrzyk, J.; Huszcza, E. Structure–Antioxidant–Antiproliferative Activity Relationships of Natural C7 and C7–C8 Hydroxylated Flavones and Flavanones. Antioxidants 2019, 8, 210. [Google Scholar] [CrossRef] [Green Version]
- Murakami, A.; Ashida, H.; Terao, J. Multitargeted cancer prevention by quercetin. Cancer Lett. 2008, 269, 315–325. [Google Scholar] [CrossRef]
- Zhu, Q.; Liu, M.; He, Y.; Yang, B. Quercetin protect cigarette smoke extracts induced inflammation and apoptosis in RPE cells. Artif. Cells Nanomed. Biotechnol. 2019, 47, 2010–2015. [Google Scholar] [CrossRef] [Green Version]
- Lisek, K.; Campaner, E.; Ciani, Y.; Walerych, D.; Del Sal, G. Mutant p53 tunes the NRF2-dependent antioxidant response to support survival of cancer cells. Oncotarget 2018, 9, 20508–20523. [Google Scholar] [CrossRef] [Green Version]
- Han, C.; Sun, T.; Xv, G.; Wang, S.; Gu, J.; Liu, C. Berberine ameliorates CCl4-induced liver injury in rats through regulation of the Nrf2-Keap1-ARE and p53 signaling pathways. Mol. Med. Rep. 2019, 20, 3095–3102. [Google Scholar] [CrossRef] [Green Version]
- Das, L.; Vinayak, M. Long Term Effect of Curcumin in Restoration of Tumour Suppressor p53 and Phase-II Antioxidant Enzymes via Activation of Nrf2 Signalling and Modulation of Inflammation in Prevention of Cancer. PLoS ONE 2015, 10, e0124000. [Google Scholar] [CrossRef]
- Zhang, S.; Qi, Y.; Xu, Y.; Han, X.; Peng, J.; Liu, K.; Sun, C.K. Protective effect of flavonoid-rich extract from Rosa laevigata Michx on cerebral ischemia–reperfusion injury through suppression of apoptosis and inflammation. Neurochem. Int. 2013, 63, 522–532. [Google Scholar] [CrossRef]
- Ahmed, O.M.; Ahmed, A.A.; Fahim, H.I.; Zaky, M.Y. Quercetin and naringenin abate diethylnitrosamine/acetylaminofluorene-induced hepatocarcinogenesis in Wistar rats: The roles of oxidative stress, inflammation and cell apoptosis. Drug Chem. Toxicol. 2019, 1–12. [Google Scholar] [CrossRef]
- Le, N.H.; Kim, C.-S.; Park, T.; Park, J.H.Y.; Sung, M.-K.; Lee, D.G.; Hong, S.-M.; Choe, S.-Y.; Goto, T.; Kawada, T.; et al. Quercetin Protects against Obesity-Induced Skeletal Muscle Inflammation and Atrophy. Mediat. Inflamm. 2014, 2014, 834294. [Google Scholar] [CrossRef]
- Perdicaro, D.J.; Rodriguez Lanzi, C.; Gambarte Tudela, J.; Miatello, R.M.; Oteiza, P.I.; Vazquez Prieto, M.A. Quercetin attenuates adipose hypertrophy, in part through activation of adipogenesis in rats fed a high-fat diet. J. Nutr. Biochem. 2020, 79, 108352. [Google Scholar] [CrossRef] [PubMed]
- Min, K.; Ebeler, S.E. Quercetin inhibits hydrogen peroxide-induced DNA damage and enhances DNA repair in Caco-2 cells. Food Chem. Toxicol. 2009, 47, 2716–2722. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, H.; Wang, H.; Shi, J. Quercetin attenuates high glucose-induced injury in human retinal pigment epithelial cell line ARPE-19 by up-regulation of miR-29b. J. Biochem. 2020, 167, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Darband, S.G.; Sadighparvar, S.; Yousefi, B.; Kaviani, M.; Ghaderi-Pakdel, F.; Mihanfar, A.; Rahimi, Y.; Mobaraki, K.; Majidinia, M. Quercetin attenuated oxidative DNA damage through NRF2 signaling pathway in rats with DMH induced colon carcinogenesis. Life Sci. 2020, 253, 117584. [Google Scholar] [CrossRef]
- Clemente-Soto, A.; Salas-Vidal, E.; Milan-Pacheco, C.; Sánchez-Carranza, J.; Peralta-Zaragoza, O.; González-Maya, L. Quercetin induces G2 phase arrest and apoptosis with the activation of p53 in an E6 expression-independent manner in HPV-positive human cervical cancer-derived cells. Mol. Med. Rep. 2019, 19, 2097–2106. [Google Scholar] [CrossRef] [Green Version]
- Neuwirthová, J.; Gál, B.; Smilek, P.; Urbánková, P. Potential of the Flavonoid Quercetin to Prevent and Treat Cancer—Current Status of Research. Klin. Onkol. 2018, 31, 184–190. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Xu, N.; Sun, W.; Zhao, Y.; Li, C.; Guo, M. Luteolin reduces inflammation in Staphylococcus aureus-induced mastitis by inhibiting NF-κB activation and MMPs expression. Oncotarget 2017, 8, 28481–28493. [Google Scholar] [CrossRef] [Green Version]
- AL-Megrin, W.A.; Alomar, S.; Alkhuriji, A.F.; Metwally, D.M.; Mohamed, S.K.; Kassab, R.B.; Abdel Moneim, A.E.; El-Khadragy, M.F. Luteolin protects against testicular injury induced by lead acetate by activating the Nrf2/ HO−1 pathway. IUBMB Life 2020, 72, 1787–1798. [Google Scholar] [CrossRef]
- Lin, Y.; Shi, R.; Wang, X.; Shen, H.-M. Luteolin, a flavonoid with potential for cancer prevention and therapy. Curr. Cancer Drug Targets 2008, 8, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yuan, T.; Yin, N.; Ma, X.; Zhang, Z.; Zhu, Z.; Shaukat, A.; Deng, G. Luteoloside Protects the Uterus from Staphylococcus aureus-Induced Inflammation, Apoptosis, and Injury. Inflammation 2018, 41, 1702–1716. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Luo, W.; Qian, Y.; Zhu, W.; Qian, J.; Li, J.; Jin, Y.; Xu, X.; Liang, G. Luteolin protects against diabetic cardiomyopathy by inhibiting NF-κB-mediated inflammation and activating the Nrf2-mediated antioxidant responses. Phytomedicine 2019, 59, 152774. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.R.M.R.; Wang, D.; Zhang, H.; Peng, S.; Shin, H.J.C.; Brandes, J.C.; Tighiouart, M.; Khuri, F.R.; Chen, Z.G.; Shin, D.M. Enhanced Anti-tumor Activity by the Combination of the Natural Compounds (−)-Epigallocatechin-3-gallate and Luteolin: Potential Role of p53. J. Biol. Chem. 2010, 285, 34557–34565. [Google Scholar] [CrossRef] [Green Version]
- Dai, C.-Q.; Luo, T.-T.; Luo, S.-C.; Wang, J.-Q.; Wang, S.-M.; Bai, Y.-H.; Yang, Y.-L.; Wang, Y.-Y. P53 and mitochondrial dysfunction: Novel insight of neurodegenerative diseases. J. Bioenerg. Biomembr. 2016, 48, 337–347. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.-Q.; Li, M.-H.; Qin, Y.-M.; Jiang, H.-Y.; Zhang, X.; Wu, M.H. Luteolin Inhibits Tumorigenesis and Induces Apoptosis of Non-Small Cell Lung Cancer Cells via Regulation of MicroRNA-34a-5p. Int. J. Mol. Sci. 2018, 19, 447. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Sánchez, A.; Barrajón-Catalán, E.; Herranz-López, M.; Castillo, J.; Micol, V. Lemon balm extract (Melissa officinalis, L.) promotes melanogenesis and prevents UVB-induced oxidative stress and DNA damage in a skin cell model. J. Dermatol. Sci. 2016, 84, 169–177. [Google Scholar] [CrossRef]
- George, V.C.; Kumar, D.R.N.; Suresh, P.K.; Kumar, S.; Kumar, R.A. Comparative Studies to Evaluate Relative in vitro Potency of Luteolin in Inducing Cell Cycle Arrest and Apoptosis in HaCaT and A375 Cells. Asian Pac. J. Cancer Prev. 2013, 14, 631–637. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.-T.; Wang, S.-K.; Huang, G.-L.; Sun, G.-J. Luteolin Induced-growth Inhibition and Apoptosis of Human Esophageal Squamous Carcinoma Cell Line Eca109 Cells in vitro. Asian Pac. J. Cancer Prev. 2012, 13, 5455–5461. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, P.; Qin, T.; Sun, D.; Zhao, X.; Zhang, B. Protective effect of epigallocatechin-3-gallate against neuroinflammation and anxiety-like behavior in a rat model of myocardial infarction. Brain Behav. 2020, 10, e01633. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.L.; Yu, Q.X.; Liang, W.C.; Leung, P.Y.; Ng, T.K.; Chu, W.K.; Pang, C.P.; Chan, S.O. Green tea extract attenuates LPS-induced retinal inflammation in rats. Sci. Rep. 2018, 8, 429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oblak, A.; Jerala, R. Toll-Like Receptor 4 Activation in Cancer Progression and Therapy. Clin. Dev. Immunol. 2011, 2011, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-Y.; Kao, C.-L.; Liu, C.-M. The Cancer Prevention, Anti-Inflammatory and Anti-Oxidation of Bioactive Phytochemicals Targeting the TLR4 Signaling Pathway. Int. J. Mol. Sci. 2018, 19, 2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, H.; Wu, N.; Liu, Z.; Zhao, H.; Zhao, M. Epigallocatechin-3-gallate alleviates paraquat-induced acute lung injury and inhibits upregulation of toll-like receptors. Life Sci. 2017, 170, 25–32. [Google Scholar] [CrossRef]
- Al-Maghrebi, M.; Alnajem, A.S.; Esmaeil, A. Epigallocatechin-3-gallate modulates germ cell apoptosis through the SAFE/Nrf2 signaling pathway. Naunyn Schmiedeberg’s Arch. Pharmacol. 2020, 393, 663–671. [Google Scholar] [CrossRef]
- He, Y.; Tan, D.; Bai, B.; Wu, Z.; Ji, S. Epigallocatechin-3-gallate attenuates acrylamide-induced apoptosis and astrogliosis in rat cerebral cortex. Toxicol. Mech. Methods 2017, 27, 298–306. [Google Scholar] [CrossRef]
- Remely, M.; Ferk, F.; Sterneder, S.; Setayesh, T.; Roth, S.; Kepcija, T.; Noorizadeh, R.; Rebhan, I.; Greunz, M.; Beckmann, J.; et al. EGCG Prevents High Fat Diet-Induced Changes in Gut Microbiota, Decreases of DNA Strand Breaks, and Changes in Expression and DNA Methylation of Dnmt1 and MLH1 in C57BL/6J Male Mice. Oxid. Med. Cell. Longev. 2017, 2017, 3079148. [Google Scholar] [CrossRef] [Green Version]
- George, V.C.; Dellaire, G.; Rupasinghe, H.P.V. Plant flavonoids in cancer chemoprevention: Role in genome stability. J. Nutr. Biochem. 2017, 45, 1–14. [Google Scholar] [CrossRef]
- George, V.C.; Ansari, S.A.; Chelakkot, V.S.; Chelakkot, A.L.; Chelakkot, C.; Menon, V.; Ramadan, W.; Ethiraj, K.R.; El-Awady, R.; Mantso, T.; et al. DNA-dependent protein kinase: Epigenetic alterations and the role in genomic stability of cancer. Mutat. Res. Rev. Mutat. Res. 2019, 780, 92–105. [Google Scholar] [CrossRef]
- De Silva, A.B.K.H.; Rupasinghe, H.P.V. Polyphenols composition and anti-diabetic properties in vitro of haskap (Lonicera caerulea L.) berries in relation to cultivar and harvesting date. J. Food Compos. Anal. 2020, 88, 103402. [Google Scholar] [CrossRef]
- Gan, Y.; Fu, Y.; Yang, L.; Chen, J.; Lei, H.; Liu, Q. Cyanidin-3-O-Glucoside and Cyanidin Protect Against Intestinal Barrier Damage and 2,4,6-Trinitrobenzenesulfonic Acid-Induced Colitis. J. Med. Food 2020, 23, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.X.; Qi, S.S.; He, J.; Hu, C.Y.; Han, H.; Jiang, H.; Li, X.S. Cyanidin-3-glucoside from Black Rice Ameliorates Diabetic Nephropathy via Reducing Blood Glucose, Suppressing Oxidative Stress and Inflammation, and Regulating Transforming Growth Factor β1/Smad Expression. J. Agric. Food Chem. 2020, 68, 4399–4410. [Google Scholar] [CrossRef]
- Liu, Z.; Hu, Y.; Li, X.; Mei, Z.; Wu, S.; He, Y.; Jiang, X.; Sun, J.; Xiao, J.; Deng, L.; et al. Nanoencapsulation of Cyanidin-3-O-glucoside Enhances Protection Against UVB-Induced Epidermal Damage through Regulation of p53-Mediated Apoptosis in Mice. J. Agric. Food Chem. 2018, 66, 5359–5367. [Google Scholar] [CrossRef] [PubMed]
- Amararathna, M.; Hoskin, D.W.; Rupasinghe, H.P.V. Anthocyanin-rich haskap (Lonicera caerulea L.) berry extracts reduce nitrosamine-induced DNA damage in human normal lung epithelial cells. Food Chem. Toxicol. 2020, 141, 111404. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Ma, Y.; Wu, S.; Chen, T.; He, Y.; Sun, J.; Jiao, R.; Jiang, X.; Huang, Y.; Deng, L.; et al. Protective Effect of Cyanidin-3-O-Glucoside against Ultraviolet B Radiation-Induced Cell Damage in Human HaCaT Keratinocytes. Front. Pharmacol. 2016, 7, 301. [Google Scholar] [CrossRef] [Green Version]
- George, V.C.; Rupasinghe, H.P.V. Apple Flavonoids Suppress Carcinogen-Induced DNA Damage in Normal Human Bronchial Epithelial Cells. Oxid. Med. Cell. Longev. 2017, 2017, 1767198. [Google Scholar] [CrossRef]
- Kaewmool, C.; Udomruk, S.; Phitak, T.; Pothacharoen, P.; Kongtawelert, P. Cyanidin-3-O-Glucoside Protects PC12 Cells Against Neuronal Apoptosis Mediated by LPS-Stimulated BV2 Microglial Activation. Neurotox. Res. 2020, 37, 111–125. [Google Scholar] [CrossRef]
- Meng, H.; Fu, G.; Shen, J.; Shen, K.; Xu, Z.; Wang, Y.; Jin, B.; Pan, H. Ameliorative Effect of Daidzein on Cisplatin-Induced Nephrotoxicity in Mice via Modulation of Inflammation, Oxidative Stress, and Cell Death. Oxid. Med. Cell. Longev. 2017, 2017, 3140680. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Naka, A.; Ohara, N.; Kondo, K.; Iida, K. Daidzein regulates proinflammatory adipokines thereby improving obesity-related inflammation through PPARγ. Mol. Nutr. Food Res. 2014, 58, 718–726. [Google Scholar] [CrossRef]
- Feng, G.; Sun, B.; Li, T. Daidzein attenuates lipopolysaccharide-induced acute lung injury via toll-like receptor 4/NF-kappaB pathway. Int. Immunopharmacol. 2015, 26, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Iovine, B.; Iannella, M.L.; Gasparri, F.; Monfrecola, G.; Bevilacqua, M.A. Synergic Effect of Genistein and Daidzein on UVB-Induced DNA Damage: An Effective Photoprotective Combination. J. Biomed. Biotechnol. 2011, 2011, 692846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iovine, B.; Garofalo, M.; Orefice, M.; Giannini, V.; Gasparri, F.; Monfrecola, G.; Bevilacqua, M.A. Isoflavones in aglycone solution enhance ultraviolet B-induced DNA damage repair efficiency. Clin. Exp. Dermatol. 2014, 39, 391–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zare, M.F.R.; Rakhshan, K.; Aboutaleb, N.; Nikbakht, F.; Naderi, N.; Bakhshesh, M.; Azizi, Y. Apigenin attenuates doxorubicin induced cardiotoxicity via reducing oxidative stress and apoptosis in male rats. Life Sci. 2019, 232, 116623. [Google Scholar] [CrossRef]
- Li, F.; Lang, F.; Zhang, H.; Xu, L.; Wang, Y.; Zhai, C.; Hao, E. Apigenin Alleviates Endotoxin-Induced Myocardial Toxicity by Modulating Inflammation, Oxidative Stress, and Autophagy. Oxid. Med. Cell. Longev. 2017, 2017, 2302896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzoeva, S.; Tong, X.; Bridgeman, B.B.; Plebanek, M.P.; Volpert, O.V. Apigenin Inhibits UVB-Induced Skin Carcinogenesis: The Role of Thrombospondin-1 as an Anti-Inflammatory Factor. Neoplasia 2018, 20, 930–942. [Google Scholar] [CrossRef]
- Ai, X.-Y.; Qin, Y.; Liu, H.-J.; Cui, Z.-H.; Li, M.; Yang, J.-H.; Zhong, W.-L.; Liu, Y.-R.; Chen, S.; Sun, T.; et al. Apigenin inhibits colonic inflammation and tumorigenesis by suppressing STAT3-NF-κB signaling. Oncotarget 2017, 8, 100216–100226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thangaiyan, R.; Robert, B.M.; Arjunan, S.; Govindasamy, K.; Nagarajan, R.P. Preventive effect of apigenin against isoproterenol-induced apoptosis in cardiomyoblasts. J. Biochem. Mol. Toxicol. 2018, 32, e22213. [Google Scholar] [CrossRef]
- Sarkaki, A.; Farbood, Y.; Mansouri, S.M.T.; Badavi, M.; Khorsandi, L.; Dehcheshmeh, M.G.; Shooshtari, M.K. Chrysin prevents cognitive and hippocampal long-term potentiation deficits and inflammation in rat with cerebral hypoperfusion and reperfusion injury. Life Sci. 2019, 226, 202–209. [Google Scholar] [CrossRef]
- Li, T.-F.; Ma, J.; Han, X.-W.; Jia, Y.-X.; Yuan, H.-F.; Shui, S.-F.; Guo, D.; Yan, L. Chrysin ameliorates cerebral ischemia/reperfusion (I/R) injury in rats by regulating the PI3K/Akt/mTOR pathway. Neurochem. Int. 2019, 129, 104496. [Google Scholar] [CrossRef]
- Del Fabbro, L.; Jesse, C.R.; De Gomes, M.G.; Borges Filho, C.; Donato, F.; Souza, L.C.; Goes, A.R.; Furian, A.F.; Boeira, S.P. The flavonoid chrysin protects against zearalenone induced reproductive toxicity in male mice. Toxicon 2019, 165, 13–21. [Google Scholar] [CrossRef] [PubMed]
- El-Marasy, S.A.; El Awdan, S.A.; Abd-Elsalam, R.M. Protective role of chrysin on thioacetamide-induced hepatic encephalopathy in rats. Chem. Biol. Interact. 2019, 299, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, O.; Mori, T.; Ito, F.; Okimura, H.; Kataoka, H.; Tanaka, Y.; Koshiba, A.; Kusuki, I.; Shigehiro, S.; Amami, T.; et al. Daidzein-rich isoflavone aglycones inhibit cell growth and inflammation in endometriosis. J. Steroid Biochem. Mol. Biol. 2018, 181, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Atiq, A.; Shal, B.; Naveed, M.; Khan, A.; Ali, J.; Zeeshan, S.; Al-Sharari, S.D.; Kim, Y.S.; Khan, S. Diadzein ameliorates 5-fluorouracil-induced intestinal mucositis by suppressing oxidative stress and inflammatory mediators in rodents. Eur. J. Pharmacol. 2019, 843, 292–306. [Google Scholar] [CrossRef]
- Yoshida, H.; Watanabe, W.; Oomagari, H.; Tsuruta, E.; Shida, M.; Kurokawa, M. Citrus flavonoid naringenin inhibits TLR2 expression in adipocytes. J. Nutr. Biochem. 2013, 24, 1276–1284. [Google Scholar] [CrossRef]
- Zhang, F.; Dong, W.; Zeng, W.; Zhang, L.; Zhang, C.; Qiu, Y.; Wang, L.; Yin, X.; Zhang, C.; Liang, W. Naringenin prevents TGF-β1 secretion from breast cancer and suppresses pulmonary metastasis by inhibiting PKC activation. Breast Cancer Res. 2016, 18, 38. [Google Scholar] [CrossRef] [Green Version]
- Chtourou, Y.; Slima, A.B.; Makni, M.; Gdoura, R.; Fetoui, H. Naringenin protects cardiac hypercholesterolemia-induced oxidative stress and subsequent necroptosis in rats. Pharmacol. Rep. 2015, 67, 1090–1097. [Google Scholar] [CrossRef]
- Tsai, M.-S.; Wang, Y.-H.; Lai, Y.-Y.; Tsou, H.-K.; Liou, G.-G.; Ko, J.-L.; Wang, S.-H. Kaempferol protects against propacetamol-induced acute liver injury through CYP2E1 inactivation, UGT1A1 activation, and attenuation of oxidative stress, inflammation and apoptosis in mice. Toxicol. Lett. 2018, 290, 97–109. [Google Scholar] [CrossRef]
- Kluska, M.; Juszczak, M.; Wysokiński, D.; Żuchowski, J.; Stochmal, A.; Woźniak, K. Kaempferol derivatives isolated from Lens culinaris Medik. reduce DNA damage induced by etoposide in peripheral blood mononuclear cells. Toxicol. Res. 2019, 8, 896–907. [Google Scholar] [CrossRef]
- Basu, A.; Das, A.S.; Sharma, M.; Pathak, M.P.; Chattopadhyay, P.; Biswas, K.; Mukhopadhyay, R. STAT3 and NF-κB are common targets for kaempferol-mediated attenuation of COX-2 expression in IL-6-induced macrophages and carrageenan-induced mouse paw edema. Biochem. Biophys. Rep. 2017, 12, 54–61. [Google Scholar] [CrossRef]
- Ansó, E.; Zuazo, A.; Irigoyen, M.; Urdaci, M.C.; Rouzaut, A.; Martínez-Irujo, J.J. Flavonoids inhibit hypoxia-induced vascular endothelial growth factor expression by a HIF-1 independent mechanism. Biochem. Pharmacol. 2010, 79, 1600–1609. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.-H.; Jeong, G.-S. Fisetin inhibits TNF-α-induced inflammatory action and hydrogen peroxide-induced oxidative damage in human keratinocyte HaCaT cells through PI3K/AKT/Nrf-2-mediated heme oxygenase-1 expression. Int. Immunopharmacol. 2015, 29, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Althunibat, O.Y.; Al Hroob, A.M.; Abukhalil, M.H.; Germoush, M.O.; Bin-Jumah, M.; Mahmoud, A.M. Fisetin ameliorates oxidative stress, inflammation and apoptosis in diabetic cardiomyopathy. Life Sci. 2019, 221, 83–92. [Google Scholar] [CrossRef]
- Hou, W.; Hu, S.; Su, Z.; Wang, Q.; Meng, G.; Guo, T.; Zhang, J.; Gao, P. Myricetin attenuates LPS-induced inflammation in RAW 264.7 macrophages and mouse models. Future Med. Chem. 2018, 10, 2253–2264. [Google Scholar] [CrossRef]
- Zhang, M.-J.; Su, H.; Yan, J.-Y.; Li, N.; Song, Z.-Y.; Wang, H.-J.; Huo, L.-G.; Wang, F.; Ji, W.-S.; Qu, X.-J.; et al. Chemopreventive effect of Myricetin, a natural occurring compound, on colonic chronic inflammation and inflammation-driven tumorigenesis in mice. Biomed. Pharmacother. 2018, 97, 1131–1137. [Google Scholar] [CrossRef]
- Chen, X.; Li, X.-F.; Chen, Y.; Zhu, S.; Li, H.-D.; Chen, S.-Y.; Wang, J.-N.; Pan, X.-Y.; Bu, F.-T.; Huang, C.; et al. Hesperetin derivative attenuates CCl4-induced hepatic fibrosis and inflammation by Gli-1-dependent mechanisms. Int. Immunopharmacol. 2019, 76, 105838. [Google Scholar] [CrossRef]
- Samie, A.; Sedaghat, R.; Baluchnejadmojarad, T.; Roghani, M. Hesperetin, a citrus flavonoid, attenuates testicular damage in diabetic rats via inhibition of oxidative stress, inflammation, and apoptosis. Life Sci. 2018, 210, 132–139. [Google Scholar] [CrossRef]
- Cheng, A.-W.; Tan, X.; Sun, J.-Y.; Gu, C.-M.; Liu, C.; Guo, X. Catechin attenuates TNF-α induced inflammatory response via AMPK-SIRT1 pathway in 3T3-L1 adipocytes. PLoS ONE 2019, 14, e0217090. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Mu, Y.; Yang, M.; Al Maruf, A.; Li, P.; Li, C.; Dai, S.; Lu, J.; Dong, Q. (+)-Catechin prevents methylglyoxal-induced mitochondrial dysfunction and apoptosis in EA.hy926 cells. Arch. Physiol. Biochem. 2017, 123, 121–127. [Google Scholar] [CrossRef]
- Knaze, V.; Zamora-Ros, R.; Luján-Barroso, L.; Romieu, I.; Scalbert, A.; Slimani, N.; Riboli, E.; Van Rossum, C.T.M.; Bueno-de-Mesquita, H.B.; Trichopoulou, A.; et al. Intake estimation of total and individual flavan-3-ols, proanthocyanidins and theaflavins, their food sources and determinants in the European Prospective Investigation into Cancer and Nutrition (EPIC) study. Br. J. Nutr. 2012, 108, 1095–1108. [Google Scholar] [CrossRef] [Green Version]
- Márquez Campos, E.; Jakobs, L.; Simon, M.-C. Antidiabetic Effects of Flavan-3-ols and Their Microbial Metabolites. Nutrients 2020, 12, 1592. [Google Scholar] [CrossRef] [PubMed]
- Thilakarathna, W.P.D.W.; Rupasinghe, H.P.V. Microbial metabolites of proanthocyanidins reduce chemical carcinogen-induced DNA damage in human lung epithelial and fetal hepatic cells in vitro. Food Chem. Toxicol. 2019, 125, 479–493. [Google Scholar] [CrossRef]
- Lei, L.; Yang, Y.; He, H.; Chen, E.; Du, L.; Dong, J.; Yang, J. Flavan-3-ols consumption and cancer risk: A meta-analysis of epidemiologic studies. Oncotarget 2016, 7, 73573–73592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gujar, K.; Wairkar, S. Nanocrystal technology for improving therapeutic efficacy of flavonoids. Phytomedicine 2020, 71, 153240. [Google Scholar] [CrossRef]
- Cassidy, A.; Bertoia, M.; Chiuve, S.; Flint, A.; Forman, J.; Rimm, E.B. Habitual intake of anthocyanins and flavanones and risk of cardiovascular disease in men. Am. J. Clin. Nutr. 2016, 104, 587–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Huang, X.; Zhang, Y.; Wang, Y.; Liu, Y.; Sun, R.; Xia, M. Anthocyanin Supplementation Improves HDL-Associated Paraoxonase 1 Activity and Enhances Cholesterol Efflux Capacity in Subjects With Hypercholesterolemia. J. Clin. Endocrinol. Metab. 2014, 99, 561–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Ling, W.; Guo, H.; Song, F.; Ye, Q.; Zou, T.; Li, D.; Zhang, Y.; Li, G.; Xiao, Y.; et al. Anti-inflammatory effect of purified dietary anthocyanin in adults with hypercholesterolemia: A randomized controlled trial. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 843–849. [Google Scholar] [CrossRef] [PubMed]
- Barchitta, M.; Maugeri, A.; La Mastra, C.; Rosa, M.C.L.; Favara, G.; Lio, R.M.S.; Agodi, A. Dietary Antioxidant Intake and Human Papillomavirus Infection: Evidence from a Cross-Sectional Study in Italy. Nutrients 2020, 12, 1384. [Google Scholar] [CrossRef]
- De Ferrars, R.M.; Czank, C.; Zhang, Q.; Botting, N.P.; Kroon, P.A.; Cassidy, A.; Kay, C.D. The pharmacokinetics of anthocyanins and their metabolites in humans: Pharmacokinetics of a (13)C-labelled anthocyanin. Br. J. Pharmacol. 2014, 171, 3268–3282. [Google Scholar] [CrossRef] [Green Version]
- Sudhakaran, M.; Doseff, A.I. The Targeted Impact of Flavones on Obesity-Induced Inflammation and the Potential Synergistic Role in Cancer and the Gut Microbiota. Molecules 2020, 25, 2477. [Google Scholar] [CrossRef]
- Zhong, Y.; Song, B.; Zheng, C.; Zhang, S.; Yan, Z.; Tang, Z.; Kong, X.; Duan, Y.; Li, F. Flavonoids from Mulberry Leaves Alleviate Lipid Dysmetabolism in High Fat Diet-Fed Mice: Involvement of Gut Microbiota. Microorganisms 2020, 8, 860. [Google Scholar] [CrossRef] [PubMed]
- Kamalifard, M.; Abbasalizadeh, S.; Mirghafourvand, M.; Bastani, P.; Gholizadeh Shamasbi, S.; Khodaei, L.; Gholizadeh, G. The effect of Seidlitzia rosmarinus (eshnan) on the prevention of recurrent cystitis in women of reproductive age: A randomized, controlled, clinical trial. Phytother. Res. 2020, 34, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Pintova, S.; Dharmupari, S.; Moshier, E.; Zubizarreta, N.; Ang, C.; Holcombe, R.F. Genistein combined with FOLFOX or FOLFOX–Bevacizumab for the treatment of metastatic colorectal cancer: Phase I/II pilot study. Cancer Chemother. Pharmacol. 2019, 84, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Curtis, P.J.; Van der Velpen, V.; Berends, L.; Jennings, A.; Feelisch, M.; Umpleby, A.M.; Evans, M.; Fernandez, B.O.; Meiss, M.S.; Minnion, M.; et al. Blueberries improve biomarkers of cardiometabolic function in participants with metabolic syndrome—Results from a 6-month, double-blind, randomized controlled trial. Am. J. Clin. Nutr. 2019, 109, 1535–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakuradze, T.; Tausend, A.; Galan, J.; Groh, I.A.M.; Berry, D.; Tur, J.A.; Marko, D.; Richling, E. Antioxidative activity and health benefits of anthocyanin-rich fruit juice in healthy volunteers. Free Radic. Res. 2019, 53, 1045–1055. [Google Scholar] [CrossRef]
- Munguia, L.; Rubio-Gayosso, I.; Ramirez-Sanchez, I.; Ortiz, A.; Hidalgo, I.; Gonzalez, C.; Meaney, E.; Villarreal, F.; Najera, N.; Ceballos, G. High Flavonoid Cocoa Supplement Ameliorates Plasma Oxidative Stress and Inflammation Levels While Improving Mobility and Quality of Life in Older Subjects: A Double-Blind Randomized Clinical Trial. J. Gerontol. Ser. A 2019, 74, 1620–1627. [Google Scholar] [CrossRef]
- Zhang, H.; Gordon, R.; Li, W.; Yang, X.; Pattanayak, A.; Fowler, G.; Zhang, L.; Catalona, W.J.; Ding, Y.; Xu, L.; et al. Genistein treatment duration effects biomarkers of cell motility in human prostate. PLoS ONE 2019, 14, e0214078. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Jia, L.; Chen, G.; Li, X.; Meng, X.; Zhao, X.; Xing, L.; Zhu, W. A prospective, three-arm, randomized trial of EGCG for preventing radiation-induced esophagitis in lung cancer patients receiving radiotherapy. Radiother. Oncol. 2019, 137, 186–191. [Google Scholar] [CrossRef]
- Ud-Din, S.; Foden, P.; Mazhari, M.; Al-Habba, S.; Baguneid, M.; Bulfone-Paus, S.; McGeorge, D.; Bayat, A. A Double-Blind, Randomized Trial Shows the Role of Zonal Priming and Direct Topical Application of Epigallocatechin-3-Gallate in the Modulation of Cutaneous Scarring in Human Skin. J. Investig. Dermatol. 2019, 139, 1680–1690. [Google Scholar] [CrossRef] [Green Version]
- Bazzucchi, I.; Patrizio, F.; Ceci, R.; Duranti, G.; Sgrò, P.; Sabatini, S.; Di Luigi, L.; Sacchetti, M.; Felici, F. The Effects of Quercetin Supplementation on Eccentric Exercise-Induced Muscle Damage. Nutrients 2019, 11, 205. [Google Scholar] [CrossRef] [Green Version]
- Karbasforooshan, H.; Hosseini, S.; Elyasi, S.; Fani Pakdel, A.; Karimi, G. Topical silymarin administration for prevention of acute radiodermatitis in breast cancer patients: A randomized, double-blind, placebo-controlled clinical trial: Topical silymarin for prevention of radiodermatitis in breast cancer. Phytother. Res. 2019, 33, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.B.; Pandita, R.K.; Eskiocak, U.; Ly, P.; Kaisani, A.; Kumar, R.; Cornelius, C.; Wright, W.E.; Pandita, T.K.; Shay, J.W. Targeting of Nrf2 induces DNA damage signaling and protects colonic epithelial cells from ionizing radiation. Proc. Natl. Acad. Sci. USA 2012, 109, E2949–E2955. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Flavonoid | Disease/Oxidative Stress Imposed | Preclinical or Cell Model | Molecular Signaling/Pathway | Inflammation/Cancer Prevention Status | Reference |
|---|---|---|---|---|---|
| Apigenin | Cardiotoxicity | Rats | Decreases caspase-3 and Bax | Inhibition of apoptosis | [175] |
| Myocardial injury | Mice | Inhibits TNF-α, IL-1β, MIP-1α, MIP-2, and NFκB | Inhibit inflammation | [176] | |
| UVB-induced skin cancer | Mice | Inhibits IL-6 and IL-12 | Inhibit inflammation | [177] | |
| Bowel disease and colitis-associated cancer | Mice and HCT-116 | Inhibits STAT3 and NF-κB | Inhibit inflammation-induced carcinogenesis | [178] | |
| Isoproterenol hydrochloride-induced apoptosis | H9C2 | Inhibits Bax, caspase-3, -8, and -9 and cytochrome c | Cancer prevention | [179] | |
| Chrysin | Cerebral ischemia | Rats | Inhibits IL-1β and TNF-α | Inhibit inflammation | [180] |
| Cerebral ischemia | Rats | Inhibits TNF-α, IL-6, and IL-1β; activates PI3K/Akt/mTOR pathway | Inhibit inflammation and apoptosis | [181] | |
| Reproductive toxicity | Mice | Inhibits IL-1β, TNF-α, IL-6, and IL-10; activates caspase 3 and 9 | Inhibit inflammation | [182] | |
| Hepatic encephalopathy | Rats | Increases glutathione level; inhibit NF-κB, TNF-α, IL-6, and TLR-4; reduces caspase-3 and hepatic necrosis | Inhibit inflammation and apoptosis | [183] | |
| Daidzein | Endometriosis | OESCs and NESCs | Inhibited IL-6, IL-8, COX-2, TNF-α-induced IκB phosphorylation and p65 | Inhibit inflammation | [184] |
| Intestinal mucositis | Mice | Inhibits TNF-α, IL-1β, and IL-6; increases CAT and GPx level | Inhibit inflammation | [185] | |
| Naringenin | Obesity | Mice | Inhibits TLRs and TNF-α | Inhibit inflammation | [186] |
| Pulmonary metastasis | Mice | Inhibits Tgf-β1 | Cancer prevention | [187] | |
| Cardiac hypercholesterolemia | Rats | Inhibits DNA damage, TNF-α, and RIP3 | Inhibits necroptosis | [188] | |
| Kaempferol | Propacetamol-induced acute liver injury | Mice | Inhibits cytochrome P450 2E1; restores SOD, GPx and CAT; inhibits Bax/Bcl-2 ratio | Prevents apoptosis | [189] |
| Etoposide-induced oxidative stress | HL-60 and PBMCs | Inhibits DNA damage (tail length) | Prevents DNA damage | [190] | |
| Paw Edema | Mice and THP-1 (together) | Inhibits Cox-2; STAT3 and NF-κB | Inhibit inflammation | [191] | |
| Brain Injury and neuroinflammation | Rats | Inhibits STAT3 and NF-κB p65 | Inhibit inflammation | [189] | |
| Fisetin | Hypoxia | NCI-H157 | Inhibit HIF 1-α and STAT3 | Prevents hypoxia | [192] |
| Hydrogen peroxide-induced oxidative stress | HaCaT | Inhibits iNOS, Cox-2, IL-1β, IL-6, and TNF-α | Inhibit inflammation | [193] | |
| Diabetic cardiomyopathy | Rats | Restores SOD and CAT; inhibits IL-1β, IL-6, and TNF-α; inhibits caspase-3, Bax, and Bax/Bcl-2 ratio | Inhibit inflammation and apoptosis | [194] | |
| Myricetin | LPS-induced Inflammation | Mice and RAW 264.7 | Inhibits NF-κB p65 and AKT activation | Inhibit inflammation | [195] |
| Colonic chronic-induced inflammation | Mice | Inhibits TNF-α, IL-1β, IL-6, NF-κB, p-NF-κB, cyclooxygenase-2 (COX-2), PCNA, and Cyclin D1 | Inhibits inflammation and tumor | [196] | |
| Hesperetin | Hepatic fibrosis | Mice and HSCs | Inhibit α-SMA, Col1α1, Col3α1 and TIMP-1 | Inhibits inflammation and induce apoptosis | [197] |
| Testicular Damage | Rats | Inhibits malondialdehyde, ROS, DNA fragmentation, and caspase 3; inhibits TNFα and IL-17 | Inhibits inflammation and apoptosis | [198] | |
| Catechin | TNF-α-induced inflammation | 3T3-L1 | Inhibit IL-1α, IL-1β, IL-6, IL-12, p35, iNOS, Cox-2, NF-κB, AMPK, FOXO3a, and SIRT1 | Inhibits inflammation | [199] |
| Methylglyoxal-induced cytotoxicity | EA.hy926 | Inhibits MMP and cytotoxicity | Inhibits apoptosis | [200] |
| Dietary Flavonoids | Condition Studied | Sample Details | Key Observations | Reference |
|---|---|---|---|---|
| Seidlitzia Rosmarinus (flavonoid-rich) | Recurrent cystitis | 126 women | Lowered symptoms of cystitis; prevent the incidence of recurrent cystitis with no side effects | [213] |
| Genistein with FOLFOX | Metastatic colorectal cancer | 13 patients | Exposure of genistein with FOLFOX was safe and tolerable | [214] |
| Anthocyanin-rich blueberry | Type 2 diabetes and CVD | 115 people | Improved endothelial function, systemic arterial stiffness and attenuated cyclic guanosine monophosphate concentrations | [215] |
| Anthocyanin-rich fruit juice | Healthy volunteers | 57 males | Demonstrated DNA-protective and anti-oxidant effects; reduction in body fat and an increase in fat-free mass with increased SOD level | [216] |
| Flavonoid-rich natural cocoa beverage | Plasma oxidative stress and inflammation | 134 people | Improved glycemia, triglyceridemia, high-density lipoprotein cholesterol, low-density lipoprotein cholesterol, triglyceridemia/HDL index, and oxidative markers | [217] |
| Genistein | Human prostate cancer | 14 males | Increased Brain Abundant Membrane Attached Signal Protein 1(BASP1) expression; decreases MMP-2 in prostate tissue | [218] |
| EGCG | Acute radiation-induced esophagitis (ARIE) | 83 patients | Lowered ARIE by reducing acute pain index (API) and acute dysphagia index (ADI) without side effects | [219] |
| EGCG | Cutaneous scarring | 62 humans | Reduced mast cells; down-regulated Vascular Endothelial Growth Factor A (VEGFA) and CD31; reduced scar thickness | [220] |
| Quercetin | Eccentric exercise-induced muscle damage | 12 males | Increased the isometric strength for contraction; lower torque and muscle fiber conduction velocity; attenuate the severity of muscle weakness | [221] |
| Silymarin | Radiation-induced dermatitis | 40 patients | Delayed in radiodermatitis development and progression | [222] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siddiqui, S.S.; Rahman, S.; Rupasinghe, H.P.V.; Vazhappilly, C.G. Dietary Flavonoids in p53—Mediated Immune Dysfunctions Linking to Cancer Prevention. Biomedicines 2020, 8, 286. https://doi.org/10.3390/biomedicines8080286
Siddiqui SS, Rahman S, Rupasinghe HPV, Vazhappilly CG. Dietary Flavonoids in p53—Mediated Immune Dysfunctions Linking to Cancer Prevention. Biomedicines. 2020; 8(8):286. https://doi.org/10.3390/biomedicines8080286
Chicago/Turabian StyleSiddiqui, Shoib Sarwar, Sofia Rahman, H.P. Vasantha Rupasinghe, and Cijo George Vazhappilly. 2020. "Dietary Flavonoids in p53—Mediated Immune Dysfunctions Linking to Cancer Prevention" Biomedicines 8, no. 8: 286. https://doi.org/10.3390/biomedicines8080286
APA StyleSiddiqui, S. S., Rahman, S., Rupasinghe, H. P. V., & Vazhappilly, C. G. (2020). Dietary Flavonoids in p53—Mediated Immune Dysfunctions Linking to Cancer Prevention. Biomedicines, 8(8), 286. https://doi.org/10.3390/biomedicines8080286