Protein Dynamics in F-like Bacterial Conjugation



Abstract
:1. Introduction
2. Dynamics of Proteins Involved in the Regulation of Bacterial Conjugation
2.1. FinO and FinP
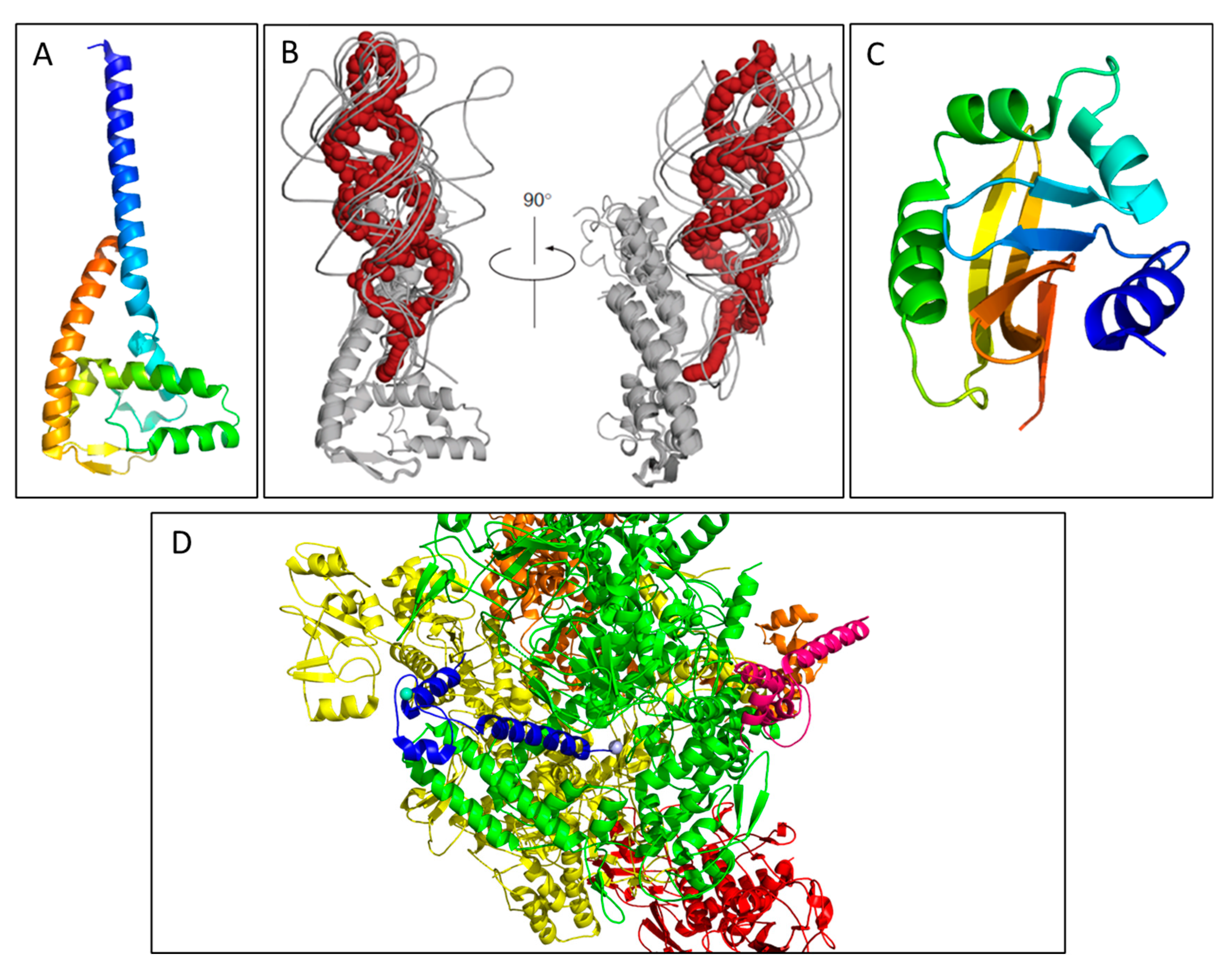
2.2. TraJ
2.3. TraR
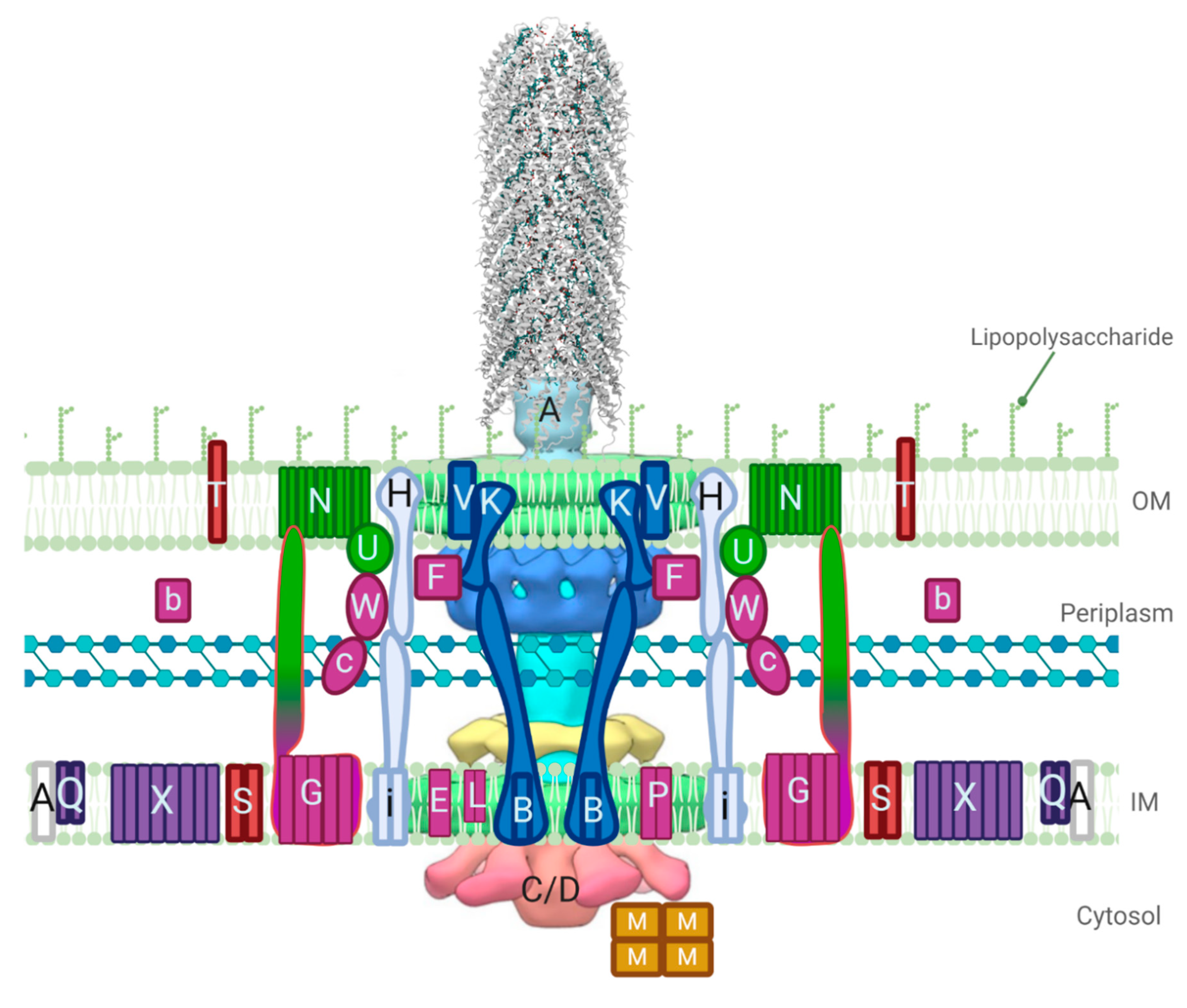
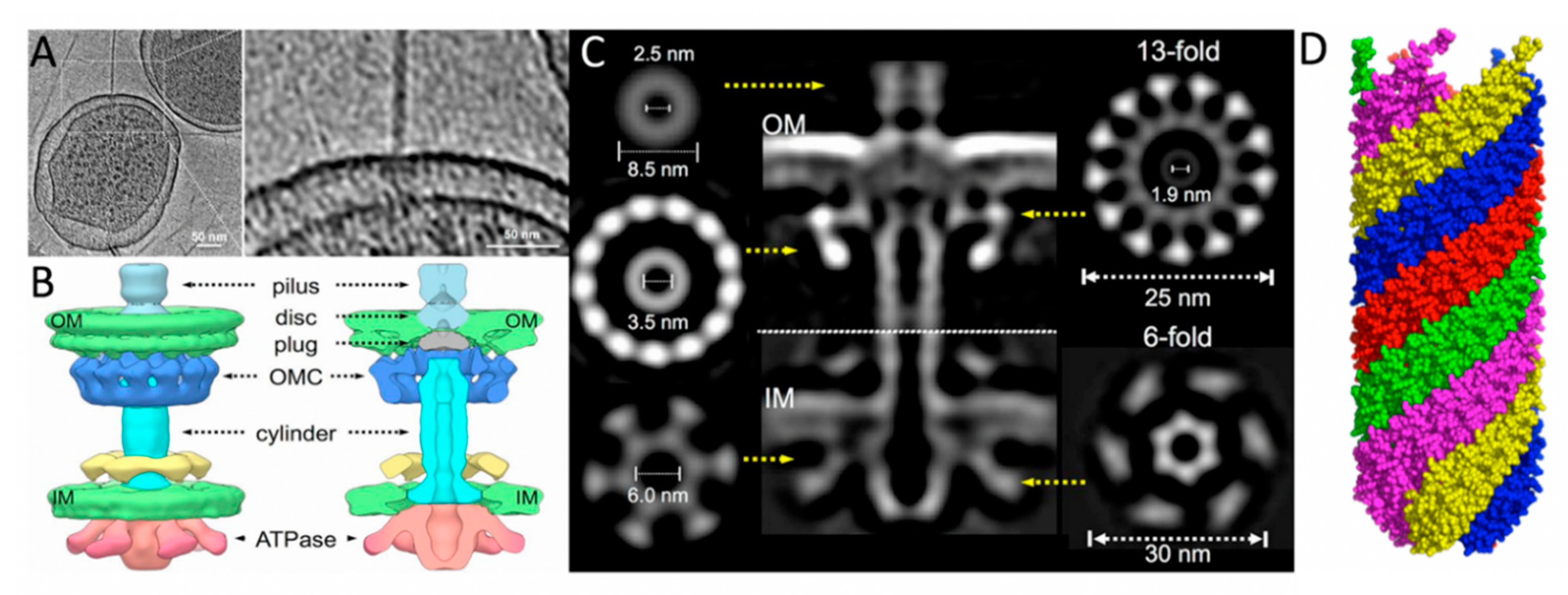
3. Structures Involved in Pilin Processing, Pilus Extension, and Retraction
3.1. Proteins Responsible for Propilin Maturation
3.1.1. The Pilin Protein TraA
3.1.2. TraQ
3.1.3. TraX
3.2. Pilus Assembly and Extension Proteins
3.2.1. Pilus Tip Formation
3.2.2. T4SS Core Proteins TraB, -K, and -V
3.2.3. Pilus Extension—TraF, TrbB, TraP, TraC, TraW, and TrbC
3.3. Pilus Retraction—TraH and TrbI
3.4. Mating Pair Formation
4. Mating Pair Stabilization Proteins and the Dynamics of their Structural Ensemble
4.1. TraN
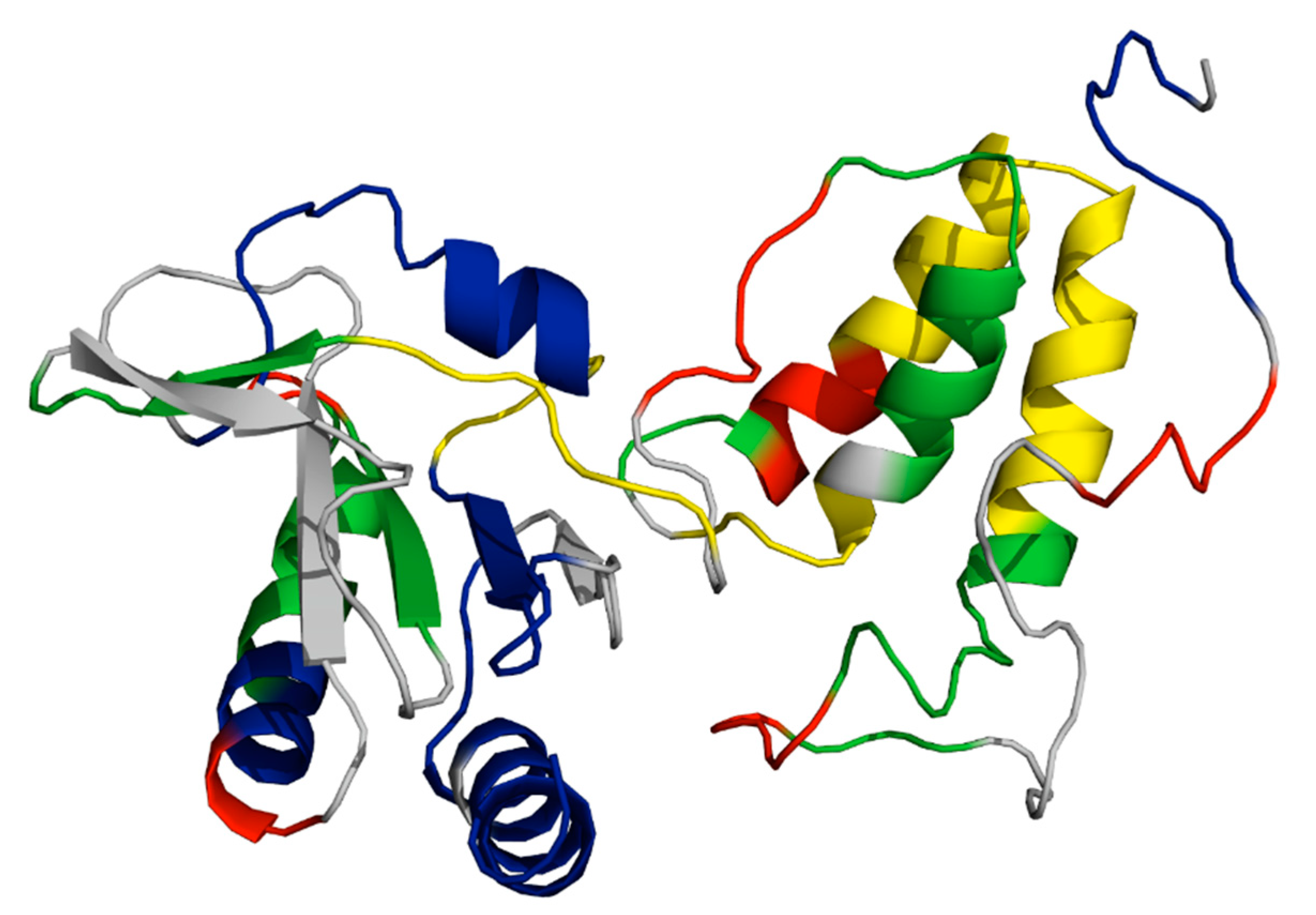
4.2. TraG and the Structural Dynamics Effectuating its Trifunctional Roles
4.3. TraU
5. The Structures of Conjugative DNA Transfer Proteins
5.1. TraY and IHF
5.2. TraI
5.3. TraM

5.4. TraD
6. Exclusion Proteins and their Low Structural Dynamicity
6.1. TraT
6.2. TraS
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lujan, S.A.; Guogas, L.M.; Ragonese, H.; Matson, S.W.; Redinbo, M.R. Disrupting antibiotic resistance propagation by inhibiting the conjugative DNA relaxase. Proc. Natl. Acad. Sci. USA 2007, 104, 12282–12287. [Google Scholar] [CrossRef] [Green Version]
- Cabezón, E.; de la Cruz, F.; Arechaga, I. Conjugation inhibitors and their potential use to prevent dissemination of antibiotic resistance genes in bacteria. Front. Microbiol. 2017, 8, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waksman, G. From conjugation to T4S systems in Gram-negative bacteria: A mechanistic biology perspective. EMBO Rep. 2019, 20, e47012. [Google Scholar] [CrossRef] [PubMed]
- Costa, T.R.D.; Felisberto-Rodrigues, C.; Meir, A.; Prevost, M.S.; Redzej, A.; Trokter, M.; Waksman, G. Secretion systems in Gram-negative bacteria: Structural and mechanistic insights. Nat. Rev. Microbiol. 2015, 13, 343–359. [Google Scholar] [CrossRef] [PubMed]
- Allen, H.K.; Donato, J.; Wang, H.H.; Cloud-Hansen, K.A.; Davies, J.; Handelsman, J. Call of the wild: Antibiotic resistance genes in natural environments. Nat. Rev. Microbiol. 2010, 8, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Wang, W.; Regev-Yochay, G.; Lipsitch, M.; Hanage, W.P. Antibiotics in agriculture and the risk to human health: How worried should we be? Evol. Appl. 2015, 8, 240–247. [Google Scholar] [CrossRef] [Green Version]
- Pelat, C.; Kardaś-Słoma, L.; Birgand, G.; Ruppé, E.; Schwarzinger, M.; Andremont, A.; Lucet, J.C.; Yazdanpanah, Y. Hand hygiene, cohorting, or antibiotic restriction to control outbreaks of multidrug-resistant enterobacteriaceae. Infect. Control Hosp. Epidemiol. 2015, 37, 272–280. [Google Scholar] [CrossRef]
- Williams, J.J.; Hergenrother, P.J. Exposing plasmids as the Achilles’ heel of drug-resistant bacteria. Curr. Opin. Chem. Biol. 2008, 12, 389–399. [Google Scholar] [CrossRef] [Green Version]
- Baron, C. Antivirulence drugs to target bacterial secretion systems. Curr. Opin. Microbiol. 2010, 13, 100–105. [Google Scholar] [CrossRef]
- Baron, C. A novel strategy to target bacterial virulence. Future Microbiol. 2013, 8, 1–3. [Google Scholar] [CrossRef]
- Arutyunov, D.; Frost, L.S. F conjugation: Back to the beginning. Plasmid 2013, 70, 18–32. [Google Scholar] [CrossRef] [PubMed]
- Novick, R.P. Plasmid incompatibility. Microbiol. Rev. 1987, 51, 381–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Lopez, R.; de Toro, M.; Moncalian, G.; Garcillan-Barcia, M.P.; de la Cruz, F. Comparative Genomics of the Conjugation Region of F-like Plasmids: Five Shades of F. Front. Mol. Biosci. 2016, 3, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawley, T.; Klimke, W.; Gubbins, M.; Frost, L. F factor conjugation is a true type IV secretion system. FEMS Microbiol. Lett. 2003, 224, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Khara, P.; Christie, P.J. Structural bases for F plasmid conjugation and F pilus biogenesis in Escherichia coli. Proc. Natl. Acad. Sci. USA 2019, 201904428. [Google Scholar] [CrossRef] [Green Version]
- Costa, T.R.D.; Ilangovan, A.; Ukleja, M.; Redzej, A.; Santini, J.M.; Smith, T.K.; Egelman, E.H.; Waksman, G. Structure of the Bacterial Sex F Pilus Reveals an Assembly of a Stoichiometric Protein-Phospholipid Complex. Cell 2016, 166, 1436.e10–1444.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCallum, M.; Burrows, L.L.; Howell, P.L. The Dynamic Structures of the Type IV Pilus. Microbiol. Spectr. 2019, 7, 1–12. [Google Scholar] [CrossRef]
- Lee, G.M.; Craik, C.S. Trapping moving targets with small molecules. Science 2009, 324, 213–215. [Google Scholar] [CrossRef] [Green Version]
- Feixas, F.; Lindert, S.; Sinko, W.; McCammon, J.A. Exploring the role of receptor flexibility in structure-based drug discovery. Biophys. Chem. 2014, 186, 31–45. [Google Scholar] [CrossRef] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Koraimann, G.; Wagner, M.A. Social behavior and decision making in bacterial conjugation. Front. Cell. Infect. Microbiol. 2014, 4, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahrl, D.; Wagner, A.; Tscherner, M.; Koraimann, G. GroEL plays a central role in stress-induced negative regulation of bacterial conjugation by promoting proteolytic degradation of the activator protein TraJ. J. Bacteriol. 2007, 189, 5885–5894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohler, V.; Keller, W.; Grohmann, E. Regulation of gram-positive conjugation. Front. Microbiol. 2019, 10, 1134. [Google Scholar] [CrossRef] [Green Version]
- Koraimann, G.; Teferle, K.; Markolin, G.; Woger, W.; Högenauer, G. The FinOP repressor system of plasmid R1: Analysis of the antisense RNA control of traJ expression and conjugative DNA transfer. Mol. Microbiol. 1996, 21, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Ghetu, A.F.; Gubbins, M.J.; Frost, L.S.; Glover, J.N. Crystal structure of the bacterial conjugation repressor finO. Nat. Struct. Biol. 2000, 7, 565–569. [Google Scholar] [CrossRef]
- Lee, S.H.; Frost, L.S.; Paranchych, W. FinOP repression of the F plasmid involves extension of the half-life of FinP antisense RNA by FinO. MGG Mol. Gen. Genet. 1992, 235, 131–139. [Google Scholar] [CrossRef]
- van Biesen, T.; Soderbom, F.; Wagner, E.G.H.; Frost, L.S. Structural and functional analyses of the FinP antisense RNA regulatory system of the F conjugative piasmid. Mol. Microbiol. 1993, 10, 35–43. [Google Scholar] [CrossRef]
- Arthur, D.C.; Edwards, R.A.; Tsutakawa, S.; Tainer, J.A.; Frost, L.S.; Glover, J.N.M. Mapping interactions between the RNA chaperone FinO and its RNA targets. Nucleic Acids Res. 2011, 39, 4450–4463. [Google Scholar] [CrossRef] [Green Version]
- Arthur, D.C.; Ghetu, A.F.; Gubbins, M.J.; Edwards, R.A.; Frost, L.S.; Glover, J.N.M. FinO is an RNA chaperone that facilitates sense-antisense RNA interactions. EMBO J. 2003, 22, 6346–6355. [Google Scholar] [CrossRef] [Green Version]
- Glover, J.N.M.; Chaulk, S.G.; Edwards, R.A.; Arthur, D.; Lu, J.; Frost, L.S. The FinO family of bacterial RNA chaperones. Plasmid 2014, 78, 79–87. [Google Scholar] [CrossRef]
- Jerome, L.J.; van Biesen, T.; Frost, L.S. Degradation of FinP antisense RNA from F-like plasmids: The RNA-binding protein, FinO, protects FinP from ribonuclease E. J. Mol. Biol. 1999, 285, 1457–1473. [Google Scholar] [CrossRef] [PubMed]
- Ghetu, A.F.; Arthur, D.C.; Kerppola, T.K.; Glover, J.N.M. Probing FinO-FinP RNA interactions by site-directed protein-RNA crosslinking and gelFRET. RNA 2002, 8, 816–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tompa, P.; Csermely, P. The role of structural disorder in the function of RNA and protein chaperones. FASEB J. 2004, 18, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Vernon, R.M.; Chong, P.A.; Bah, A.; Lin, H.; Farber, P.; Tsang, B.; Kim, T.H.; Forman-Kay, J.D. Pi-Pi contacts are an overlooked protein feature relevant to phase separation. Elife 2018, 7, 1–48. [Google Scholar] [CrossRef]
- The Pymol Molecular Graphics System, Version 2.0 Schrödinger LLC. 2020.
- Ziegelin, G.; Furste, J.P.; Lanka, E. TraJ protein of plasmid RP4 binds to a 19-base pair invert sequence repetition within the transfer origin. J. Biol. Chem. 1989, 264, 11989–11994. [Google Scholar] [PubMed]
- Frost, L.S.; Ippen-Ihler, K.; Skurray, R.A. Analysis of the sequence and gene products of the transfer region of the F sex factor. Microbiol. Rev. 1994, 58, 162–210. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Maillard, J.M.; Arutyunov, D.; Frost, L.S. The F plasmid transfer activator TraJ is a dimeric helix-turn-helix DNA-binding protein. FEMS Microbiol. Lett. 2010, 310, 112–119. [Google Scholar] [CrossRef] [Green Version]
- Silverman, P.M.; Wickersham, E.; Rainwater, S.; Harris, R. Regulation of the F plasmid tra Y promoter in Escherichia coli K12 as a function of sequence context. J. Mol. Biol. 1991, 220, 271–279. [Google Scholar] [CrossRef]
- Will, W.R.; Lu, J.; Frost, L.S. The role of H-NS in silencing F transfer gene expression during entry into stationary phase. Mol. Microbiol. 2004, 54, 769–782. [Google Scholar] [CrossRef]
- Lu, J.; Wu, R.; Adkins, J.N.; Joachimiak, A.; Glover, J.N.M. Crystal Structures of the F and pSLT Plasmid TraJ N-Terminal Regions Reveal Similar Homodimeric PAS Folds with Functional Interchangeability. Biochemistry 2014, 53, 5810–5819. [Google Scholar] [CrossRef] [Green Version]
- Strohmaier, H.; Noiges, R.; Kotschan, S.; Sawers, G.; Högenauer, G.; Zechner, E.L.; Koraimann, G. Signal transduction and bacterial conjugation: Characterization of the role of ArcA in regulating conjugative transfer of the resistance plasmid R1. J. Mol. Biol. 1998, 277, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Peng, Y.; Wan, S.; Frost, L.S.; Raivio, T.; Mark Glover, J.N. Cooperative function of TraJ and ArcA in regulating the F plasmid tra operon. J. Bacteriol. 2019, 201, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Arutyunov, D.; Rodriguez-Maillard, J.M.; Frost, L.S. A PAS domain within F plasmid TraJ is critical for its function as a transcriptional activator. Biochem. Cell Biol. 2011, 89, 396–404. [Google Scholar] [CrossRef]
- Taylor, B.L.; Zhulin, I.B. PAS Domains: Internal Sensors of Oxygen, Redox Potential, and Light. Microbiol. Mol. Biol. Rev. 1999, 63, 479–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Peng, Y.; Arutyunov, D.; Frost, L.S.; Glover, J.N.M. Error-Prone PCR Mutagenesis Reveals Functional Domains of a Bacterial Transcriptional Activator, TraJ. J. Bacteriol. 2012, 194, 3670–3677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, M.A.; Bischof, K.; Kati, D.; Koraimann, G. Silencing and activating type IV secretion genes of the F-like conjugative resistance plasmid R1. Microbiology 2013, 159, 2481–2491. [Google Scholar] [CrossRef]
- Molodtsov, V.; Sineva, E.; Zhang, L.; Huang, X.; Cashel, M.; Ades, S.E.; Murakami, K.S. Allosteric Effector ppGpp Potentiates the Inhibition of Transcript Initiation by DksA. Mol. Cell 2018, 69, 828–839. [Google Scholar] [CrossRef] [Green Version]
- Blankschien, M.D.; Potrykus, K.; Grace, E.; Choudhary, A.; Vinella, D.; Cashel, M.; Herman, C. TraR, a homolog of a RNAP secondary channel interactor, modulates transcription. PLoS Genet. 2009, 5, e1000345. [Google Scholar] [CrossRef] [Green Version]
- Maneewannakul, K.; Ippen-Ihler, K. Construction and analysis of F plasmid traR, trbJ, and trbH mutants. J. Bacteriol. 1993, 175, 1528–1531. [Google Scholar] [CrossRef] [Green Version]
- Moore, D.; Wu, J.H.; Kathir, P.; Hamilton, C.M.; Ippen-Ihler, K. Analysis of transfer genes and gene products within the traB-traC region of the Escherichia coli fertility factor, F. J. Bacteriol. 1987, 169, 3994–4002. [Google Scholar] [CrossRef] [Green Version]
- Gopalkrishnan, S.; Ross, W.; Chen, A.Y.; Gourse, R.L.; Roberts, J.W. TraR directly regulates transcription initiation by mimicking the combined effects of the global regulators DksA and ppGpp. Proc. Natl. Acad. Sci. USA 2017, 114, E5539–E5548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekine, S.I.; Murayama, Y.; Svetlov, V.; Nudler, E.; Yokoyama, S. The ratcheted and ratchetable structural states of RNA polymerase underlie multiple transcriptional functions. Mol. Cell 2015, 57, 408–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Gopalkrishnan, S.; Chiu, C.; Chen, A.Y.; Campbell, E.A.; Gourse, R.L.; Ross, W.; Darst, S.A. E. coli TraR allosterically regulates transcription initiation by altering RNA polymerase conformation. Elife 2019, 8, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Christie, P.J. The Mosaic Type IV Secretion Systems. EcoSal Plus 2016, 7, 1–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christie, P.J.; Whitaker, N.; González-Rivera, C. Mechanism and structure of the bacterial type IV secretion systems. Biochim. Biophys. Acta 2014, 1843, 1578–1591. [Google Scholar] [CrossRef] [Green Version]
- Bergeron, J.R.C.; Sgourakis, N.G. Type IV Pilus: One Architectural Problem, Many Structural Solutions. Structure 2015, 23, 253–255. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.A.; Yu, X.; Silverman, P.M.; Harris, R.L.; Egelman, E.H. The Structure of F-Pili. J. Mol. Biol. 2009, 385, 22–29. [Google Scholar] [CrossRef]
- Manchak, J.; Anthony, K.G.; Frost, L.S. Mutational analysis of F-pilin reveals domains for pilus assembly, phage infection and DNA transfer. Mol. Microbiol. 2002, 43, 195–205. [Google Scholar] [CrossRef]
- Majdalani, N.; Ippen-Ihler, K. Membrane insertion of the F-pilin subunit is Sec independent but requires leader peptidase B and the proton motive force. J. Bacteriol. 1996, 178, 3742–3747. [Google Scholar] [CrossRef] [Green Version]
- Zechner, E.L.; Lang, S.; Schildbach, J.F. Assembly and mechanisms of bacterial type IV secretion machines. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 1073–1087. [Google Scholar] [CrossRef]
- Frost, L.S.; Paranchych, W.; Willetts, N.S. The complete sequence of the F traALE region that includes the gene for F pilin. J. Bacteriol. 1984, 160, 395–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverman, P.M. Towards a structural biology of bacterial conjugation. Mol. Microbiol. 1997, 23, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Firth, N.; Ippen-ihler, K.; Skurray, R.A. Structure and Function of the F Factor and Mechanism of Conjugation. In Escherichia coli and Salmonella: Cellular and Molecular Biology, 2nd ed.; ASM Press: Washington, DC, USA, 1996; pp. 2377–2401. [Google Scholar]
- Harris, R.L.; Sholl, K.A.; Conrad, M.N.; Dresser, M.E.; Silverman, P.M. Interaction between the F plasmid TraA (F-pilin) and TraQ proteins. Mol. Microbiol. 1999, 34, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.H.; Ippen-Ihler, K. Nucleotide sequence of traQ and adjacent loci in the Escherichia coli K-12 F-plasmid transfer operon. J. Bacteriol. 1989, 171, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, D.; Hamilton, C.M.; Maneewannakul, K.; Mintz, Y.; Frost, L.S.; Ippen-Ihler, K. The Escherichia coli K-12 F plasmid gene traX is required for acetylation of F pilin. J. Bacteriol. 1993, 175, 1375–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maneewannakul, K.; Maneewannakul, S.; Ippen-Ihler, K. Characterization of traX, the F plasmid locus required for acetylation of F-pilin subunits. J. Bacteriol. 1995, 177, 2957–2964. [Google Scholar] [CrossRef] [Green Version]
- Cram, D.S.; Loh, S.M.; Cheah, K.C.; Skurray, R.A. Sequence and conservation of genes at the distal end of the transfer region on plasmids F and R6-5. Gene 1991, 104, 85–90. [Google Scholar] [CrossRef]
- Clarke, M.; Maddera, L.; Harris, R.L.; Silverman, P.M. F-pili dynamics by live-cell imaging. Proc. Natl. Acad. Sci. USA 2008, 105, 17978–17981. [Google Scholar] [CrossRef] [Green Version]
- Skerker, J.M.; Berg, H.C. Direct observation of extension and retraction of type IV pili. Proc. Natl. Acad. Sci. USA 2001, 98, 6901. [Google Scholar] [CrossRef] [Green Version]
- Silverman, P.M.; Clarke, M.B. New insights into F-pilus structure, dynamics, and function. Integr. Biol. 2010, 2, 25–31. [Google Scholar] [CrossRef]
- Folkhard, W.; Leonard, K.R.; Malsey, S.; Marvin, D.A.; Dubochet, J.; Engel, A.; Achtman, M.; Helmuth, R. X-ray diffraction and electron microscope studies on the structure of bacterial F pili. J. Mol. Biol. 1979, 130, 145–160. [Google Scholar] [CrossRef]
- Deltoro, D.; Ortiz, D.; Ordyan, M.; Sippy, J.; Oh, C.S.; Keller, N.; Feiss, M.; Catalano, C.E.; Smith, D.E. Walker-A Motif Acts to Coordinate ATP Hydrolysis with Motor Output in Viral DNA Packaging. J. Mol. Biol. 2016, 428, 2709–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirasu, K.; Kado, C.I. Membrane location of the Ti plasmid VirB proteins involved in the biosynthesis of a pilin-like conjugative structure on Agrobacterium tumefaciens. FEMS Microbiol. Lett. 1993, 111, 287–293. [Google Scholar] [CrossRef]
- Anthony, K.G.; Kathir, P.; Moore, D.; Ippen-Ihler, K.; Frost, L.S. Analysis of the traLEKBP sequence and the TraP protein from three F-like plasmids: F, R100-1 and ColB2. J. Bacteriol. 1996, 178, 3194–3200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willetts, N.S. Characterization of the F transfer cistron, traL. Genet. Res. Camb. 1973, 21, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Firth, N.; Skurray, R. Characterization of the F plasmid bifunctional conjugation gene, traG. Mol. Gen. Genet. 1992, 232, 145–153. [Google Scholar] [CrossRef]
- Audette, G.F.; Manchak, J.; Beatty, P.; Klimke, W.A.; Frost, L.S. Entry exclusion in F-like plasmids requires intact TraG in the donor that recognizes its cognate TraS in the recipient. Microbiology 2007, 153, 442–451. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.L.; Hombs, V.; Silverman, P.M. Evidence that F-plasmid proteins TraV, TraK and TraB assemble into an envelope-spanning structure in Escherichia coli. Mol. Microbiol. 2001, 42, 757–766. [Google Scholar] [CrossRef]
- Anthony, K.G.; Klimke, W.A.; Manchak, J.; Frost, L.S. Comparison of proteins involved in pilus synthesis and mating pair stabilization from the related plasmids F and R100-1: Insights into the mechanism of conjugation. J. Bacteriol. 1999, 181, 5149–5159. [Google Scholar] [CrossRef] [Green Version]
- Deng, W.L.; Huang, H.C. Cellular locations of Pseudomonas syringae pv. syringae HrcC and HrcJ proteins, required for harpin secretion via the type III pathway. J. Bacteriol. 1999, 181, 2298–2301. [Google Scholar] [CrossRef] [Green Version]
- Lawley, T.D.; Gilmour, M.W.; Gunton, J.E.; Tracz, D.M.; Taylor, D.E. Functional and mutational analysis of conjugative transfer region 2 (Tra2) from the IncHI1 plasmid R27. J. Bacteriol. 2003, 185, 581–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron, C.; OCallaghan, D.; Lanka, E. Bacterial secrets of secretion: EuroConference on the biology of type IV secretion processes. Mol. Microbiol. 2002, 43, 1359–1365. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.L.; Silverman, P.M. Tra proteins characteristic of F-like type IV secretion systems constitute an interaction group by yeast two-hybrid analysis. J. Bacteriol. 2004, 186, 5480–5485. [Google Scholar] [CrossRef] [Green Version]
- Lento, C.; Ferraro, M.; Wilson, D.; Audette, G.F. HDX-MS and deletion analysis of the type 4 secretion system protein TraF from the Escherichia coli F plasmid. FEBS Lett. 2016, 590, 376–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arutyunov, D.; Arenson, B.; Manchak, J.; Frost, L.S. F Plasmid TraF and TraH Are Components of an Outer Membrane Complex Involved in Conjugation. J. Bacteriol. 2010, 192, 1730–1734. [Google Scholar] [CrossRef] [Green Version]
- Hemmis, C.W.; Schildbach, J.F. Thioredoxin-like proteins in F and other plasmid systems. Plasmid 2013, 70, 168–189. [Google Scholar] [CrossRef] [Green Version]
- Hemmis, C.W.; Berkmen, M.; Eser, M.; Schildbach, J.F. TrbB from Conjugative Plasmid F Is a Structurally Distinct Disulfide Isomerase That Requires DsbD for Redox State Maintenance. J. Bacteriol. 2011, 193, 4588–4597. [Google Scholar] [CrossRef] [Green Version]
- Elton, T.C.; Holland, S.J.; Frost, L.S.; Hazes, B. F-like type IV secretion systems encode proteins with thioredoxin folds that are putative DsbC homologues. J. Bacteriol. 2005, 187, 8267–8277. [Google Scholar] [CrossRef] [Green Version]
- Schandel, K.A.; Muller, M.M.; Webster, R.E. Localization of TraC, a protein involved in assembly of the F conjugative pilus. J. Bacteriol. 1992, 174, 3800–3806. [Google Scholar] [CrossRef] [Green Version]
- Maneewannakul, S.; Kathir, P.; Moore, D.; Le, L.A.; Wu, J.H.; Ippen-Ihler, K. Location of F plasmid transfer operon genes traC and traW and identification of the traW product. J. Bacteriol. 1987, 169, 5119–5124. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N. Intrinsically disordered proteins in overcrowded milieu: Membrane-less organelles, phase separation, and intrinsic disorder. Curr. Opin. Struct. Biol. 2017, 44, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Markossian, K.A.; Kurganov, B.I. Review: Protein folding, misfolding, and aggregation. Formation of inclusion bodies and aggresomes. Biokhimiya 2004, 69, 1196–1212. [Google Scholar]
- Walldén, K.; Williams, R.; Yan, J.; Lian, P.W.; Wang, L.; Thalassinos, K.; Orlova, E.V. Structure of the VirB4 ATPase, alone and bound to the core complex of a type IV secretion system. Proc. Natl. Acad. Sci. USA 2012, 109, 11348–11353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maneewannakul, S.; Maneewannakul, K.; Ippen-Ihler, K. Characterization, localization, and sequence of F transfer region products: The pilus assembly gene product TraW and a new product, TrbI. J. Bacteriol. 1992, 174, 5567–5574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shala-Lawrence, A.; Bragagnolo, N.; Nowroozi-Dayeni, R.; Kheyson, S.; Audette, G.F. The interaction of TraW and TrbC is required to facilitate conjugation in F-like plasmids. Biochem. Biophys. Res. Commun. 2018, 503, 2386–2392. [Google Scholar] [CrossRef]
- Maneewannakul, S.; Maneewannakul, K.; Ippen-Ihler, K. Characterization of trbC, a new F plasmid tra operon gene that is essential to conjugative transfer. J. Bacteriol. 1991, 173, 3872–3878. [Google Scholar] [CrossRef] [Green Version]
- Moore, D.; Sowa, B.A.; Ippen-Ihler, K. The effect of tra mutations on the synthesis of the F-pilin membrane polypeptide. Mol. Gen. Genet. 1981, 184, 260–264. [Google Scholar] [CrossRef]
- Smillie, C.; Garcillán-Barcia, M.P.; Francia, M.V.; Rocha, E.P.C.; de la Cruz, F. Mobility of Plasmids. Microbiol. Mol. Biol. Rev. 2010, 74, 434–452. [Google Scholar] [CrossRef] [Green Version]
- Schröder, G.; Lanka, E. The mating pair formation system of conjugative plasmids—A versatile secretion machinery for transfer of proteins and DNA. Plasmid 2005, 54, 1–25. [Google Scholar] [CrossRef]
- Klimke, W.A.; Rypien, C.D.; Klinger, B.; Kennedy, R.A.; Rodriguez-Maillard, J.M.; Frost, L.S. The mating pair stabilization protein, TraN, of the F plasmid is an outer-membrane protein with two regions that are important for its function in conjugation. Microbiology 2005, 151, 3527–3540. [Google Scholar] [CrossRef] [Green Version]
- Dunny, G.M.; Leonard, B.A.B. Cell-cell communication in gram-positive bacteria. Annu. Rev. Microbiol. 1997, 51, 527–564. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, T.; Komano, T. Mutational analysis of plasmid R64 thin pilus prepilin: The entire prepilin sequence is required for processing by type IV prepilin peptidase. J. Bacteriol. 1998, 180, 4613–4620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achtman, M.; Kennedy, N.; Skurray, R. Cell-cell interactions in conjugating Escherichia coli: Role of traT protein in surface exclusion. Proc. Natl. Acad. Sci. USA 1977, 74, 5104–5108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klimke, W.A.; Frost, L.S. Genetic analysis of the role of the transfer gene, traN, of the F and R100-1 plasmids in mating pair stabilization during conjugation. J. Bacteriol. 1998, 180, 4036–4043. [Google Scholar] [CrossRef] [Green Version]
- Fairman, J.W.; Noinaj, N.; Buchanan, S.K. The structural biology of β-barrel membrane proteins: A summary of recent reports. Curr. Opin. Struct. Biol. 2011, 21, 523–531. [Google Scholar] [CrossRef] [Green Version]
- Holtz, K.M.; Robinson, P.S.; Matthews, E.E.; Hashimoto, Y.; McPherson, C.E.; Khramtsov, N.; Reifler, M.J.; Meghrous, J.; Rhodes, D.G.; Cox, M.M.; et al. Modifications of cysteine residues in the transmembrane and cytoplasmic domains of a recombinant hemagglutinin protein prevent cross-linked multimer formation and potency loss. BMC Biotechnol. 2014, 14, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Maneewannakul, S.; Kathir, P.; Ippen-Ihler, K. Characterization of the F plasmid mating aggregation gene traN and of a new F transfer region locus trbE. J. Mol. Biol. 1992, 225, 299–311. [Google Scholar] [CrossRef]
- Lento, C.; Wilson, D.J.; Audette, G.F. Dimerization of the type IV pilin from Pseudomonas aeruginosa strain K122-4 results in increased helix stability as measured by time-resolved hydrogen-deuterium exchange. Struct. Dyn. 2016, 3, 012001. [Google Scholar] [CrossRef] [Green Version]
- Willetts, N.; Achtman, M. Genetic analysis of transfer by the Escherichia coli sex factor F, using P1 transductional complementation. J. Bacteriol. 1972, 110, 843–851. [Google Scholar] [CrossRef] [Green Version]
- Silhavy, T.J.; Kahne, D.; Walker, S. The bacterial cell envelope. Cold Spring Harb. Perspect. Biol. 2010, 2, a000414. [Google Scholar] [CrossRef]
- Marrero, J.; Waldor, M.K. Interactions between Inner Membrane Proteins in Donor and Recipient Cells Limit Conjugal DNA Transfer. Dev. Cell 2005, 8, 963–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valvekens, D.; Van Montagu, M.; Van Lijsebettens, M. Agrobacterium tumefaciens-mediated transformation of Arabidopsis thaliana root explants by using kanamycin selection. Proc. Natl. Acad. Sci. USA 1988, 85, 5536–5540. [Google Scholar] [CrossRef] [Green Version]
- Christie, P.J. Type IV secretion: The Agrobacterium VirB/D4 and related conjugation systems. Biochim. Biophys. Acta Mol. Cell Res. 2004, 1694, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.; Ward, D.; Middleton, R.; Grossmann, J.G.; Zambryski, P.C. Agrobacterium tumefaciens VirB8 structure reveals potential protein-protein interaction sites. Proc. Natl. Acad. Sci. USA 2006, 103, 2582–2587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.A.; Coinon, M.; Paschos, A.; Jolicoeur, B.; Lavallée, P.; Sygusch, J.; Baron, C. Identification of the binding site of Brucella VirB8 interaction inhibitors. Chem. Biol. 2012, 19, 1041–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillespie, J.J.; Phan, I.Q.H.; Scheib, H.; Subramanian, S.; Edwards, T.E.; Lehman, S.S.; Piitulainen, H.; Sayeedur Rahman, M.; Rennoll-Bankert, K.E.; Staker, B.L.; et al. Structural Insight into How Bacteria Prevent Interference between Multiple Divergent Type IV Secretion Systems. MBio 2015, 6, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Fercher, C.; Probst, I.; Kohler, V.; Goessweiner-Mohr, N.; Arends, K.; Grohmann, E.; Zangger, K.; Meyer, N.H.; Keller, W. VirB8-like protein TraH is crucial for DNA transfer in Enterococcus faecalis. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Casu, B.; Mary, C.; Sverzhinsky, A.; Fouillen, A.; Nanci, A.; Baron, C. VirB8 homolog TraE from plasmid pKM101 forms a hexameric ring structure and interacts with the VirB6 homolog TraD. Proc. Natl. Acad. Sci. USA 2018, 115, 5950–5955. [Google Scholar] [CrossRef] [Green Version]
- Moore, D.; Maneewannakul, K.; Maneewannakul, S.; Wu, J.H.; Ippen-Ihler, K.; Bradley, D.E. Characterization of the F-plasmid conjugative transfer gene traU. J. Bacteriol. 1990, 172, 4263–4270. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.H.; Kathir, P.; Ippen-Ihler, K. The product of the F plasmid transfer operon gene, traF, is a periplasmic protein. J. Bacteriol. 1988, 170, 3633–3639. [Google Scholar] [CrossRef] [Green Version]
- Fekete, R.A.; Frost, L.S. Mobilization of chimeric oriT plasmids by F and R100-1: Role of relaxosome formation in defining plasmid specificity. J. Bacteriol. 2000, 182, 4022–4027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragonese, H.; Haisch, D.; Villareal, E.; Choi, J.H.; Matson, S.W. The F plasmid-encoded TraM protein stimulates relaxosome-mediated cleavage at oriT through an interaction with TraI. Mol. Microbiol. 2007, 63, 1173–1184. [Google Scholar] [CrossRef]
- Karl, W.; Bamberger, M.; Zechner, E.L. Transfer protein TraY of plasmid R1 stimulates TraI-catalyzed oriT cleavage in vivo. J. Bacteriol. 2001, 183, 909–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dostál, L.; Schildbach, J.F. Single-stranded DNA binding by F TraI relaxase and helicase domains is coordinately regulated. J. Bacteriol. 2010, 192, 3620–3628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Gao, Q.; Deonier, R.C. Mutational and physical analysis of F plasmid traY protein binding to oriT. Mol. Microbiol. 1994, 11, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.T.; Nelson, W.C.; Matson, S.W. Stepwise assembly of a relaxosome at the F plasmid origin of transfer. J. Biol. Chem. 1995, 270, 28381–28386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haft, R.J.F.; Palacios, G.; Nguyen, T.; Mally, M.; Gachelet, E.G.; Zechner, E.L.; Traxler, B. General Mutagenesis of F Plasmid TraI Reveals Its Role in Conjugative Regulation. J. Bacteriol. 2006, 188, 6346–6353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matson, S.W.; Ragonese, H. The F-plasmid Tral protein contains three functional domains required for conjugative DNA strand transfer. J. Bacteriol. 2005, 187, 697–706. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.J.W.; Lu, J.; Glover, J.N.M. Relaxosome function and conjugation regulation in F-like plasmids—A structural biology perspective. Mol. Microbiol. 2012, 85, 602–617. [Google Scholar] [CrossRef]
- Guogas, L.M.; Kennedy, S.A.; Lee, J.H.; Redinbo, M.R. A Novel Fold in the TraI Relaxase-Helicase C-Terminal Domain Is Essential for Conjugative DNA Transfer. J. Mol. Biol. 2009, 386, 554–568. [Google Scholar] [CrossRef] [Green Version]
- Dostal, L.; Shao, S.; Schildbach, J.F. Tracking F plasmid TraI relaxase processing reactions provides insight into F plasmid transfer. Nucleic Acids Res. 2011, 39, 2658–2670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, S.; Larkin, C.; Schildbach, J.F. Structural Insights into Single-Stranded DNA Binding and Cleavage by F Factor TraI. Structure 2003, 11, 1369–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, C.; Datta, S.; Harley, M.J.; Anderson, B.J.; Ebie, A.; Hargreaves, V.; Schildbach, J.F. Inter- and Intramolecular Determinants of the Specificity of Single-Stranded DNA Binding and Cleavage by the F Factor Relaxase. Structure 2005, 13, 1533–1544. [Google Scholar] [CrossRef] [PubMed]
- Boer, R.; Russi, S.; Guasch, A.; Lucas, M.; Blanco, A.G.; Pérez-Luque, R.; Coll, M.; de la Cruz, F. Unveiling the Molecular Mechanism of a Conjugative Relaxase: The Structure of TrwC Complexed with a 27-mer DNA Comprising the Recognition Hairpin and the Cleavage Site. J. Mol. Biol. 2006, 358, 857–869. [Google Scholar] [CrossRef]
- Wong, J.J.W.; Lu, J.; Edwards, R.A.; Frost, L.S.; Glover, J.N.M. Structural basis of cooperative DNA recognition by the plasmid conjugation factor, TraM. Nucleic Acids Res. 2011, 39, 6775–6788. [Google Scholar] [CrossRef]
- Disqué-Kochem, C.; Dreiseikelmann, B. The cytoplasmic DNA-binding protein TraM binds to the inner membrane protein TraD in vitro. J. Bacteriol. 1997, 179, 6133–6137. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Wong, J.J.W.; Edwards, R.A.; Manchak, J.; Frost, L.S.; Glover, J.N.M. Structural basis of specific TraD-TraM recognition during F plasmid-mediated bacterial conjugation. Mol. Microbiol. 2008, 70, 89–99. [Google Scholar] [CrossRef]
- Beranek, A.; Zettl, M.; Lorenzoni, K.; Schauer, A.; Manhart, M.; Koraimann, G. Thirty-eight C-terminal amino acids of the coupling protein TraD of the F-like conjugative resistance plasmid R1 are required and sufficient to confer binding to the substrate selector protein TraM. J. Bacteriol. 2004, 186, 6999–7006. [Google Scholar] [CrossRef] [Green Version]
- Gomis-Rüth, F.X.; Moncalián, G.; Pérez-Luque, R.; González, A.; Cabezón, E.; De La Cruz, F.; Coll, M. The bacterial conjugation protein TrwB resembles ring helicases and F1-atpase. Nature 2001, 409, 637–641. [Google Scholar] [CrossRef]
- Garcillán-Barcia, M.P.; de la Cruz, F. Why is entry exclusion an essential feature of conjugative plasmids? Plasmid 2008, 60, 1–18. [Google Scholar] [CrossRef]
- Achtman, M.; Manning, P.A.; Kusecek, B.; Schwuchow, S.; Neil, W. Genetic analysis of F sex factor cistrons needed for surface exclusion in Escherichia coli. J. Mol. Biol. 1980, 138, 779–795. [Google Scholar] [CrossRef]
- Ou, J.T. Role of surface exclusion genes in lethal zygosis in Escherichia coli K12 mating. MGG Mol. Gen. Genet. 1980, 178, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Skurray, R.A.; Reeves, P. Characterization of lethal zygosis associated with conjugation in Escherichia coli K-12. J. Bacteriol. 1973, 113, 58–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perumal, N.B.; Minkley, E.G.J. The product of the F sex factor traT surface exclusion gene is a lipoprotein. J. Biol. Chem. 1984, 259, 5357–5360. [Google Scholar] [PubMed]
- Sukupolvi, S.; O’Connor, C.D. TraT lipoprotein, a plasmid-specified mediator of interactions between gram-negative bacteria and their environment. Microbiol. Rev. 1990, 54, 331–341. [Google Scholar] [CrossRef]
- Minkley, E.G. Purification and characterization of pro-TraTp, the signal sequence-containing precursor of a secreted protein encoded by the F sex factor. J. Bacteriol. 1984, 158, 464–473. [Google Scholar] [CrossRef] [Green Version]
- Minkley, E.G.; Willetts, N.S. Overproduction, purification and characterization of the F traT protein. Mol. Gen. Genet. MGG 1984, 196, 225–235. [Google Scholar] [CrossRef]
- Manning, P.A.; Timmis, J.K.; Moll, A.; Timmis, K.N. Mutants that overproduce TraTp, a plasmid-specified major outer membrane protein of Escherichia coli. MGG Mol. Gen. Genet. 1982, 187, 426–431. [Google Scholar] [CrossRef]
- Harrison, J.L.; Taylor, I.M.; Platt, K.; O’Connor, C.D. Surface exclusion specificity of the TraT lipoprotein is determined by single alterations in a five-amino-acid region of the protein. Mol. Microbiol. 1992, 6, 2825–2832. [Google Scholar] [CrossRef]
- Riede, I.; Eschbach, M.L. Evidence that TraT interacts with OmpA of Escherichia coli. FEBS Lett. 1986, 205, 241–245. [Google Scholar] [CrossRef] [Green Version]
- Anthony, K.G.; Sherburne, C.; Sherburne, R.; Frost, L.S. The role of the pilus in recipient cell recognition during bacterial conjugation mediated by F-like plasmids. Mol. Microbiol. 1994, 13, 939–953. [Google Scholar] [CrossRef] [PubMed]
- Achtman, M. Mating aggregates in Escherichia coli conjugation. J. Bacteriol. 1975, 123, 505–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalajakumari, M.B.; Guidolin, A.; Buhk, H.J.; Manning, P.A.; Ham, L.M.; Hodgson, A.L.M.; Cheah, K.C.; Skurray, R.A. Surface exclusion genes traS and traT of the F sex factor of Escherichia coli K-12. Determination of the nucleotide sequence and promoter and terminator activities. J. Mol. Biol. 1987, 198, 1–11. [Google Scholar] [CrossRef]
- Achtman, M.; Manning, P.A.; Edelbluth, C.; Herrlich, P. Export without proteolytic processing of inner and outer membrane proteins encoded by F sex factor tra cistrons in Escherichia minicells. Proc. Natl. Acad. Sci. USA 1979, 76, 4837–4841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oluwadare, M.; Lee, M.D.; Grim, C.J.; Lipp, E.K.; Cheng, Y.; Maurer, J.J. The Role of the Salmonella spvB IncF Plasmid and Its Resident Entry Exclusion Gene traS on Plasmid Exclusion. Front. Microbiol. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Uversky, V.N.; Kurgan, L. Disordered nucleiome: Abundance of intrinsic disorder in the DNA- and RNA-binding proteins in 1121 species from Eukaryota, Bacteria and Archaea. Proteomics 2016, 16, 1486–1498. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Perumal, N.B.; Oldfield, C.J.; Su, E.W.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder in transcription factors. Biochemistry 2006, 45, 6873–6888. [Google Scholar] [CrossRef] [Green Version]
- Nash, R.P.; McNamara, D.E.; Keith Ballentine, W.; Matson, S.W.; Redinbo, M.R. Investigating the impact of bisphosphonates and structurally related compounds on bacteria containing conjugative plasmids. Biochem. Biophys. Res. Commun. 2012, 424, 697–703. [Google Scholar] [CrossRef] [Green Version]
- Garcillán-Barcia, M.P.; Jurado, P.; González-Pérez, B.; Moncalián, G.; Fernández, L.A.; De La Cruz, F. Conjugative transfer can be inhibited by blocking relaxase activity within recipient cells with intrabodies. Mol. Microbiol. 2007, 63, 404–416. [Google Scholar] [CrossRef]
- Casu, B.; Smart, J.; Hancock, M.A.; Smith, M.; Sygusch, J.; Baron, C. Structural Analysis and inhibition of TraE from the pKM101 type IV secretion system. J. Biol. Chem. 2016, 291, 23817–23829. [Google Scholar] [CrossRef] [Green Version]
- Graf, F.E.; Palm, M.; Warringer, J.; Farewell, A. Inhibiting conjugation as a tool in the fight against antibiotic resistance. Drug Dev. Res. 2019, 80, 19–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ripoll-Rozada, J.; García-Cazorla, Y.; Getino, M.; Machón, C.; Sanabria-Ríos, D.; de la Cruz, F.; Cabezón, E.; Arechaga, I. Type IV traffic ATPase TrwD as molecular target to inhibit bacterial conjugation. Mol. Microbiol. 2016, 100, 912–921. [Google Scholar] [CrossRef]
- Holliger, P.; Riechmann, L.; Williams, R.L. Crystal structure of the two N-terminal domains of g3p from filamentous phage fd at 1.9 Å: Evidence for conformational lability. J. Mol. Biol. 1999, 288, 649–657. [Google Scholar] [CrossRef] [Green Version]
- Novotny, C.; Knight, W.S.; Brinton, C.C. Inhibition of bacterial conjugation by ribonucleic acid and deoxyribonucleic acid male-specific bacteriophages. J. Bacteriol. 1968, 95, 314–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, A.; Jimenez, J.; Derr, J.; Vera, P.; Manapat, M.L.; Esvelt, K.M.; Villanueva, L.; Liu, D.R.; Chen, I.A. Inhibition of bacterial conjugation by phage M13 and its protein g3p: Quantitative analysis and model. PLoS ONE 2011, 6, e19991. [Google Scholar] [CrossRef]
- Shaffer, C.L.; Good, J.A.D.; Kumar, S.; Syam Krishnan, K.; Gaddy, J.A.; Loh, J.T.; Chappell, J.; Almqvist, F.; Cover, T.L.; Hadjifrangiskou, M. Peptidomimetic small molecules disrupt type IV secretion system activity in diverse bacterial pathogens. MBio 2016, 7, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y. Protein structure prediction: When is it useful? Curr. Opin. Struct. Biol. 2009, 19, 145–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tompa, P.; Fuxreiter, M. Fuzzy complexes: Polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 2008, 33, 2–8. [Google Scholar] [CrossRef]
- Hegyi, H.; Schad, E.; Tompa, P. Structural disorder promotes assembly of protein complexes. BMC Struct. Biol. 2007, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Highest Confidence Model | |||||||
|---|---|---|---|---|---|---|---|
| Function | Protein | Localization | Size (aa) | % Disordered | Sequences | % Identity | PDB |
| Transcription Regulation | FinO | Cytoplasm | 186 | 42% | 33–184 | 100% | 1DVO |
| TraJ | Cytoplasm | 229 | 21% | 15–128 | 100% | 4KQD | |
| TraR | Cytoplasm | 73 | 19% | 4–73 | 100% | 5W1S | |
| ProPilin Maturation | TraA | IM, ECM | 70 | 20% | 21–41 | 29% | 2L8S |
| TraQ | IM | 94 | 22% | 32–64 | 46% | 3MP7 | |
| TraX | IM | 248 | 13% | 28–37 | 24% | 2KP6 | |
| T4SS Core Proteins | TraB | IM, Periplasm | 475 | 44% | 203–412 | 14% | 6GYB |
| TraK | OM, Periplasm | 242 | 34% | 26–107 | 8% | 6GYB | |
| TraV | IM | 171 | 61% | 25–36 | 42% | 5V8K | |
| Pilus Assembly/Extension | TraP | IM | 196 | 24% | 57–118 | 18% | 2V4J |
| TraE | IM | 188 | 11% | 93–182 | 16% | 5KPE | |
| TraL | IM | 91 | 13% | 18–47 | 30% | 3GEB | |
| TraC | Cytoplasm, IM | 875 | 15% | 448–819 | 18% | 4AG5 | |
| TraW | Periplasm | 210 | 16% | 100–171 | 20% | 2FU3 | |
| TrbC | Periplasm | 212 | 29% | 103–190 | 13% | 1HYU | |
| TraF | Periplasm | 247 | 17% | 146–256 | 19% | 6GC1 | |
| TrbB | Periplasm | 181 | 31% | 33–175 | 19% | 2HYX | |
| Pilus Retraction | TraH | OM, Periplasm | 458 | 25% | 90–144 | 24% | 6SZ9 |
| TrbI | IM, Periplasm | 128 | 23% | 47–120 | 81% | 1U2M | |
| Mating Pair Stabilization | TraN | OM, Periplasm | 602 | 25% | 127–408 | 98% | 4XBM |
| TraG | IM, Periplasm | 938 | 43% | 774–930 | 93% | 3CWG | |
| TraU | Periplasm | 330 | 26% | 31–39 | 26% | 3TOW | |
| Relaxase | TraI | Cytoplasm | 1756 | 20% | 1–1473 | 98% | 5N8O |
| Relaxosome Accessory | TraY | Cytoplasm | 131 | 25% | 1–51 | 25% | 1U9P |
| TraM | Cytoplasm | 127 | 33% | 2–121 | 39% | 3ON0 | |
| TraD | Cytoplasm, IM | 717 | 29% | 145–574 | 30% | 1E9R | |
| Surface Exclusion | TraT | OM, ECM | 244 | 34% | 52–98 | 15% | 3BF2 |
| Entry Exclusion | TraS | IM | 173 | 8% | 22–35 | 38% | 2M4V |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bragagnolo, N.; Rodriguez, C.; Samari-Kermani, N.; Fours, A.; Korouzhdehi, M.; Lysenko, R.; Audette, G.F. Protein Dynamics in F-like Bacterial Conjugation. Biomedicines 2020, 8, 362. https://doi.org/10.3390/biomedicines8090362
Bragagnolo N, Rodriguez C, Samari-Kermani N, Fours A, Korouzhdehi M, Lysenko R, Audette GF. Protein Dynamics in F-like Bacterial Conjugation. Biomedicines. 2020; 8(9):362. https://doi.org/10.3390/biomedicines8090362
Chicago/Turabian StyleBragagnolo, Nicholas, Christina Rodriguez, Naveed Samari-Kermani, Alice Fours, Mahboubeh Korouzhdehi, Rachel Lysenko, and Gerald F. Audette. 2020. "Protein Dynamics in F-like Bacterial Conjugation" Biomedicines 8, no. 9: 362. https://doi.org/10.3390/biomedicines8090362
APA StyleBragagnolo, N., Rodriguez, C., Samari-Kermani, N., Fours, A., Korouzhdehi, M., Lysenko, R., & Audette, G. F. (2020). Protein Dynamics in F-like Bacterial Conjugation. Biomedicines, 8(9), 362. https://doi.org/10.3390/biomedicines8090362